
Abstract
Marine sponges of the class Hexactinellida (glass sponges) are among the most understudied groups of Porifera, and molecular approaches to investigating their evolution have only recently emerged. Although these first results appeared reliable as they largely corroborated morphology-based hypotheses, they were almost exclusively based on ribosomal RNA genes (rDNA) and should, therefore, be further tested with independent types of genetic data, such as protein-coding genes. To this end, we established the mitochondrial-encoded cytochrome oxidase subunit I gene (COI) as an additional marker, and conducted phylogenetic analyses on DNA- and amino-acid level, as well as a supermatrix analysis based on combined COI DNA and rDNA alignments. Furthermore, we increased taxon sampling compared to previous studies by adding seven additional species. The COI-based phylogenies were largely congruent with the rDNA-based phylogeny but suffered from poor bootstrap support for many nodes. However, addition of the COI sequences to the rDNA data set increased resolution of the overall molecular phylogeny. Thus, although obtaining COI sequences from glass sponges turned out to be quite challenging, this gene appears to be a valuable supplement to rDNA data for molecular evolutionary studies of this group. Some implications of our extended phylogeny for the evolution and systematics of Hexactinellida are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A robust and comprehensive reconstruction of the poriferan Tree of Life is of prime importance for sponge science (and beyond), because all aspects of sponge biology can be best understood in light of the evolutionary context in which the past and current diversity of these animals emerged. Molecular phylogenetics certainly constitutes the most promising approach for attaining this goal, and progress in this field has been rapid over the last two decades or so (Erpenbeck & Wörheide, 2007). However, many gaps in phylogenetic knowledge remain to be filled, and it is a further, much greater challenge to fully reconcile morphology-based taxonomy with molecular phylogenies of Porifera.
With respect to the latter, one of the most under-studied groups, the glass sponges (class Hexactinellida; see Leys et al., 2007 for a comprehensive review), may provide the best chances for establishing a systematics that integrates both morphological and molecular information. This is because the glass sponges appear to be an exceptional case, as compared to other sponges (e.g., Dohrmann et al., 2006), where the first published molecular systematic results were largely in line with morphological predictions (Dohrmann et al., 2008, 2009). Nonetheless, some results remain ambiguous, and the monophyly of certain taxa could not be tested due to the lack of sequence data for more than one species. Furthermore, these phylogenies were exclusively based on three ribosomal RNA genes (rDNA)—nuclear 18S, partial nuclear 28S, and partial mitochondrial 16S—and should, therefore, be tested with independent molecular markers.
While a number of protein-coding sequences have been published for a few hexactinellid species in nonphylogenetic studies (e.g., Gundacker et al., 2001; Bebenek et al., 2004; Manuel et al., 2004; Conejo et al., 2008; Rosengarten et al., 2008), molecular phylogenetic studies including this kind of data from glass sponges are scarce (e.g., Borchiellini et al., 1998; Rokas et al., 2003; Haen et al., 2007; Philippe et al., 2009; Sperling et al., 2009), and based on a very limited taxon sampling of Hexactinellida since they did not aim at reconstructing the internal relationships of this group. We, therefore, decided to establish the mitochondrial-encoded cytochrome oxidase subunit I gene (COI) as an additional marker, because (a) this gene is widely regarded as an easily amplifiable “standard” gene for molecular evolutionary studies, and (b) COI sequence data might be useful for other applications besides systematics, such as molecular species identification (“barcoding”; see Bucklin et al., 2011 for a recent review). We also increased taxonomic sampling of hexactinellids by including seven previously unsampled species, and discuss our new results in light of the current taxonomy of the group.
Materials and methods
We added nine additional specimens, seven of which belong to previously unsampled species (Table 1), to the taxon set reported in Dohrmann et al. (2009). 18S, 28S, and 16S rDNA sequences were obtained as previously described (Dohrmann et al., 2008). COI sequences spanning the “Folmer-“ and the I3-M11 regions (cf. Erpenbeck et al., 2006; ca. 1.3 kb) were amplified using various combinations of mostly degenerate primers (Supplementary Table S1), Promega’s GoTaq (reaction mixes as in Dohrmann et al., 2008), and “touch-down” thermal regimes with final annealing temperatures of 45 or 30°C. Since amplification of this complete region was only rarely successful, 5’- and 3’-halves had to be amplified separately in most cases. To obtain sequences, amplicons were further processed as described (Dohrmann et al., 2008). COI sequences from Regadrella sp., Acanthascus dawsoni, and Oopsacas minuta were taken from ongoing mitochondrial genome sequencing projects (Haen & Lavrov, in prep.); those of Iphiteon panicea, Sympagella nux, and Aphrocallistes vastus were downloaded from GenBank and served as initial templates for primer design (cf. Table S1). Supplementary Table S2 gives an overview of the data set and accession numbers for the newly generated sequences.
Ribosomal DNA sequences were manually aligned to previous alignments (Dohrmann et al., 2009), aided by RNA secondary structure in case of 18S and 28S (cf. Dohrmann et al., 2008); ambiguous regions were excluded from the phylogenetic analysis. COI sequences were pre-aligned in ClustalX 2.0 (Larkin et al., 2007), followed by manual refinement. The COI alignment was largely unambiguous, but contained several instances of single species, 1-bp insertions that were either sequencing errors or putative +1 translational frameshifts (Haen et al., 2007; Rosengarten et al., 2008); these sites were removed.
Preliminary analyses recovered essentially the same relationships among nonbilaterian animals as reported in Dohrmann et al. (2008); however, topology and support values for Hexactinellida were not markedly affected when the outgroups were excluded (results not shown). Therefore, we did not include any nonhexactinellid sequences in the final analyses, instead designating the six amphidiscophorans as a multi-species outgroup. This is justified because monophyly of Hexactinellida and its two subclasses, Amphidiscophora and Hexasterophora, is beyond doubt (see Dohrmann et al., 2008), and the deep divergence between the latter two taxa makes them ideal outgroups for each other. Furthermore, investigating relationships between the major nonbilaterian animal lineages is beyond the scope of this article and should better be approached with different, e.g. phylogenomic, data sets (see Philippe et al., 2009; Pick et al., 2010).
Phylogenetic analyses of the COI DNA alignment, the concatenated rDNA alignment, and a supermatrix (cf. de Queiroz & Gatesy, 2007) of all four partitions were conducted in a maximum-likelihood (ML) framework as implemented in RAxML (Stamatakis, 2006) 7.2.6 (http://wwwkramer.in.tum.de/exelixis/software.html), using the Pthreads-parallelized version on a 64-bit Linux cluster at the Molecular Geo- and Palaeobiology Lab, LMU Munich. For the combined rDNA (3328 bp) and supermatrix (4582 bp) analyses, the markers were concatenated in SeaView 4.0 (Gouy et al., 2010) and analyzed under mixed substitution models. Because, in contrast to the previously used Bayesian Markov Chain Monte Carlo (BMCMC) application PHASE (see Dohrmann et al., 2008, 2009), computational limitations are not an issue with RAxML, the least simplifying models could be explored, namely the 16-state paired-sites model (cf. Savill et al., 2001) S16 for 18S + 28S double-stranded regions (stems), and independent GTR models (Lanave et al., 1984) for 18S single-stranded regions (loops), 28S loops, 16S, and COI. However, using the 7- and 6-state paired-sites models S7D and S6B, which do not fully account for mismatch pairs (see Savill et al., 2001), but were found best-fitting in the BMCMC framework among the models tested by Dohrmann et al. (2008, 2009), lead to essentially the same results (not shown). Among-site rate variation was modeled for each partition independently using discrete gamma distributions with four rate categories (+G4; Yang, 1994). We also analyzed the COI data on the amino-acid (aa) level, with DNA sequences translated using the hexactinellid-specific mitochondrial genetic code (Haen et al., 2007), and employing the MtRev+F+G4 model of aa replacement, as suggested by ProtTest 2.4 (Abascal et al., 2005) under the Akaike Information Criterion (AIC; Akaike, 1974). In all analyses, clade stability was assessed by rapid bootstrapping (Felsenstein, 1985; Stamatakis et al., 2008) based on 1000 pseudoreplicates.
The final supermatrix and the associated structure-, partition-, and tree files are available at Open Data LMU (http://dx.doi.org/10.5282/ubm/data.40).
Results and discussion
Contrary to our expectations (see Introduction), and for reasons that remain somewhat elusive, obtaining COI sequence data from hexactinellid specimens turned out to be rather challenging. Extremely low annealing temperatures were required to obtain amplicons (see Materials and methods), and in many cases, only very faint bands of target sequences were observed or PCR failed completely for one or both of the fragments (cf. Table S2). Also, different primer combinations worked for different specimens, necessitating that PCRs be optimized individually and no standard protocol could be established after an initial optimization step. Further problems included amplification of nontarget DNA (e.g. prokaryotes; cf. Siddall et al., 2009), multiple bands making gel extraction mandatory, and poor sequence reads leading to nonoverlapping of fragments (cf. Table S2) and requiring increased use of the IUPAC code for ambiguous base calls. Despite these practical difficulties, however, COI proved to be a useful addition to the three established rDNA markers (Dohrmann et al., 2008), as discussed below.
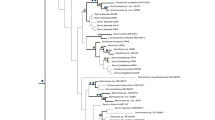
Phylogenies reconstructed from the COI alignments and the combined rDNA alignment, respectively, are largely congruent (Figs. 1, 2, 3), i.e. there are no conflicting clades with high bootstrap support (BS). Despite the overall congruence, many nodes are poorly (BS < 70%) supported in the COI phylogenies, especially in the aa tree (Fig. 2). Strikingly, this is also the case to a lesser extent in the rDNA phylogeny, which appears less robust than the Bayesian trees presented in Dohrmann et al. (2009), even when the different significance levels of bootstrap versus posterior probability values (cf. Hillis & Bull, 1993; Huelsenbeck & Rannala, 2004) are taken into account. For example, support for order Lyssacinosida is very weak and the topology within family Rossellidae is less resolved compared to our previous study. We suspect that these results are due to further methodological and/or implementational differences between RAxML and PHASE, an issue that will be explored elsewhere.

Maximum likelihood phylogeny of Hexactinellida inferred from COI DNA sequence alignment (RAxML; GTR+G4 substitution model). Bootstrap support values (1000 replicates) given at nodes. Previously unsampled species are highlighted in bold. Scale bar, expected number of substitutions per site. See “Materials and methods” section for further details

Maximum likelihood phylogeny of Hexactinellida inferred from COI amino-acid sequence alignment (RAxML; MtRev+F+G4 substitution model). Bootstrap support values (1000 replicates) given at nodes. Previously unsampled species highlighted in bold. Scale bar, expected number of substitutions per site. See “Materials and methods” section for further details

Maximum likelihood phylogeny of Hexactinellida inferred from concatenated 18S, 28S, and 16S rDNA sequence alignments (RAxML; independent GTR+G4 substitution models for 18S loops, 28S loops, and 16S; S16+G4 paired-sites model for 18S+28S stems). Bootstrap support values (1000 replicates) given at nodes. Previously unsampled species highlighted in bold. Scale bar, expected number of substitutions per site. See “Materials and methods” section for further details
Compared to the results of the separate analyses, robustness and resolution is increased when the two data sets are analyzed together (Fig. 4). For example, support for monophyly of Lyssacinosida is only 58 and 55% in the COI DNA and the rDNA phylogeny, respectively, but rises to 75% in the combined tree, which is significant according to Hillis & Bull (1993). Below, we discuss the placement of the previously unsampled species, as well as some other new results, on the basis of the supermatrix tree (Fig. 4). A more in-depth discussion of the phylogeny of the dictyonal, sceptrule-bearing glass sponges (Sceptrulophora), and implications for spicule evolution is provided elsewhere (Dohrmann et al., 2011).

Maximum likelihood phylogeny of Hexactinellida inferred from concatenated rDNA and COI DNA sequence alignments (RAxML; independent GTR+G4 substitution models for 18S loops, 28S loops, 16S, and COI; S16+G4 paired-sites model for 18S+28S stems). Bootstrap support values (1000 replicates) given at nodes. Previously unsampled species are highlighted in bold. Scale bar, expected number of substitutions per site. See “Materials and methods” section for further details. Bold numbers at nodes refer to the following putatively apomorphic morphological characters (for terminology, see Tabachnick & Reiswig, 2002). 1 triaxonic spicules, syncytial soft tissue, 2 amphidiscs, 3 sceptres, 4 hexasters, capability of spicule fusion, 5 sceptrules, euretoid dictyonal frameworks, 6 schizorhyses, bundled arrangement of dermal uncinates, 7 diarhyses, 8 clavules, farreoid dictyonal frameworks, 9 floricomes, 10 hypodermal pentactins
Within Sceptrulophora, we find that Sarostegia oculata does not group with the remaining Farreidae, which form a well-supported clade sister to Aphrocallistidae. Interestingly, Sarostegia is the only farreid with a euretoid dictyonal framework and lacks clavules, a spicule type that is typical for, and restricted to, Farreidae. Although this species does not group with the representative of Euretidae (but see Fig. 3), topology-tests indicate that our supermatrix data are consistent with such a placement, and we thus suggest resurrection of Sarostegia’s earlier classification in Euretidae (Dohrmann et al., 2011).
Although poorly supported here (BS = 64%), the position of Psilocalyx wilsoni as sister to the other two tretodictyids, Hexactinella and Tretodictyum, receives significant support (BS > 75%) when the taxon set is restricted to dictyonal sponges, allowing for the inclusion of additional rDNA positions (Dohrmann et al., 2011). Thus, monophyly of Tretodictyidae (Dohrmann et al., 2008) is further corroborated. It is particularly noteworthy that this morphologically well-characterized taxon (Mehl, 1992; Reiswig, 2002) was not resolved in the COI and rDNA trees, respectively (Figs. 1, 2, 3). These results indicate that considerable numbers of molecular characters may be required to support certain groups, and demonstrate the beneficial effect of supplementing rDNA evidence with COI sequence data.
Within order Lyssacinosida, we were able to resolve the phylogenetic position of Clathrochone clathroclada (see Dohrmann et al., 2009), as sister to Leucopsacidae+Rossellidae [note that this is also recovered in the rDNA tree (Fig. 3) with somewhat weaker support, but is strongly supported in the COI DNA tree (Fig. 1)]. This result rejects our earlier proposal that this species might belong to Leucopsacidae (Dohrmann et al., 2008), and corroborates the hypothesis that it represents an independent evolutionary lineage not belonging to any of the three described families of Lyssacinosida (Tabachnick, 2002a).
Within family Rossellidae, inclusion of Sympagella nux allowed us to test monophyly of subfamily Lanuginellinae, which was so far only represented by a single species, Lophocalyx profundum. Since Lanuginellinae is morphologically well defined by the presence of strobiloplumicomes (Tabachnick, 2002b), we expected that these two species would group together in the molecular phylogeny. Surprisingly, this hypothesis is not supported because Sympagella is resolved as sister to a Caulophacus/Caulophacella/Lophocalyx clade. We consider the convergent evolution of strobiloplumicomes to be unlikely, and speculate that this spicule type was lost in Caulophacus and Caulophacella. Interestingly, a closer relationship of Sympagella, Caulophacus, and Caulophacella is consistent with earlier classification schemes of Rossellidae (see historical discussion in Tabachnick, 2002b; morphological characters supporting this grouping include the presence of a stalk and pinular hexactins or pentactins, with the latter also found among Lophocalyx spp.; however, these features are not unique to these genera). Thus, our results suggest that some abandoned taxonomic hypotheses have to be reconsidered.
Within family Euplectellidae, placement of Regadrella sp. (Corbitellinae) in a nested position within Euplectellinae (here: Euplectella, Docosaccus, Acoelocalyx, and Malacosaccus) again challenges monophyly of the latter subfamily (see Dohrmann et al., 2008, 2009). Thus, none of the three euplectellid subfamilies are currently supported by molecular data, which suggests that the features used to discriminate these taxa, namely the mode of attachment to the substrate (Tabachnick, 2002c), are highly plastic and of limited phylogenetic value (see also discussion in Dohrmann et al., 2009).
Conclusions
Given the technical difficulties we faced in generating COI sequence data from glass sponges, we consider it unlikely that this gene will play a major role in barcoding hexactinellids, since this approach to species identification relies heavily on easily applicable standard protocols that can be used in a high-throughput context (e.g., Ivanova et al., 2009). However, the additional sequence data proved a valuable supplement to rRNA genes for molecular phylogenetics, so the extra-effort that was required for most specimens certainly paid off. The good congruence between COI and rDNA phylogenies indicates that ribosomal RNA- and protein-coding genes harbor the same phylogenetic signal, thus increasing the reliability of molecular approaches for investigating organismal evolution of glass sponges. Combined analysis of the two data sets led to a more robust and resolved tree, providing a basis for further evolutionary studies such as reconstructing morphological character evolution or estimating divergence times. Finally, the increased taxon sampling of the present study provided some further hints as to where the current Linnean classification needs improvement; we are confident that continued addition of key taxa will ultimately help to resolve remaining taxonomic ambiguities, resulting in a system of Hexactinellida that is as natural as possible.
References
Abascal, F., R. Zardoya & D. Posada, 2005. ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21: 2104–2105.
Akaike, H., 1974. A new look at the statistical model identifications. IEEE Transactions on Automatic Control 19: 716–723.
Bebenek, I. G., R. D. Gates, J. Morris, V. Hartenstein & D. K. Jacobs, 2004. sine oculis in basal Metazoa. Development Genes and Evolution 214: 342–351.
Borchiellini, C., N. Boury-Esnault, J. Vacelet & Y. Le Parco, 1998. Phylogenetic analysis of the Hsp70 sequences reveals the monophyly of Metazoa and specific phylogenetic relationships between animals and fungi. Molecular Biology and Evolution 15: 647–655.
Bucklin, A., D. Steinke & L. Blanco-Bercial, 2011. DNA barcoding of marine Metazoa. Annual Review of Marine Science 3: 471–508.
Conejo, M., M. Bertin, S. A. Pomponi & W. R. Ellington, 2008. The early evolution of the phosphagen kinases – insights from choanoflagellate and poriferan arginine kinases. Journal of Molecular Evolution 66: 11–20.
de Queiroz, A. & J. Gatesy, 2007. The supermatrix approach to systematics. Trends in Ecology and Evolution 22: 34–41.
Dohrmann, M., O. Voigt, D. Erpenbeck & G. Wörheide, 2006. Non-monophyly of most supraspecific taxa of calcareous sponges (Porifera, Calcarea) revealed by increased taxon sampling and partitioned Bayesian analysis of ribosomal DNA. Molecular Phylogenetics and Evolution 40: 830–843.
Dohrmann, M., D. Janussen, J. Reitner, A. G. Collins & G. Wörheide, 2008. Phylogeny and evolution of glass sponges (Porifera, Hexactinellida). Systematic Biology 57: 388–405.
Dohrmann, M., A. G. Collins & G. Wörheide, 2009. New insights into the phylogeny of glass sponges (Porifera, Hexactinellida): monophyly of Lyssacinosida and Euplectellinae, and the phylogenetic position of Euretidae. Molecular Phylogenetics and Evolution 52: 257–262.
Dohrmann, M., C. Göcke, D. Janussen, J. Reitner, C. Lüter & G. Wörheide, 2011. Systematics and spicule evolution in dictyonal sponges (Hexactinellida: Sceptrulophora) with description of two new species. Zoological Journal of the Linnean Society (in press).
Erpenbeck, D. & G. Wörheide, 2007. On the molecular phylogeny of sponges (Porifera). Zootaxa 1668: 107–126.
Erpenbeck, D., J. N. A. Hooper & G. Wörheide, 2006. CO1 phylogenies in diploblasts and the ‘Barcoding of Life’ – are we sequencing a suboptimal partition? Molecular Ecology Notes 6: 550–553.
Felsenstein, J., 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39: 783–791.
Göcke, C. & D. Janussen, 2011. ANT XXIV/2 (SYSTCO) Hexactinellida (Porifera) and bathymetric traits of Antarctic glass sponges (incorporating ANDEEP-material); including an emendation of the rediscovered genus Lonchiphora. Deep-Sea Research Part II: in press.
Gouy, M., S. Guindon & O. Gascuel, 2010. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Molecular Biology and Evolution 27: 221–224.
Gundacker, D., S. P. Leys, H. C. Schröder, I. M. Müller & W. E. G. Müller, 2001. Isolation and cloning of a C-type lectin from the hexactinellid sponge Aphrocallistes vastus: a putative aggregation factor. Glycobiology 11: 21–29.
Haen, K. M., B. F. Lang, S. A. Pomponi & D. V. Lavrov, 2007. Glass sponges and bilaterian animals share derived mitochondrial genomic features: a common ancestry or parallel evolution? Molecular Biology and Evolution 24: 1518–1527.
Hillis, D. M. & J. J. Bull, 1993. An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analysis. Systematic Biology 42: 182–192.
Huelsenbeck, J. P. & B. Rannala, 2004. Frequentist properties of Bayesian posterior probabilities of phylogenetic trees under simple and complex substitution models. Systematic Biology 53: 904–913.
Ivanova, N. V., A. V. Borisenko & P. D. N. Hebert, 2009. Express barcodes: racing from specimen to identification. Molecular Ecology Resources 9(Supplement 1): 35–41.
Lanave, C., G. Preparata, C. Saccone & G. Serio, 1984. A new method for calculating evolutionary substitution rates. Journal of Molecular Evolution 20: 86–93.
Larkin, M. A., G. Blackshields, N. P. Brown, R. Chenna, P. A. McGettigan, H. McWilliam, F. Valentin, I. M. Wallace, A. Wilm, R. Lopez, J. D. Thompson, T. J. Gibson & D. G. Higgins, 2007. ClustalW and ClustalX version 2.0. Bioinformatics 23: 2947–2948.
Leys, S. P. G. O., G. O. Mackie & H. M. Reiswig, 2007. The biology of glass sponges. Advances in Marine Biology 52: 1–145.
Manuel, M., Y. Le Parco & C. Borchiellini, 2004. Comparative analysis of Brachyury T-domains, with the characterization of two new sponge sequences, from a hexactinellid and a calcisponge. Gene 340: 291–301.
Mehl, D., 1992. Die Entwicklung der Hexactinellida seit dem Mesozoikum. Paläobiologie, Phylogenie und Evolutionsökologie. Berliner Geowissenschaftliche Abhandlungen Reihe E: Paläobiologie 2: 1–164.
Philippe, H., R. Derelle, P. Lopez, K. Pick, C. Borchiellini, N. Boury-Esnault, J. Vacelet, E. Renard, E. Houliston, E. Quéinnec, C. Da Silva, P. Wincker, H. Le Guyader, S. Leys, D. J. Jackson, F. Schreiber, D. Erpenbeck, B. Morgenstern, G. Wörheide & M. Manuel, 2009. Phylogenomics revives traditional views on deep animal relationships. Current Biology 19: 706–712.
Pick, K. S., H. Philippe, F. Schreiber, D. Erpenbeck, D. J. Jackson, P. Wrede, M. Wiens, A. Alié, B. Morgenstern, M. Manuel & G. Wörheide, 2010. Improved phylogenomic taxon sampling noticeably affects non-bilaterian relationships. Molecular Biology and Evolution 27: 1983–1987.
Reiswig, H. M., 2002. Family Tretodictyidae Schulze, 1886. In Hooper, J. N. A. & R. W. M. van Soest (eds), Systema Porifera. A Guide to the Classification of Sponges. Plenum, New York: 1341–1354.
Rokas, A., N. King, J. Finnerty & S. B. Carroll, 2003. Conflicting phylogenetic signals at the base of the metazoan tree. Evolution and Development 5: 346–359.
Rosengarten, R. D., E. A. Sperling, M. A. Moreno, S. P. Leys & S. L. Dellaporta, 2008. The mitochondrial genome of the hexactinellid sponge Aphrocallistes vastus: evidence for programmed translational frameshifting. BMC Genomics 9: 33.
Savill, N. J., D. C. Hoyle & P. G. Higgs, 2001. RNA sequence evolution with secondary structure constraints: comparison of substitution rate models using maximum-likelihood methods. Genetics 157: 399–411.
Siddall, M. E., F. M. Fontanella, S. C. Watson, S. Kvist & C. Erséus, 2009. Barcoding bamboozled by bacteria: convergence to metazoan mitochondrial primer targets by marine microbes. Systematic Biology 58: 445–451.
Sperling, E. A., K. J. Peterson & D. Pisani, 2009. Phylogenetic-signal dissection of nuclear housekeeping genes supports the paraphyly of sponges and the monophyly of Eumetazoa. Molecular Biology and Evolution 26: 2261–2274.
Stamatakis, A., 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688–2690.
Stamatakis, A., P. Hoover & J. Rougemont, 2008. A rapid bootstrap algorithm for the RAxML web servers. Systematic Biology 57: 758–771.
Tabachnick, K. R., 2002a. Lyssacinosida incertae sedis. In Hooper, J. N. A. & R. W. M. van Soest (eds), Systema Porifera. A Guide to the Classification of Sponges. Plenum, New York: 1506–1509.
Tabachnick, K. R., 2002b. Family Rossellidae Schulze, 1885. In Hooper, J. N. A. & R. W. M. van Soest (eds), Systema Porifera. A Guide to the Classification of Sponges. Plenum, New York: 1441–1505.
Tabachnick, K. R., 2002c. Family Euplectellidae Gray, 1867. In Hooper, J. N. A. & R. W. M. van Soest (eds), Systema Porifera. A Guide to the Classification of Sponges. Plenum, New York: 1388–1434.
Tabachnick, K. R. & H. M. Reiswig, 2002. Dictionary of Hexactinellida. In Hooper, J. N. A. & R. W. M. van Soest (eds), Systema Porifera. A Guide to the Classification of Sponges. Plenum, New York: 1224–1229.
Yang, Z., 1994. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. Journal of Molecular Evolution 39: 306–314.
Acknowledgments
Work at the Wörheide lab was supported by the Deutsche Forschungsgemeinschaft (DFG; project Wo896/5-3). The DFG is further acknowledged for funding the Deep Down Under expedition (Project Wo896/7-1; www.deepdownunder.de). During writing of this manuscript, MD was supported by a Postdoctoral Fellowship of the Smithsonian Institution. Work at the Lavrov lab was supported by the National Science Foundation (NSF) through the Poriferan Tree of Life project (DEB-0828783; https://www.portol.org/). Shirley Pomponi (HBOI), Sally Leys, and Jean Vacelet are acknowledged for providing additional specimens. Our warmest thanks go to the organizers of the 8th World Sponge Conference for making this great meeting possible. Allen Collins and three anonymous reviewers provided comments that led to improvement of this manuscript. Finally, we would like to thank Alexandros Stamatakis for implementing paired-sites models in his amazing program, RAxML.
Author information
Authors and Affiliations
Corresponding author
Additional information
Guest editors: M. Maldonado, X. Turon, M. A. Becerro & M. J. Uriz / Ancient animals, new challenges: developments in sponge research
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Dohrmann, M., Haen, K.M., Lavrov, D.V. et al. Molecular phylogeny of glass sponges (Porifera, Hexactinellida): increased taxon sampling and inclusion of the mitochondrial protein-coding gene, cytochrome oxidase subunit I. Hydrobiologia 687, 11–20 (2012). https://doi.org/10.1007/s10750-011-0727-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10750-011-0727-z