
Abstract
Calcium ion (Ca2+) cycle plays a crucial role in the contraction and relaxation of cardiomyocytes. The sarcoplasmic reticulum (SR) acts as an organelle for storing Ca2+, which mediated the release and re-uptake of Ca2+ during contraction and relaxation. Disorders of SR function lead to the dysfunction of Ca2+ cycle and myocardial cell function. The sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 2a (SERCA2a) acts as a subtype of SERCA expressed in the heart, which mediates the contraction of cardiomyocytes and Ca2+ in the cytoplasm to re-enter into the SR. The rate of uptake of Ca2+ by the SR determines the rate of myocardial relaxation. The regulation of SERCA2a activity controls the contractility and relaxation of the heart, affecting cardiac function. The expression and activity of SERCA2a are reduced in failing hearts. Gene therapy by increasing the expression of SERCA2a in the heart has been proven effective. In addition, SERCA2a is regulated by a variety of factors, including transmembrane micropeptides, protein kinases, and post-translational modifications (PTMs). In this review, we discuss the regulatory factors of SERCA2a and provide new insights into future treatments and the direction of heart failure research. In addition, gene therapy for SERCA2a has recently emerged as therapeutic option and hence will be discussed in this review.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
With the aging population and the rising risk of cardiovascular disease, heart failure (HF) has become a global public health problem, which affects nearly 30 million people worldwide [1]. HF is the end stage of various heart diseases, which is called “the final battlefield” in the cardiovascular field. Drugs for clinical treatment of HF mainly include diuretics, angiotensin-converting enzyme inhibitors (ACE inhibitors) or angiotensin II receptor antagonists, and beta blockers [2]. However, heart failure still has high hospitalization and mortality rate.
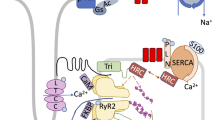
Ca2+ acts as a second messenger in the cellular process to mediate a variety of signaling pathways, and its role in the heart has been widely known [3,4,5]. In the heart, the sarcoplasmic reticulum (SR) is a Ca2+ reservoir and the intracellular L-type Ca2+ channel (LTCC), the ryanodine receptor 2 (RyR2), and the sarcoplasmic reticulum calcium pump (SERCA) maintain intracellular Ca2+ homeostasis, which converts electrical signals into cardiac contractility [6]. Depolarization of the cell membrane allows extracellular Ca2+ to enter the cytosol through the LTCC located in the T-tubule, and the gap between LTCC and SR is narrow, which provides conditions for the transient increase of intracellular Ca2+. Extracellular Ca2+ triggers RyR2 on the SR membrane to release a large amount of Ca2+ from SR, and the cytosol Ca2+ concentration rises to a critical level, which triggers Ca2+ binding to troponin C and promotes coarse/thin filament slip. These series of reactions trigger a cardiac contraction effect. The diastolic effect of the heart is caused by SERCA2a (the major subtype of SERCA in the heart) and the Na/Ca2+ exchanger on the cell membrane that takes Ca2+ back into the SR (more than about 75% Ca2+) and excreted from the cell [7] (Fig. 1). The activity and function of SERCA2a are impaired in HF. On the one hand, the decrease in SERCA2a activity reduces the Ca2+ content of SR, thereby reducing the amount of calcium ions released in the sarcoplasmic reticulum and causing systolic dysfunction. On the other hand, loss of SERCA2a activity also reduces the amount and rate of calcium removal from the cytoplasm, which inhibits myocardial relaxation and diastolic dysfunction to some extent [8]. SERCA2a is regulated by a variety of transmembrane micropeptides. In addition to the previously studied phospholamban (PLB) and sarcolipin (SLN), some newly identified micropeptides such as dwarf open reading frame (DWORF) [9], another-regulin (ALN) [10], and myoregulin (MLN) [11] also regulate the activity of SERCA2a. With the development of proteomics, post-translational modification (PTM) has also been found to serve as a new target for the regulation of SERCA2a activity. For example, acetylation [12], SUMOylation [13], and nitrification [14] and their corresponding PTMs will be discussed.

Calcium cycle in cardiomyocytes. During systole, in the presence of membrane depolarization, calcium ions enter the cytosol through LTCCs, which triggers a large amount of Ca2+ in the SR to enter the cytoplasm through RyR2. After reaching the critical concentration, Ca2+ bind to troponin C and the myofilament slides. During diastole, a large amount of Ca2+ re-uptake into the SR through SERCA2a, and the remaining Ca2+ are excreted from the cells through NCX. Ca2+ are represented by red balls; Na+ are represented by purple balls. Two-color arrows indicate the contraction process and blue arrows indicate the diastolic process. LTCC, voltage-dependent L-type Ca2+ channel; RyR2, ryanodine receptor 2; SR, sarcoplasmic reticulum; SERCA2, sarcoplasmic/endoplasmic reticulum calcium ATPase 2; PLB, phospholamban; NCX, Na+/Ca2+ exchanger
Specifically, in this review, we will describe SERCA2a and its related regulatory factors, with emphasis on the therapeutic potential of PTMs. In addition, we will also cover the recent gene therapy of SERCA2a. Recent studies have used SERCA2a as a strategic prospect for the treatment of heart failure.
SERCA2a is a Ca2+ refluxor
The mammalian SERCA family has several subtypes encoded by SERCA1, SERCA2, and SERCA3, wherein SERCA2a (encoded by the ATP2A2 gene) and SERCA1a (encoded by the ATP2A1 gene) have strong similarities and 84% protein sequence identity [15]. Although the expression position is not consistent, SERCA2a is mainly expressed in cardiomyocytes and type I skeletal muscle, while SERCA1a is mainly expressed in type II skeletal muscle, but the consistency of structure and sequence indicates that the Ca2+ transport mechanism may be highly conserved [16]. SERCA2a is a key protein in the calcium cycle of cardiomyocytes, which is used to re-uptake Ca2+ in the cytoplasm into the sarcoplasmic reticulum to relax the cardiomyocytes, thereby maintaining Ca2+ homeostasis in cells. After the cardiomyocytes complete a contraction action, SERCA2a takes up Ca2+ and returns to SR [17]. This process involves the mutual transformation of the two structures of the SERCA2a protein: E1 and E2. The E1 structure of SERCA2a has a higher affinity for Ca2+, and the binding site is exposed to SR. However, the E2 structure has a lower affinity for Ca2+, which is located on the lumen of the SR. After relaxation of cardiomyocytes, Ca2+ is released from troponin into the cytosol and binds to E1 (the binding site is at the aspartic acid residue), after which phosphorylation of E1 occurs with the participation of ATP (Ca2+-E1-P), which then causes a conformational change from E1 to E2 (CA2+-E2-P), and the E2 structure becomes an ADP-insensitive intermediate [18, 19]. This process results in the localization of the Ca2+ binding site in the SR. Due to the low affinity of E2 for Ca2+, Ca2+ is released into the SR after uncoupling with E2, accompanied by E2 converted to E1 and re-entering the next cycle (Fig. 2).

The mechanism of action of SERCA2a. After relaxation of the heart, Ca2+ bind to SERCA2a in the E1 state, and with the participation of ATP, the conformational change of E1 is converted to E2. Ca2+ are transported into SR, and intracellular Ca2+ concentration restores resting level. Ca2+ are represented by green balls; Pi are represented by red balls. SR, sarcoplasmic reticulum; E1, E1 structure of SERCA2a; E2, E2 structure of SERCA2a
SERCA2a and its regulator—transmembrane micropeptide
PLB and SLN have been shown to bind to the cytoplasmic/transmembrane domains of SERCA2a, which inhibits the affinity of SERCA2a for Ca2+ [20]. As a major regulator of SERCA2a activity, PLB is the only regulatory protein of SERCA2a directly involved in the development of cardiac disease, including HF.
PLB
PLB is a 52–amino acid single transmembrane protein expressed in the SR, which exists as a pentamer and regulates the activity of SERCA2a [21]. It was first discovered by James and his colleagues that PLB interacts with SERCA2a and that non-phosphorylated PLB inhibits SERCA2a activity [22], which can be phosphorylated by cAMP-dependent protein kinase (PKA) and calcium/calmodulin-dependent protein kinase II (CaMKII). SERCA2a undergoes conformational change (become a calcium binding state) to relieve the inhibition of PLB [23]. Subsequent research focused on the location of SERCA2a in response to PLB inhibition. SERCA2 has two key domains: one is the region between amino acids 336 and 412 in the phosphorylation domain, which responds to the action of PLB; the other is the amino acids 467 and 762 in the nucleotide-binding domain, which mainly determines the strength of SERCA2 for Ca2+ affinity. These two domains collectively respond to phosphorylated PLB to shift the conformation of SERCA2 from a non-calcium-bound state to a calcium-bound state, thereby enhancing the Ca2+ affinity of SERCA2. Toyofuku et al. further revealed that the cytoplasmic 1A domain of PLB plays a key role in the function of SERCA2a through mutations in PLB residues [24].
The regulation of SERCA2a activity by PLB involves two steps: first, the dissociation of PLB from a pentamer to a monomer, which is a reversible process. The monomeric PLB then combines with SERCA2a to form a PLB-SERCA2a complex, thereby exerting an inhibitory effect. The increase in SERCA2a activity is regulated by phosphorylation of PLB and Ca2+ [25] (Fig. 3). On the one hand, PLB phosphorylation dissociates from the PLB-SERCA2a complex, which abolishes the inhibitory effect of PLB on SERCA2a. In addition, the increase of cytosolic Ca2+ concentration directly regulates the binding ability of SERCA2a to Ca2+, which enhances the capacity of sarcoplasmic reticulum to regress Ca2+ and induce myocardial cell relaxation.

Interaction mechanism of PLB-SERCA2a. The PLB pentamer can be depolymerized into a monomer, which is a reversible process. PLB monomer can inhibit the binding of SERCA2a; there are two cases that can inhibit the inhibition of SERCA2a by PLB: the first case is phosphorylation of PLB and the second case is calcium with increasing Ca2+ concentration. Ca2+ binding to SERCA2a will relieve PLB inhibition. SERCA2, sarcoplasmic/endoplasmic reticulum calcium ATPase 2; PLB, phospholamban
In recent years, a lot of research has been done on PLB and SERCA2a. Mazzocchi et al. [26] used PLB ablation to improve Ca2+ uptake by SERCA2a. Interestingly, it prevented arrhythmias caused by Ca2+ leakage triggered by excessive phosphorylation of RyR2. In addition, ablation of PLB has a cardioprotective effect on heart failure mice and reduces mortality in heart failure mice [27]. Another study found that PLB-deficient mice prevented arrhythmia during ischemia-reperfusion. It is worth noting that ablation of PLB significantly increased the infarct size of the heart, suggesting that ablation of PLB exacerbates ischemia-reperfusion injury [28]. The inconsistency between the tendency of reduced arrhythmia and the further deterioration of cardiac damage caused by ischemia-reperfusion justifies the involvement of many different regulatory mechanisms. In addition, PLB is related to genetic predispositions. In a study conducted by Schmitt and colleagues [29], they found that the phosphorylation of PLB was impaired and the Ca2+ binding ability of SERCA2a was weakened after the missense mutation of arginine at position 9 of PLB amino acid residue was changed to cysteine. Studies show abnormal Ca2+ treatment and impaired cardiac function. With the development of proteomics technology, the cognitive molecules of PLB are more widely recognized. Protein phosphatase 1 (PP1) is an endogenous dephosphorylation enzyme of PLB, and the newly identified PP1 regulatory subunit (PPP1R3A) binds to PLB. PPP1R3A knockout mice generated by deletion of exon fragments by Alsina et al. found that PP1-mediated targeting of RyR2 and PLB dephosphorylation was impaired, and excessive SERCA2a and RyR2 activity was highly likely to be triggered by atrial fibrillation [30]. In recent years, the emergence of near-infrared fluorescence nanoprobe has also provided us with a more efficient and convenient way to identify transmembrane micropeptides. Zhan et al. [31] designed specific probes for PLB and identified a range of properties, including sensitivity, specificity, and effects on PLB, which contribute to early diagnosis of heart failure. The emergence of new technologies will guide us in the far-reaching function of PLB.
SLN
SLN is another transmembrane micropeptide present on SR, consisting of 31 amino acids, which is mainly expressed in the atria. SLN shares the same protein family as PLB. The SLN contains a short dynamic helix and a rigid helix, and two short unstructured terminuses: one at the N-terminal 1–6 residues and the other at the C-terminal 27–31 residues [32]. SLN was found later than PLB, which was first identified on the skeletal muscle SR of rabbits. Therefore, earlier studies have tended to study the role of SLN in SERCA1 [33, 34]. Similar to the effect of PLB on SERCA2a, SLN can inhibit SERCA1a [35]. Initially, Asahi and colleagues [36] used mutations in SLN and PLB and co-immunoprecipitation of SERCA to show that SLN can bind to PLB, which in turn forms a SLN-PLB-SERCA complex that has a higher inhibitory effect on SERCA. Moreover, the SLN-PLB complex prevents the polymerization of PLB (cannot be converted from a monomeric form to a pentamer), which has a sustained inhibitory effect on SERCA. Paradoxically, SLN still has an inhibitory effect on SERCA in tissues (atrial, slow-shrinking skeletal muscle) with low PLB expression or no expression. Michio et al. [37] found in mice with cardiac-specific overexpression of SLN that SERCA2a has reduced affinity for Ca2+. In addition, adenovirus-mediated SLN overexpression prolonged 50% relaxation and Ca2+ decay time with impaired cardiac function [38]. Ablation of SLN does not alter PLB protein expression and phosphorylation, and enhances SERCA affinity for Ca2+ and the maximum rate of Ca2+ uptake [39, 40]. These studies show that SLN does not rely on PLB to function but directly binds to SERCA to exert inhibition. Interestingly, SLN has a different effect on SERCA than PLB. Under high concentration of Ca2+, SLN still plays a role in the inhibition of SERCA [41], which is contrary to the inhibition of SERCA by PLB at high concentration of Ca2+. SLN mainly reduces the maximal rate of Ca2+ uptake by interacting with SERCA, which is different from the regulation mechanism of PLB on ATPase activity of SERCA. The regulation of SLN is not affected by Ca2+ concentration [42]. In addition, SLN is also regulated by phosphorylation, and CaMKII phosphorylates the N-terminal threonine-5 residue of SLN [43]. Serine/threonine phosphokinase 16 also exerts phosphorylation of SLN, which in turn significantly reduces the inhibitory effect of SLN on SERCA [44]. In conclusion, SLN is an important regulator of myocardial SERCA, and its physiological function and structure require further research. Specific drugs for SLN have not yet been discovered, but various studies have demonstrated that it is a strategic approach to improve the Ca2+ cycle disorder in failing hearts by regulating SLN. This requires a combination of basic experiments and clinical validation.
Potential micropeptides of SERCA2a
In recent years, some newly discovered transmembrane micropeptides have been identified, which play a novel role in the regulation of SERCA2a.
DWORF removes the inhibitors on SERCA2a
A 34–amino acid peptide was identified: DWORF, which was previously considered a non-coding RNA. DWORF can eliminate the inhibition of SERCA2a by replacing PLB, SLN, thereby enhancing the activity of SERCA2a. It was confirmed in the study of mouse cardiomyocytes and type I skeletal muscle that DWORF significantly affects intracellular Ca2+ treatment and enhances calcium pump activity [9]. DWORF has a higher affinity for SERCA than PLB, and overexpression of DWORF significantly improved cardiac function in dilated heart disease mice and improved dysregulated Ca2+ circulation [45]. These findings give us a preliminary understanding of DWORF. On the one hand, DWORF can be used as a biomarker in heart failure. The decrease in its level means that the contractility of the myocardium is affected. In addition, DWORF can be used as a target for new drugs. It is a strategic approach to increase the activity of SERCA2a by increasing the amount of DWORF in the heart, which will break through the limitations of modern positive inotropic drugs.
MLN/ELN/ALN: additional inhibitor of SERCA2a
Myoregulin (MLN) is a newly discovered functional micropeptide encoded by skeletal muscle–specific RNA, a micropeptide with 46 amino acids. MLN and PLB have similar effects on SERCA, and MLN is widely expressed in skeletal muscle as a suppressor of SERCA1. Deletion of MLN in mice revealed an increase in SERCA activity and improved Ca2+ handling [11]. In addition, endoregulin (ELN) and ALN are newly identified two transmembrane micropeptides whose N-terminal domain in the cytosol contains conserved sequences of serine and threonine residues. ELN mainly inhibits SERCA3 present in endothelial cells and epithelial cells. Notably, the N-terminal domain of ALN contains a motif that can be phosphorylated by PKA, which is similar to the phosphorylation motif of PLN. It is suggested that the inhibition of SERCA2b by ALN may be related to the inhibition of SERCA2a by PLN through the same conserved mechanism [10]. Interestingly, the C-terminal transmembrane helix of ELN, ALN, and MLN is similar to PLB and SLN, suggesting that these new transmembrane micropeptides are a conserved mechanism for Ca2+ regulation. However, there are few studies on the role of ELN, ALN, and MLN in the myocardium. Currently, enough evidence is unavailable concerning whether these new micropeptides have similar effects on SERCA2a like PLB and SLN. Whether ELN, ALN, and MLN have the same effect on SERCA2a and the combination of PLN or SLN is worthy of further study.
Multiple PTMs regulate the activity of SERCA2a
PTM refers to the covalent attachment of chemical small molecule groups to the amino acid side chain of a protein, significantly increasing the complexity and diversity of the protein [46]. More than 400 PTMs have been identified, PTM can occur in almost all proteins, and multiple PTMs can occur in the same protein [47]. More importantly, the PTM of the protein significantly changes the physicochemical properties and conformation of the protein, thereby directly changing the binding ability and function of the protein. Therefore, even if the expression level of the protein does not change, the state of the post-translational modification changes the function of the protein significantly [48]. A number of studies have demonstrated that SERCA2a is capable of PTM and different types of PTM occur at different sites, which affects the activity and function of SERCA2a.
SUMOylation
The small ubiquitin-like modifier (SUMO) is an important member of the ubiquitin-like protein family, and its molecular structure is similar to that of ubiquitin, but the biological significance of these two types of protein modification is different. Ubiquitination mediates protein degradation, while the SUMO family is widely involved in cellular activities such as protein structural stability, nuclear translocation [49], and regulation of transcriptional activity [50]. The SUMO family mainly includes SUMO1, SUMO2, and SUMO3. Since SUMO2 and SUMO3 have a high degree of sequence identity, they are called SUMO2/3 [51], SUMO1 is mainly involved in delayed response, and SUMO2/3 is involved in acute stress response. SUMO was originally an immature precursor, which required the splicing of SUMO proteases (SENPs) to become activated forms. Activation of splicing of SUMO molecules is terminated by Gly residues [52]. SUMO modification involves a cascade of related enzymes: First, mature SUMO was first activated by SUMO-activating enzyme (E1), and ATP participated in this process. E1 is a heterodimeric protein complex composed of Uba2 and Aosl. The cysteine residues on SUMO and Uba2 were bound by thioester bonds with the participation of SUMO adenosine intermediates. Uba2 is only known as SUMO-binding enzyme (E2). The binding of SUMO to the substrate protein is accomplished by the action of SUMO ligase (E3). SUMO was linked to the substrate protein lysine side chain to form an isopeptide bond and exert PTM action [53]. SUMO modification is a reversible dynamic process. Under the enzymatic cleavage of SENPs, SUMO molecules dissociate from the substrate and re-enter the SUMO cycle.
In 2011, Kho et al. [54] found that the expression of SUMO1 and the SUMOylation of SERCA2a were significantly decreased in patients and mice subjected to heart failure. They used small hairpin RNA (shRNA) to downregulate SUMO1, which accelerated the deterioration of cardiac function and demonstrated that SUMOylation is essential for the activity and stability of SERCA2a. Overexpression of SUMO1 significantly improved cardiac function, including improvement of Ca2+ cycle in cardiomyocytes, and functional verification of SUMOylation of SERCA2a, followed by regulation of SUMOylation of SERCA2a as a research target for heart failure treatment. Kho [55] further characterized the small molecule N106 and found that N106 increased the SUMOylation level of SERCA2a, and a series of studies on this small molecule compound found that N106 indirectly triggered the endogenous SUMOylation of SERCA2a by directly activating E1. Treatment with N106 improves the contractility of cardiomyocytes and improves cardiac function in heart failure mice. This further demonstrates that SUMOylation by increased SERCA2a is a promising treatment, which also provides more new indicators for drug development. It is worth noting that the level of SUMO1 in the heart increased slightly during hypertrophy, but the level of heart failure decreased sharply [56]. The staged suggestion of SUMO1 levels has a more beneficial effect on gene therapy with SUMO1 in the compensatory phase. In addition, gene therapy of SUMO1 [57] has also been shown to be effective in a porcine model of ischemic heart, which is characterized by improved cardiac function (including ejection fraction, maximum rate of increase in pressure) and contractile relaxation effect of cardiomyocytes. Oh et al. [58] observed a strong correlation between miR-146a and SUMO1 in the heart of depleted mice and humans, and further studies of both showed elevated levels of miR-146a in the failing heart, which reduced the expression of SUMO1 and negatively regulated the SUMO1-SERCA2a axis. Additionally, a research by Du and co-researchers [59] used the cardiac ischemia-reperfusion mouse model to find the SUMOylation of SERCA2a and the decrease of SUMO1 in the heart. Further studies found that the SUMOylation receptor sites of mouse SERCA2a are lysine 585, 480, and 571. Treatment with luteolin improved heart function in heart failure mice and reduced infarct size in the heart, and demonstrated that luteolin regulates SERCA2a through SUML 585 to reduce myocardial ischemia-reperfusion injury. These studies suggest that improving cardiac function in heart failure by mediating SUMOylation of SERCA2a should be one of the notable targets in the development of new drugs.
Acetylation
The acetylation modification is the transfer of the acetyl group of the donor to the amino acid residue of the protein of interest by an enzymatic or spontaneous reaction, which modulates the function of the target protein. Protein acetylation is another PTM that regulates protein activity and function [60]. With the development of gene manipulation and proteomics, various functions following protein acetylation have attracted more attention. Acetylation is involved in transcriptional regulation, metabolic regulation, and signaling pathway regulation, and plays an important role in controlling protein conformation, activity, and stability [61]. For acetylation modification, current research hotspots focus on acetylation modification on histones, which is crucial in the process of apparent regulation [62]. However, the broader function of acetylation modification is not limited to the nucleus, and the acetylation modification is also extensive in a large number of proteins in the cytoplasm and other sub-cells, called non-histone acetylation modification. Acetylation modification is mainly divided into two categories: one is acetylation at the amino acid residue at the end of the receptor protein and the other is acetylation at the lysine in the receptor protein chain [63]. The former is a covalent modification that occurs mostly in nascent proteins of eukaryotes and is important for the maturation and cellular localization of nascent proteins, and is responsible for N-acetyltransferases (NATs), which is a reversible acetylation modification. The acetyl-CoA acetyl group is mainly transferred to the ε-amino side chain of lysine by lysine acetyltransferase (KATs) to exert a regulatory effect and can be reversed by lysine deacetylase (KDACs). The reversible changes in protein or histone acetylation regulated by KATs and KDACs are associated with many common diseases, including heart disease [64], diabetes [65], neurodegenerative diseases [66], and cancer [67], as well as some rare diseases such as mitochondrial diseases [68].
In a recent study from humans, pigs, and mice [12], the researchers found a significant increase in the level of acetylation of SERCA2a in failing hearts, a phenomenon attributed to a decrease in the level of SIRT1 (a class III histone deacetylase). The acetylation of SERCA2a had the opposite effect as SUMOylation, and the increase in acetylation of SERCA2a significantly reduced the activity of SERCA2a, which caused a decrease in the uptake of Ca2+ and impaired cardiac function of SERCA2a. The acetylation level of lysine 492 (K492) was found to be responsible for SERCA2a dysfunction by measuring the acetylation site. The histone acetyltransferase p300–mediated K492 acetylation reduced the ATP binding ability of SERCA2a. This study points us to new research ideas, on the one hand, blocking the acetylation of SERCA2a mediated by p300, thereby reducing the level of acetylation of SERCA2a, which indirectly improves the function of damaged SERCA2a in failing hearts. On the other hand, the acetylation level of SERCA2a is lowered by increasing the level of SIRT1, which has been shown to have high acetylation of SERCA2a. It is a strategic approach to develop a new drug or small molecule that mediates the reduction of SECCA2a acetylation levels from the source and on the way of SERCA2a [69]. It is worth noting that acetylation mediates a variety of physiological effects and widely expressed in vivo, and the side effects produced after activation of SIRT1 will need to be considered. In addition, drug development by combining SUMOylation and acetylation of SERCA2a may result in better balance and fewer side effects.
Phosphorylation and O-GlcNAc modification
In addition to SUMO and acetylation, SERCA2a is also affected by other PTMs. Phosphorylation of proteins is the most widely studied and plays an important role in protein PTM, which is involved in the regulation of various important molecular activities and functions. Striated muscle–specific protein kinase (SPEG), as a member of the myosin light chain kinase (MLCK) subgroup of the CaMK Ser/Thr protein kinase family, is closely related to phosphorylation of SERCA2a. Quick et al. [70] used proteomics to discover that SERCA2a interacts with SPEG, and they further used cell lines and primary cardiomyocytes of neonatal rats to demonstrate that the second kinase domain of SPEG acts on SERCA2a, which relies on SPEG to directly phosphorylated the Thr484 site of SERCA2a, thereby enhancing the oligomerization of SERCA2a and increased the Ca2+ transport capacity of SERCA2a. Further study [71] of SPEG-SERCA2a through used SPEG cardiac-specific knockout mice revealed that phosphorylation of the Thr484 site of SERCA2a and the level of oligomerization of SERCA2a were significantly reduced in the hearts of mice deficient in SPEG, which inhibited the activity of SERCA2a and impaired Ca2+ uptake in the sarcoplasmic reticulum of the cardiomyocytes, which was subsequently accompanied by maladaptive cardiac function in mice leading to the death of these mice. In addition, Li and subordinates [14] found that iron supplementation stimulated cardiomyocyte hypertrophy and led to increased formation of cardiac protein carbonyl tyrosine, which resulted in abnormal myocardial calcium homeostasis in diabetic rats. Further studies in vitro have revealed that tyrosine nitration of SERCA2a played an important role in the activity of SERCA2a, which reduced the ability of SERCA2a to modulate Ca2+ and damaged SERCA2a activity. Intracellular protein translation was followed by N-acetylglucosamine (O-GlcNAc) modification, catalyzed by O-GlcNAC glycosyltransferase, which is involved in the regulation of many important biological processes in cells, and application in human diseases and treatment [72]. O-GlcNAc has been shown to modify SERCA2 protein levels and prevent Ca2+ treatment of SERCA2a. In addition to modifying SERCA2a protein levels, O-GlcNAc can also modify SERCA2a activity by regulating phosphorylation of PLB [73]. In conclusion, O-GlcNAc regulates the activity of SERCA2a either directly or indirectly. In addition, NO is a physiological regulator that stimulates SERCA2a to accelerate the reduction of intracellular Ca2+ concentration, which relaxes the heart muscle, skeletal muscle, and smooth muscle. NO-derived intermediates can also play a role in protein modification. Lancel et al. [74] clarified the molecular mechanism by which nitroxyl partially exerts positive inotropic and relaxing effects in the myocardium by enhancing the activity of SERCA2a. They found that nitroxyl passes S-glutathionylation at cysteine 674 and increased the activity of SERCA2a, which enhanced the contractile relaxation effect of cardiomyocytes.
SERCA2a is affected by a variety of PTMs, and it may be a promising direction to mediate PTM in clinical or experimental settings to improve reduced activity in heart failure. It is worth noting that the effects of PTM changes on other functional proteins have not been revealed, which requires us to study more in depth.
Gene therapy approaches to increase SERCA2a pump activity—stimulating the calcium pumps
With the development of molecular biology, transgenic technology, and vector technology, gene therapy has gradually become a new breakthrough in heart failure treatment. Decreased expression and activity of SERCA2a in heart failure are key links in the Ca2+ cycle. Therefore, overexpression of SERCA2a by gene transduction technology has become a key point in gene therapy for heart failure. Studies of genetic alteration models have identified the functional role of the SERCA pump in Ca2+ handling and cardiac physiology. A series of cell models, rodent models, and large animal models have shown that the introduction of SERCA2a gene into viral cardiomyocytes with viral vectors can improve systolic and diastolic effects [75,76,77]. After occluding the proximal left anterior descending balloon of the pig and then reperfusion for 1 month, adeno-associated vector type 1 (AAV1)–mediated transgenic therapy of SERCA2a significantly improved cardiac function and prevented expansion of left ventricular volume [57]. In 2007, the first clinical trial of heart failure to SERCA2a gene therapy began in the USA [78]. This study used AAV1 with SERCA2a to treat patients with advanced heart failure by direct intracoronary injection, which was used to determine the effectiveness and safety of different doses of AAV/SERCA2a. The study found that in the high-dose AAV1-SERCA2a group, the improvement in cardiac function and the 6-min walking distance were significantly better than in the placebo group [79]. After 3 years of follow-up, the major adverse cardiovascular events (including recurrent myocardial infarction, worsening HF, and rehospitalization) were significantly lower than in the placebo group [80]. Based on previous findings, a larger sample size study was conducted in 2012, which included 250 patients with advanced heart failure. Surprisingly, the AAV1-SERCA2a group showed a major endpoint compared with the placebo group. No significant gains were found in the secondary endpoints [81]. After that, RT-PCR analysis of tissue samples from patients undergoing heart transplantation or left ventricular assist device implantation and death found that the amount of vector DNA in the cardiomyocytes of patients in the second clinical trial was much lower than that in the first phase. Despite the small number of tissue samples, this suggested that the failure of the second clinical trial may be due to the low transfection rate of SERCA2a. Although the large sample clinical trial failed, the trial confirmed that the SERCA2a gene is safe for heart failure. Gene therapy remains the most promising treatment for diseases. Problems encountered in SERCA2a gene therapy are expected to be resolved in the future. At present, it has been found that the purpose of improving the efficiency of intracoronary injection can be improved by establishing a closed loop system for recycling [82]. In addition, the use of immunosuppressive drugs or plasmapheresis methods may reduce the effects of AAV neutralizing antibodies, or the development of vector engineering and recombinant technology to improve viral vector safety and transfection efficiency. With the development of gene transduction technology and novel gene therapy vector system, gene therapy will make breakthrough progress in the near future.
Perspectives
A variety of evidences demonstrate the importance of SERCA2a in maintaining normal heart function (Table 1). Several studies have shown that gene therapy methods for SERCA2a expression are effective in clinical trials [79, 83]. However, subsequent large-scale clinical trials have not been validated [84, 85]. Although no evidence of improved prognosis was found at the AAV1/SERCA2a dose studied, these trials spurred further studies using gene therapy to treat HF. A variety of means for regulating SERCA2a-associated transmembrane micropeptides are also being investigated. It is worth considering that the regulation of SERCA2a by myriads of emerging PTMs cannot be ignored. SERCA2a-mediated gene therapy increases SERCA2a expression but does not alter SERCA2a PTM. The discovery of small molecules, such as N106, improves the activity of SERCA2a by increasing the level of SUMOylation of SERCA2a [55]. Gene therapy for SUMO1 has also been shown to be effective in the heart of pigs [57]. In addition, elevated acetylation of SERCA2a in newly discovered heart failure patients may also be a new strategy by reducing the level of acetylation of SERCA2a in failing hearts [12]. Drug discovery targeting the PTM of SERCA2a will be an impeccable therapeutic option for people with heart failure in the near future.
Abbreviations
- AAV1:
-
adeno-associated vector type 1
- ALN:
-
another-regulin
- Ca2+ :
-
calcium ion
- CaMKII:
-
calcium/calmodulin-dependent protein kinase II
- DWORF:
-
dwarf open reading frame
- ELN:
-
endoregulin
- HF:
-
heart failure
- KATs:
-
lysine acetyltransferases
- KDACs:
-
lysine deacetylase
- LTCC:
-
L-type Ca2+ channel
- MLN:
-
myoregulin
- NATs:
-
N-acetyltransferases
- PLB:
-
phospholamban
- PKA:
-
cAMP-dependent protein kinase
- PP1:
-
protein phosphatase 1
- PTM:
-
post-translational modification
- RyR2:
-
ryanodine receptor 2
- SERCA2a:
-
sarco/endoplasmic reticulum Ca2+ ATPase 2a
- SLN:
-
sarcolipin
- SPEG:
-
striated muscle–specific protein kinase
- SR:
-
sarcoplasmic reticulum
- SUMO:
-
small ubiquitin-like modifier
References
Bloom MW, Greenberg B, Jaarsma T, Januzzi JL, Lam CSP, Maggioni AP, Trochu JN, Butler J (2017) Heart failure with reduced ejection fraction. Nat Rev Dis Primers 3:17058. https://doi.org/10.1038/nrdp.2017.58
Chen H, Liu S, Zhao C, Zong Z, Ma C, Qi G (2018) Cardiac contractility modulation improves left ventricular systolic function partially via miR-25 mediated SERCA2A expression in rabbit trans aortic constriction heart failure model. J Thorac Dis 10(6):3899–3908. https://doi.org/10.21037/jtd.2018.06.22
Eisner D, Caldwell J, Trafford A (2013) Sarcoplasmic reticulum Ca-ATPase and heart failure 20 years later. Circ Res 113(8):958–961. https://doi.org/10.1161/CIRCRESAHA.113.302187
Eisner DA, Caldwell JL, Kistamas K, Trafford AW (2017) Calcium and excitation-contraction coupling in the heart. Circ Res 121(2):181–195. https://doi.org/10.1161/CIRCRESAHA.117.310230
Kho C, Lee A, Hajjar RJ (2012) Altered sarcoplasmic reticulum calcium cycling--targets for heart failure therapy. Nat Rev Cardiol 9(12):717–733. https://doi.org/10.1038/nrcardio.2012.145
Marks AR (2013) Calcium cycling proteins and heart failure: mechanisms and therapeutics. J Clin Invest 123(1):46–52. https://doi.org/10.1172/JCI62834
Owens AT, Brozena SC, Jessup M (2016) New management strategies in heart failure. Circ Res 118(3):480–495. https://doi.org/10.1161/CIRCRESAHA.115.306567
Samuel TJ, Rosenberry RP, Lee S, Pan Z (2018) Correcting calcium dysregulation in chronic heart failure using SERCA2a gene therapy. Int J Mol Sci 19(4). https://doi.org/10.3390/ijms19041086
Nelson BR, Makarewich CA, Anderson DM, Winders BR, Troupes CD, Wu F, Reese AL, McAnally JR, Chen X, Kavalali ET, Cannon SC, Houser SR, Bassel-Duby R, Olson EN (2016) A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science 351(6270):271–275. https://doi.org/10.1126/science.aad4076
Anderson DM, Makarewich CA, Anderson KM, Shelton JM, Bezprozvannaya S, Bassel-Duby R, Olson EN (2016) Widespread control of calcium signaling by a family of SERCA-inhibiting micropeptides. Sci Signal 9(457):ra119. https://doi.org/10.1126/scisignal.aaj1460
Anderson DM, Anderson KM, Chang CL, Makarewich CA, Nelson BR, McAnally JR, Kasaragod P, Shelton JM, Liou J, Bassel-Duby R, Olson EN (2015) A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 160(4):595–606. https://doi.org/10.1016/j.cell.2015.01.009
Gorski PA, Jang SP, Jeong D, Lee A, Lee P, Oh JG, Chepurko V, Yang DK, Kwak TH, Eom SH, Park ZY, Yoo YJ, Kim DH, Kook H, Sunagawa Y, Morimoto T, Hasegawa K, Sadoshima J, Vangheluwe P, Hajjar RJ, Park WJ, Kho C (2019) Role of SIRT1 in modulating acetylation of the sarco-endoplasmic reticulum Ca(2+)-ATPase in heart failure. Circ Res 124(9):e63–e80. https://doi.org/10.1161/CIRCRESAHA.118.313865
Barry J, Lock RB (2011) Small ubiquitin-related modifier-1: wrestling with protein regulation. Int J Biochem Cell Biol 43(1):37–40. https://doi.org/10.1016/j.biocel.2010.09.022
Li X, Li W, Gao Z, Li H (2016) Association of cardiac injury with iron-increased oxidative and nitrative modifications of the SERCA2a isoform of sarcoplasmic reticulum Ca(2+)-ATPase in diabetic rats. Biochimie 127:144–152. https://doi.org/10.1016/j.biochi.2016.05.011
Gianni D, Chan J, Gwathmey JK, del Monte F, Hajjar RJ (2005) SERCA2a in heart failure: role and therapeutic prospects. J Bioenerg Biomembr 37(6):375–380. https://doi.org/10.1007/s10863-005-9474-z
Sitsel A, De Raeymaecker J, Drachmann ND, Derua R, Smaardijk S, Andersen JL, Vandecaetsbeek I, Chen J, De Maeyer M, Waelkens E, Olesen C, Vangheluwe P, Nissen P (2019) Structures of the heart specific SERCA2a Ca(2+)-ATPase. EMBO J 38(5). https://doi.org/10.15252/embj.2018100020
Bublitz M, Musgaard M, Poulsen H, Thogersen L, Olesen C, Schiott B, Morth JP, Moller JV, Nissen P (2013) Ion pathways in the sarcoplasmic reticulum Ca2+-ATPase. J Biol Chem 288(15):10759–10765. https://doi.org/10.1074/jbc.R112.436550
Toyoshima C, Iwasawa S, Ogawa H, Hirata A, Tsueda J, Inesi G (2013) Crystal structures of the calcium pump and sarcolipin in the Mg2+-bound E1 state. Nature 495(7440):260–264. https://doi.org/10.1038/nature11899
Winther AM, Bublitz M, Karlsen JL, Moller JV, Hansen JB, Nissen P, Buch-Pedersen MJ (2013) The sarcolipin-bound calcium pump stabilizes calcium sites exposed to the cytoplasm. Nature 495(7440):265–269. https://doi.org/10.1038/nature11900
Asahi M, Nakayama H, Tada M, Otsu K (2003) Regulation of sarco(endo)plasmic reticulum Ca2+ adenosine triphosphatase by phospholamban and sarcolipin: implication for cardiac hypertrophy and failure. Trends Cardiovasc Med 13(4):152–157
Arkin IT, Adams PD, MacKenzie KR, Lemmon MA, Brunger AT, Engelman DM (1994) Structural organization of the pentameric transmembrane alpha-helices of phospholamban, a cardiac ion channel. EMBO J 13(20):4757–4764
James P, Inui M, Tada M, Chiesi M, Carafoli E (1989) Nature and site of phospholamban regulation of the Ca2+ pump of sarcoplasmic reticulum. Nature 342(6245):90–92. https://doi.org/10.1038/342090a0
Simmerman HK, Jones LR (1998) Phospholamban: protein structure, mechanism of action, and role in cardiac function. Physiol Rev 78(4):921–947. https://doi.org/10.1152/physrev.1998.78.4.921
Toyofuku T, Kurzydlowski K, Tada M, MacLennan DH (1994) Amino acids Glu2 to Ile18 in the cytoplasmic domain of phospholamban are essential for functional association with the Ca(2+)-ATPase of sarcoplasmic reticulum. J Biol Chem 269(4):3088–3094
MacLennan DH, Kranias EG (2003) Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol 4(7):566–577. https://doi.org/10.1038/nrm1151
Mazzocchi G, Sommese L, Palomeque J, Felice JI, Di Carlo MN, Fainstein D, Gonzalez P, Contreras P, Skapura D, McCauley MD, Lascano EC, Negroni JA, Kranias EG, Wehrens XH, Valverde CA, Mattiazzi A (2016) Phospholamban ablation rescues the enhanced propensity to arrhythmias of mice with CaMKII-constitutive phosphorylation of RyR2 at site S2814. J Physiol 594(11):3005–3030. https://doi.org/10.1113/JP271622
Kaneko M, Hashikami K, Yamamoto S, Matsumoto H, Nishimoto T (2016) Phospholamban ablation using CRISPR/Cas9 system improves mortality in a murine heart failure model. PLoS One 11(12):e0168486. https://doi.org/10.1371/journal.pone.0168486
Valverde CA, Mazzocchi G, Di Carlo MN, Ciocci Pardo A, Salas N, Ragone MI, Felice JI, Cely-Ortiz A, Consolini AE, Portiansky E, Mosca S, Kranias EG, Wehrens XHT, Mattiazzi A (2019) Ablation of phospholamban rescues reperfusion arrhythmias but exacerbates myocardium infarction in hearts with Ca2+/calmodulin kinase II constitutive phosphorylation of ryanodine receptors. Cardiovasc Res 115(3):556–569. https://doi.org/10.1093/cvr/cvy213
Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, Kranias EG, MacLennan DH, Seidman JG, Seidman CE (2003) Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 299(5611):1410–1413. https://doi.org/10.1126/science.1081578
Alsina KM, Hulsurkar M, Brandenburg S, Kownatzki-Danger D, Lenz C, Urlaub H, Abu-Taha I, Kamler M, Chiang DY, Lahiri SK, Reynolds JO, Quick AP, Scott L Jr, Word TA, Gelves MD, Heck AJR, Li N, Dobrev D, Lehnart SE, Wehrens XHT (2019) Loss of protein phosphatase 1 regulatory subunit PPP1R3A promotes atrial fibrillation. Circulation. https://doi.org/10.1161/CIRCULATIONAHA.119.039642
Zhan R, Li X, Guo W, Liu X, Liu Z, Xu K, Tang B (2019) An aptamer-based near-infrared fluorescence nanoprobe for detecting and imaging of phospholamban micropeptide in cardiomyocytes. ACS Sens 4(3):733–739. https://doi.org/10.1021/acssensors.9b00026
Buffy JJ, Buck-Koehntop BA, Porcelli F, Traaseth NJ, Thomas DD, Veglia G (2006) Defining the intramembrane binding mechanism of sarcolipin to calcium ATPase using solution NMR spectroscopy. J Mol Biol 358(2):420–429. https://doi.org/10.1016/j.jmb.2006.02.005
Odermatt A, Taschner PE, Scherer SW, Beatty B, Khanna VK, Cornblath DR, Chaudhry V, Yee WC, Schrank B, Karpati G, Breuning MH, Knoers N, MacLennan DH (1997) Characterization of the gene encoding human sarcolipin (SLN), a proteolipid associated with SERCA1: absence of structural mutations in five patients with Brody disease. Genomics 45(3):541–553. https://doi.org/10.1006/geno.1997.4967
Odermatt A, Becker S, Khanna VK, Kurzydlowski K, Leisner E, Pette D, MacLennan DH (1998) Sarcolipin regulates the activity of SERCA1, the fast-twitch skeletal muscle sarcoplasmic reticulum Ca2+-ATPase. J Biol Chem 273(20):12360–12369. https://doi.org/10.1074/jbc.273.20.12360
Asahi M, Kurzydlowski K, Tada M, MacLennan DH (2002) Sarcolipin inhibits polymerization of phospholamban to induce superinhibition of sarco(endo)plasmic reticulum Ca2+-ATPases (SERCAs). J Biol Chem 277(30):26725–26728. https://doi.org/10.1074/jbc.C200269200
Asahi M, Sugita Y, Kurzydlowski K, De Leon S, Tada M, Toyoshima C, MacLennan DH (2003) Sarcolipin regulates sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) by binding to transmembrane helices alone or in association with phospholamban. Proc Natl Acad Sci U S A 100(9):5040–5045. https://doi.org/10.1073/pnas.0330962100
Asahi M, Otsu K, Nakayama H, Hikoso S, Takeda T, Gramolini AO, Trivieri MG, Oudit GY, Morita T, Kusakari Y, Hirano S, Hongo K, Hirotani S, Yamaguchi O, Peterson A, Backx PH, Kurihara S, Hori M, MacLennan DH (2004) Cardiac-specific overexpression of sarcolipin inhibits sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA2a) activity and impairs cardiac function in mice. Proc Natl Acad Sci U S A 101(25):9199–9204. https://doi.org/10.1073/pnas.0402596101
Babu GJ, Zheng Z, Natarajan P, Wheeler D, Janssen PM, Periasamy M (2005) Overexpression of sarcolipin decreases myocyte contractility and calcium transient. Cardiovasc Res 65(1):177–186. https://doi.org/10.1016/j.cardiores.2004.08.012
Babu GJ, Bhupathy P, Timofeyev V, Petrashevskaya NN, Reiser PJ, Chiamvimonvat N, Periasamy M (2007) Ablation of sarcolipin enhances sarcoplasmic reticulum calcium transport and atrial contractility. Proc Natl Acad Sci U S A 104(45):17867–17872. https://doi.org/10.1073/pnas.0707722104
Xie LH, Shanmugam M, Park JY, Zhao Z, Wen H, Tian B, Periasamy M, Babu GJ (2012) Ablation of sarcolipin results in atrial remodeling. Am J Phys Cell Phys 302(12):C1762–C1771. https://doi.org/10.1152/ajpcell.00425.2011
Periasamy M, Bhupathy P, Babu GJ (2008) Regulation of sarcoplasmic reticulum Ca2+ ATPase pump expression and its relevance to cardiac muscle physiology and pathology. Cardiovasc Res 77(2):265–273. https://doi.org/10.1093/cvr/cvm056
Shaikh SA, Sahoo SK, Periasamy M (2016) Phospholamban and sarcolipin: are they functionally redundant or distinct regulators of the sarco(endo)plasmic reticulum calcium ATPase? J Mol Cell Cardiol 91:81–91. https://doi.org/10.1016/j.yjmcc.2015.12.030
Bhupathy P, Babu GJ, Ito M, Periasamy M (2009) Threonine-5 at the N-terminus can modulate sarcolipin function in cardiac myocytes. J Mol Cell Cardiol 47(5):723–729. https://doi.org/10.1016/j.yjmcc.2009.07.014
Gramolini AO, Trivieri MG, Oudit GY, Kislinger T, Li W, Patel MM, Emili A, Kranias EG, Backx PH, Maclennan DH (2006) Cardiac-specific overexpression of sarcolipin in phospholamban null mice impairs myocyte function that is restored by phosphorylation. Proc Natl Acad Sci U S A 103(7):2446–2451. https://doi.org/10.1073/pnas.0510883103
Makarewich CA, Munir AZ, Schiattarella GG, Bezprozvannaya S, Raguimova ON, Cho EE, Vidal AH, Robia SL, Bassel-Duby R, Olson EN (2018) The DWORF micropeptide enhances contractility and prevents heart failure in a mouse model of dilated cardiomyopathy. Elife:7. https://doi.org/10.7554/eLife.38319
Liddy KA, White MY, Cordwell SJ (2013) Functional decorations: post-translational modifications and heart disease delineated by targeted proteomics. Genome Med 5(2):20. https://doi.org/10.1186/gm424
Qing G, Lu Q, Xiong Y, Zhang L, Wang H, Li X, Liang X, Sun T (2017) New opportunities and challenges of smart polymers in post-translational modification proteomics. Adv Mater 29(20). https://doi.org/10.1002/adma.201604670
Nussinov R, Tsai CJ, Xin F, Radivojac P (2012) Allosteric post-translational modification codes. Trends Biochem Sci 37(10):447–455. https://doi.org/10.1016/j.tibs.2012.07.001
Zhao X (2018) SUMO-mediated regulation of nuclear functions and signaling processes. Mol Cell 71(3):409–418. https://doi.org/10.1016/j.molcel.2018.07.027
Sarangi P, Zhao X (2015) SUMO-mediated regulation of DNA damage repair and responses. Trends Biochem Sci 40(4):233–242. https://doi.org/10.1016/j.tibs.2015.02.006
Hendriks IA, Vertegaal AC (2016) A comprehensive compilation of SUMO proteomics. Nat Rev Mol Cell Biol 17(9):581–595. https://doi.org/10.1038/nrm.2016.81
Hickey CM, Wilson NR, Hochstrasser M (2012) Function and regulation of SUMO proteases. Nat Rev Mol Cell Biol 13(12):755–766. https://doi.org/10.1038/nrm3478
Gareau JR, Lima CD (2010) The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol 11(12):861–871. https://doi.org/10.1038/nrm3011
Kho C, Lee A, Jeong D, Oh JG, Chaanine AH, Kizana E, Park WJ, Hajjar RJ (2011) SUMO1-dependent modulation of SERCA2a in heart failure. Nature 477(7366):601–605. https://doi.org/10.1038/nature10407
Kho C, Lee A, Jeong D, Oh JG, Gorski PA, Fish K, Sanchez R, DeVita RJ, Christensen G, Dahl R, Hajjar RJ (2015) Small-molecule activation of SERCA2a SUMOylation for the treatment of heart failure. Nat Commun 6:7229–7211. https://doi.org/10.1038/ncomms8229
Lee A, Jeong D, Mitsuyama S, Oh JG, Liang L, Ikeda Y, Sadoshima J, Hajjar RJ, Kho C (2014) The role of SUMO-1 in cardiac oxidative stress and hypertrophy. Antioxid Redox Signal 21(14):1986–2001. https://doi.org/10.1089/ars.2014.5983
Tilemann L, Lee A, Ishikawa K, Aguero J, Rapti K, Santos-Gallego C, Kohlbrenner E, Fish KM, Kho C, Hajjar RJ (2013) SUMO-1 gene transfer improves cardiac function in a large-animal model of heart failure. Sci Transl Med 5(211):211ra159. https://doi.org/10.1126/scitranslmed.3006487
Oh JG, Watanabe S, Lee A, Gorski PA, Lee P, Jeong D, Liang L, Liang Y, Baccarini A, Sahoo S, Brown BD, Hajjar RJ, Kho C (2018) miR-146a suppresses SUMO1 expression and induces cardiac dysfunction in maladaptive hypertrophy. Circ Res 123(6):673–685. https://doi.org/10.1161/CIRCRESAHA.118.312751
Du Y, Liu P, Xu T, Pan D, Zhu H, Zhai N, Zhang Y, Li D (2018) Luteolin modulates SERCA2a leading to attenuation of myocardial ischemia/reperfusion injury via sumoylation at lysine 585 in mice. Cell Physiol Biochem 45(3):883–898. https://doi.org/10.1159/000487283
Ali I, Conrad RJ, Verdin E, Ott M (2018) Lysine acetylation goes global: from epigenetics to metabolism and therapeutics. Chem Rev 118(3):1216–1252. https://doi.org/10.1021/acs.chemrev.7b00181
Menzies KJ, Zhang H, Katsyuba E, Auwerx J (2016) Protein acetylation in metabolism - metabolites and cofactors. Nat Rev Endocrinol 12(1):43–60. https://doi.org/10.1038/nrendo.2015.181
Shen Y, Wei W, Zhou DX (2015) Histone acetylation enzymes coordinate metabolism and gene expression. Trends Plant Sci 20(10):614–621. https://doi.org/10.1016/j.tplants.2015.07.005
Verdin E, Ott M (2015) 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol 16(4):258–264. https://doi.org/10.1038/nrm3931
McKinsey TA (2012) Therapeutic potential for HDAC inhibitors in the heart. Annu Rev Pharmacol Toxicol 52:303–319. https://doi.org/10.1146/annurev-pharmtox-010611-134712
Yang Q, Vijayakumar A, Kahn BB (2018) Metabolites as regulators of insulin sensitivity and metabolism. Nat Rev Mol Cell Biol 19(10):654–672. https://doi.org/10.1038/s41580-018-0044-8
Chuang DM, Leng Y, Marinova Z, Kim HJ, Chiu CT (2009) Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci 32(11):591–601. https://doi.org/10.1016/j.tins.2009.06.002
Lane AA, Chabner BA (2009) Histone deacetylase inhibitors in cancer therapy. J Clin Oncol 27(32):5459–5468. https://doi.org/10.1200/JCO.2009.22.1291
Lu Z, Scott I, Webster BR, Sack MN (2009) The emerging characterization of lysine residue deacetylation on the modulation of mitochondrial function and cardiovascular biology. Circ Res 105(9):830–841. https://doi.org/10.1161/CIRCRESAHA.109.204974
Chen X, Zhang X, Gross S, Houser SR, Soboloff J (2019) Acetylation of SERCA2a, another target for heart failure treatment? Circ Res 124(9):1285–1287. https://doi.org/10.1161/CIRCRESAHA.119.315017
Quick AP, Wang Q, Philippen LE, Barreto-Torres G, Chiang DY, Beavers D, Wang G, Khalid M, Reynolds JO, Campbell HM, Showell J, McCauley MD, Scholten A, Wehrens XH (2017) SPEG (striated muscle preferentially expressed protein kinase) is essential for cardiac function by regulating junctional membrane complex activity. Circ Res 120(1):110–119. https://doi.org/10.1161/CIRCRESAHA.116.309977
Quan C, Li M, Du Q, Chen Q, Wang H, Campbell D, Fang L, Xue B, MacKintosh C, Gao X, Ouyang K, Wang HY, Chen S (2019) SPEG controls calcium reuptake into the sarcoplasmic reticulum through regulating SERCA2a by its second kinase-domain. Circ Res 124(5):712–726. https://doi.org/10.1161/CIRCRESAHA.118.313916
Yang X, Qian K (2017) Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol 18(7):452–465. https://doi.org/10.1038/nrm.2017.22
Stammers AN, Susser SE, Hamm NC, Hlynsky MW, Kimber DE, Kehler DS, Duhamel TA (2015) The regulation of sarco(endo)plasmic reticulum calcium-ATPases (SERCA). Can J Physiol Pharmacol 93(10):843–854. https://doi.org/10.1139/cjpp-2014-0463
Lancel S, Zhang J, Evangelista A, Trucillo MP, Tong X, Siwik DA, Cohen RA, Colucci WS (2009) Nitroxyl activates SERCA in cardiac myocytes via glutathiolation of cysteine 674. Circ Res 104(6):720–723. https://doi.org/10.1161/CIRCRESAHA.108.188441
Kawase Y, Ly HQ, Prunier F, Lebeche D, Shi Y, Jin H, Hadri L, Yoneyama R, Hoshino K, Takewa Y, Sakata S, Peluso R, Zsebo K, Gwathmey JK, Tardif JC, Tanguay JF, Hajjar RJ (2008) Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J Am Coll Cardiol 51(11):1112–1119. https://doi.org/10.1016/j.jacc.2007.12.014
Lyon AR, Sato M, Hajjar RJ, Samulski RJ, Harding SE (2008) Gene therapy: targeting the myocardium. Heart 94(1):89–99. https://doi.org/10.1136/hrt.2007.116483
Watanabe S, Ishikawa K, Plataki M, Bikou O, Kohlbrenner E, Aguero J, Hadri L, Zarragoikoetxea I, Fish K, Leopold JA, Hajjar RJ (2018) Safety and long-term efficacy of AAV1.SERCA2a using nebulizer delivery in a pig model of pulmonary hypertension. Pulm Circ 8(4):2045894018799738. https://doi.org/10.1177/2045894018799738
Hajjar RJ, Zsebo K, Deckelbaum L, Thompson C, Rudy J, Yaroshinsky A, Ly H, Kawase Y, Wagner K, Borow K, Jaski B, London B, Greenberg B, Pauly DF, Patten R, Starling R, Mancini D, Jessup M (2008) Design of a phase 1/2 trial of intracoronary administration of AAV1/SERCA2a in patients with heart failure. J Card Fail 14(5):355–367. https://doi.org/10.1016/j.cardfail.2008.02.005
Jaski BE, Jessup ML, Mancini DM, Cappola TP, Pauly DF, Greenberg B, Borow K, Dittrich H, Zsebo KM, Hajjar RJ, Calcium up-regulation by percutaneous administration of gene therapy in cardiac disease trial I (2009) calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J Card Fail 15(3):171–181. https://doi.org/10.1016/j.cardfail.2009.01.013
Zsebo K, Yaroshinsky A, Rudy JJ, Wagner K, Greenberg B, Jessup M, Hajjar RJ (2014) Long-term effects of AAV1/SERCA2a gene transfer in patients with severe heart failure: analysis of recurrent cardiovascular events and mortality. Circ Res 114(1):101–108. https://doi.org/10.1161/CIRCRESAHA.113.302421
Greenberg B, Butler J, Felker GM, Ponikowski P, Voors AA, Desai AS, Barnard D, Bouchard A, Jaski B, Lyon AR, Pogoda JM, Rudy JJ, Zsebo KM (2016) Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet 387(10024):1178–1186. https://doi.org/10.1016/S0140-6736(16)00082-9
Fargnoli AS, Katz MG, Yarnall C, Isidro A, Petrov M, Steuerwald N, Ghosh S, Richardville KC, Hillesheim R, Williams RD, Kohlbrenner E, Stedman HH, Hajjar RJ, Bridges CR (2013) Cardiac surgical delivery of the sarcoplasmic reticulum calcium ATPase rescues myocytes in ischemic heart failure. Ann Thorac Surg 96(2):586–595. https://doi.org/10.1016/j.athoracsur.2013.04.021
Jessup M, Greenberg B, Mancini D, Cappola T, Pauly DF, Jaski B, Yaroshinsky A, Zsebo KM, Dittrich H, Hajjar RJ, Calcium upregulation by percutaneous administration of gene therapy in cardiac disease I (2011) Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation 124(3):304–313. https://doi.org/10.1161/CIRCULATIONAHA.111.022889
Greenberg B, Yaroshinsky A, Zsebo KM, Butler J, Felker GM, Voors AA, Rudy JJ, Wagner K, Hajjar RJ (2014) Design of a phase 2b trial of intracoronary administration of AAV1/SERCA2a in patients with advanced heart failure: the CUPID 2 trial (calcium up-regulation by percutaneous administration of gene therapy in cardiac disease phase 2b). JACC Heart Fail 2(1):84–92. https://doi.org/10.1016/j.jchf.2013.09.008
Hulot JS, Salem JE, Redheuil A, Collet JP, Varnous S, Jourdain P, Logeart D, Gandjbakhch E, Bernard C, Hatem SN, Isnard R, Cluzel P, Le Feuvre C, Leprince P, Hammoudi N, Lemoine FM, Klatzmann D, Vicaut E, Komajda M, Montalescot G, Lompre AM, Hajjar RJ, Investigators A-H (2017) Effect of intracoronary administration of AAV1/SERCA2a on ventricular remodelling in patients with advanced systolic heart failure: results from the AGENT-HF randomized phase 2 trial. Eur J Heart Fail 19(11):1534–1541. https://doi.org/10.1002/ejhf.826
Funding
This study is supported by grants from the Tianjin Outstanding Youth Science Foundation (17JCJQJC46200), the National Natural Science Foundation of China (NSFC 81774050), the Natural Science Foundation of Tianjin (17JCYBJC29000), and the Ministry of Education of People’s Republic of China “Program for Innovative Research Team in University” (No. IRT_16R54).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhihao, L., Jingyu, N., Lan, L. et al. SERCA2a: a key protein in the Ca2+ cycle of the heart failure. Heart Fail Rev 25, 523–535 (2020). https://doi.org/10.1007/s10741-019-09873-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-019-09873-3