
Abstract
Chronic thromboembolic pulmonary hypertension (CTEPH) is an established long-term complication of pulmonary thromboembolism (PTE). However, studies have shown that many patients with a definitive CTEPH diagnosis have no history of symptomatic PTE, suggesting that PTE is not the only cause of CTEPH. Despite extensive progress in research on pulmonary hypertension in recent years, due to a lack of relevant studies on the pathophysiology of CTEPH, implementing pulmonary endarterectomy (PEA) in patients has many challenges, and the prognosis of patients with CTEPH is still not optimistic. Therefore, revealing the pathogenesis of CTEPH would be of great significance for understanding the occurrence and development of CTEPH, developing relevant drug treatment studies and formulating intervention strategies, and may provide new preventive measures. This article summarizes the current research progress in CTEPH pathogenesis from the perspective of risk factors related to medical history, abnormal coagulation and fibrinolytic mechanisms, inflammatory mechanisms, genetic susceptibility factors, angiogenesis, in situ thrombosis, vascular remodeling, and other aspects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Definition and clinical features of CTEPH
Chronic thromboembolism pulmonary hypertension (CTEPH) is classified as group 4 in the current classification of pulmonary hypertension (PH) [1]. CTEPH is a precapillary PH, and the diagnosis of CTEPH is based on findings obtained after at least 3 months of effective anticoagulation to discriminate this condition from “subacute” pulmonary embolism (PE). These findings indicate a pulmonary arterial pressure (mPAP) ≥ 25 mmHg and a pulmonary wedge pressure ≤ 15 mmHg, as measured by right cardiac catheterization, mismatched perfusion defects on lung scan, and specific diagnostic signs for CTEPH seen by multidetector CT angiography, MR imaging, or conventional pulmonary cineangiography, such as ring-like stenoses, webs/slits, and chronic total occlusions [2].
CTEPH is a rare and progressive pulmonary vascular disease, caused by pulmonary artery and pulmonary arteriole obstruction by near-end thrombus embolism or distal vascular remodeling, increasing right heart load and pulmonary vascular resistance, leading to failure of the right heart, seriously affecting the quality of life of patients. Disease detection requires clinical suspicion followed by a thorough evaluation including radiographic and hemodynamic assessments. Once diagnosed, consultation with a CTEPH expert is recommended to formulate a management plan and to assess for operability with pulmonary endarterectomy [3]. In a review study based on published literature, and hospital databases, it was anticipated that the occurrence of CTEPH in all PE survivors was approximately 4% (0.1–9.1%) [4]. These results are similar to those of the INFORM Study (3.8%) [5]. A prospective long-term follow-up study by Pengo monitored 223 patients after acute PE but without prior venous thromboembolism (VTE), for up to 10 years. The cumulative incidence rates of symptomatic CTEPH were 1.0%, 3.1%, and 3.8% at 6 months, 1 year, and 2 years, respectively. No cases occurred among the patients after 2 years. Based on these data, the annual incidence of CTEPH can be estimated at 3 to 5 cases per 100,000 in the USA and Europe, with less than one third of cases (7–29%) being diagnosed based on reviewed databases [6] and the vast majority of CTEPH cases being detected within 2 years of an acute thromboembolic event [6, 7]. A Spanish registration study showed that the prevalence and incidence of CTEPH were 3.2 cases per million and 0.9 cases each year [8]. In recent years, a study suggested that there are 3–30 people with CTEPH per one million people in the general population and conservatively estimated that 300,000 people in the USA have acute pulmonary thromboembolism (PTE) each year, presumably causing nearly 3000 people to exhibit CTEPH each year [9]. Compared with the USA and Europe, the incidence of PE in the Asian population is reported to be lower [10]. According to a multicenter registration study, which records pulmonary embolism incidence and fatality trends in Chinese hospitals from 1997 to 2008, the incidence of PE of all hospitalized patients increased from 0.02% in 1997 to 0.14% in 2003 and then remained at 0.1%, while the mortality decreased dramatically from 25.1% in 1997 to 8.6% in 2008. In addition, the incidence of PE is reported to be higher in northern China than in southern China, whereas the mortality is lower [11]. Another consecutive cohort study showed that the incidence rates of CTEPH of Chinese patients after acute PE were 0.8% in the first year, 1.3% in the second year, and 1.7% in the third year [7]. These reports confirm that the Chinese PE as well as CTEPH incidence are lower than in the USA and Europe. In the International CTEPH Registry from 2007 to 2009, of 679 newly diagnosed (patients < 6 months after diagnosis), 75% had a previous PE and most were diagnosed in the sixth decade of life [12]. Most frequently, patients reported New York Heart Association (NYHA) functional class III/IV symptoms at the time of diagnosis [13]. CTEPH is a treatable and potentially curable form of PH; however, due to a lack of research on the pathophysiology of CTEPH, its diagnosis is often delayed and is frequently missed. Moreover, the implementation of pulmonary endarterectomy (PEA) in patients has strict requirements, and the prognosis of patients with CTEPH is still not optimistic. Without intervention, the rate of survival is very low and proportional to the degree of PH at the time of diagnosis. In a study by Riedel, the survival rates at 5 years were 30% among patients with that exceeded 40 mmHg and only 10% among those with an mPAP value that exceeded 50 mmHg at the time of diagnosis [14]. In one study by Lewczuk, an mPAP of 30 mmHg appeared to be the threshold for a poor prognosis [15]. In conclusion, CTEPH is a serious disease that threatens human life, and understanding the pathophysiological mechanism is necessary for its early identification and treatment.
Relationship of CTEPH with PTE
CTEPH is an established long-term complication of PE, and the European CTEPH Registration study found that 74.8% of CTEPH patients had a history of pulmonary thrombosis and that 56.1% had a history of deep venous thrombosis (DVT) [12], indicating that CTEPH is closely associated with thrombi. Many people have tried to establish animal models to illustrate the disease’s mechanism. Among these, pigs, dogs, rabbits, and mice have been the most intensively studied animal models. However, studies have found that repeated administration of thromboembolic substances does not cause CTEPH [16] in animal models; the embolization of large thrombi through catheters to the pulmonary artery causes only a mild increase in pulmonary arterial pressure and does not cause long-term cardiovascular effects, with the exception of hypoxemia. This phenomenon was reversed when methotrexate was used to inhibit thrombotic fibrin dissolution or ligation of the lateral pulmonary artery [17]. Further studies of pigs have found that only the use of spring rings with tissue adhesives to embolize their pulmonary arteries can produce histological features similar to those in humans with CTEPH [17]. Combined with the reports that lysis of pulmonary emboli is much faster in dogs [18] and pigs [19] than in human beings due to the higher activity of u-PA as well as a closer association of u-PA activity with animals’ platelets and pulmonary artery endothelial cells, it is clear that unresolved pulmonary thrombi play an essential role in the development of CTEPH. CTEPH arises from unresolved pulmonary thrombi, which lead to chronic obstruction of the pulmonary artery tree, small-vessel arteriopathy, and high pulmonary vascular resistance [20]. Clinical observation also confirmed that patients suffering from residual pulmonary obstruction after acute pulmonary embolism have a significantly higher risk of CTEPH than who do not [21].
Pulmonary residual thrombosis after acute PTE is common, but it is uncommon for acute thrombosis rich in fibrin to transform into intravascular scars, which leads to persistent obstruction of the pulmonary artery and increased pulmonary vascular resistance, which in turn further develops into CTEPH [22]. In previous studies, the incidence rates of CTEPH in patients with symptomatic PTE were 0.1% to 0.5% [20]; however, recent studies have shown that the incidence rates of CTEPH in these patients are higher, ranging from 0.1% to 9.1% [23]. CTEPH as a direct consequence of a symptomatic PTE is relatively rare, with a large number of clearly diagnosed CTEPH patients not having a symptomatic history of PTE [24]. The incidence rates of CTEPH in a prospective study of 614 patients in China who were followed up for 3.3 years after the onset of acute PE were 0.8%, 1.3%, and 1.7% in years 1, 2, and 3, respectively; 3 years later, there were no CTEPH patients [7]. In this study, conducted by Yang in China, patients diagnosed with a first episode of acute PE were included, patients with previously confirmed CTEPH or PH at the time of acute PE were excluded, and patients with other medical conditions that could have caused non-thromboembolic PH were excluded. Patients with transient risk factors (including trauma, recent surgery, > 3 days of immobilization for medical reasons, recent prolonged immobility, deep venous catheterization, pregnancy or being in the peripartum period, and the use of oral contraceptives or hormone replacement therapy) or permanent risk factors (including obesity, active malignancy, chronic heart or respiratory failure, thrombophilia, cerebrovascular disease, and varicose veins) for PE were classified as having “provoked” PE. PE occurring in the absence of risk factors was defined as “unprovoked” PE. The numbers of cases of unprovoked PE, PE with transient PE risk factors, and PE with permanent PE risk factors were 204 (33.2%), 125 (20.4%), 285 (46.4%), and 316 (51.5%) patients who had concomitant DVT. History of varicose veins of the lower limbs (HR = 4.3, 95% CI = 1.2–15.4; p = 0.024), SPAP > 50 mmHg when acute PE occurs (HR = 23.5, 95% CI = 2.7–207.6; p = 0.005), moderately dangerous PE (HR = 1.2, 95% CI = 1.0–1.4; p = 0.030), and a CT blocking index of more than 30% after 3 months of acute PE (HR = 42.5, 95% CI = 4.4–409.8; p = 0.001) increased the risk of CTEPH. In another prospective cohort study by Miniati, 834 patients were prospectively evaluated for suspected PE for a median period of 2.1 years (range, 0–4.8 years); the survival rates of patients with PE (n = 320) were compared with those of patients without PE (n = 514) [25], and all patients underwent continuous lung scans. The incidence of CTEPH was 1.3%, 60% of patients with CTEPH had a history of moderate PE, and no recurrence of PE was found in continuous lung scans. Thus, it is suggested that PTE is not the only cause of CTEPH (Table 1).
Pathogenesis of CTEPH
CTEPH is mainly caused by undissolved thrombosis or repeated embolism of the pulmonary vasculature, which results in the dysfunction of the pulmonary vascular endothelium, secretory imbalance of vascular active substances and cytokines, and pulmonary vascular constriction, causing increased pulmonary vascular resistance, pulmonary vascular remodeling, and an irreversible increase in pulmonary artery pressure. This process eventually results in severe right heart failure. Unlike pulmonary arterial hypertension (PAH), which mainly involves blood vessels that are less than 300 μm, CTEPH mainly involves large vessels. In this process, the variety of factors involved in the formation of CTEPH mainly includes the following: risk factors related to patient history, abnormal coagulation and fibrinolytic mechanisms, inflammatory mechanisms, genetic susceptibility factors, angiogenesis, in situ thrombosis, and vascular remodeling. This article summarizes the current research progress on the pathogenesis of CTEPH from these aspects.
Risk factors related to medical history
Non-type O blood
A study found that the occurrence of CTEPH is related to blood type, and CTEPH is more common in non-type O blood. Bonderman found that the prevalence of non-type O blood is significantly higher than that of type O blood in CTEPH patients (OR = 2.09, 95% CI = 1.12~3.94). The study confirmed that non-type O blood increases the risk of VTE and CTEPH. The possible mechanistic factors, including von Willebrand factor, factor VIII, P-selectin, and tumor necrosis factor (TNF), were elevated in non-type O blood, and these factors were associated with the occurrence of VTE [9].
Chronic inflammatory diseases
Studies have confirmed that chronic inflammatory diseases including inflammatory bowel disease, osteomyelitis and anticardiolipin antibody syndrome (APS), chronic venous ulcers, and chronic infections of indwelling venous catheters may increase the risk of CTEPH [33,34,35].
Malignant tumors
A history of cancer is more common in CTEPH patients than in patients with other types of PH [9]. It is well known that cancer results in a high coagulation state that increases the risk of thrombosis, which is associated with the type of cancer and anticancer regimen (including the choice of chemotherapy drugs, the use of high-dose steroids, and the application of exogenous erythropoietin) [9]. The mechanism of cancer-induced thrombosis remains unclear and may include the following: [1] The patient has no corresponding clinical symptoms, resulting in a delay between the occurrence of PTE and the standard anticoagulant. Cronin found that asymptomatic PTE occurs in 3.3% of cancer patients evaluated using enhanced CT scans. [2] Verso found that 27~67% of patients have asymptomatic catheter-related DVT because cancer patients often need to have an indwelling vein catheter. [3] Neutrophils play an important role in the initial thrombosis, and the reduction in neutrophils delays the removal of thrombosis and causes up to three times the normal deposition of collagen fibers in the wall of blood vessels, which was found through a rat thrombosis model. Due to chemotherapy, neutropenia is common in cancer patients, and it is speculated that cancer patients may have a delay in the removal of thrombi due to neutropenia [9]. Bonderman et al. [34] studied 433 patients with CTEPH and 254 patients without CTEPH and confirmed the correlation between CTEPH and malignancy (OR = 3.76, 95% CI = 1.47–10.43, p = 0.005).
Hypothyroidism
In the general population, hypothyroidism and thyroid hormone replacement therapy account for approximately 9.5% of patients, but the incidence of hypothyroidism and thyroid hormone replacement is significantly increased in CTEPH patients [9]. The series results of three European multicenter studies showed that 19.9% of CTEPH patients had simultaneous thyroid hormone replacement therapy and that 6.2% of CTEPH patients had a history of hypothyroidism [9]. Whether thyroid dysfunction or thyroid hormone replacement therapy increases the risk of CTEPH remains unknown. Hypothyroidism may be a high coagulation state, with an increased risk of venous thrombosis in patients with hypothyroidism compared that in the control group (RR = 1.64, 95% CI = 1.59~1.60) [9].
Special medical history
Many studies have evaluated the incidence and related risk factors of CTEPH after acute PTE. Studies have shown that merging certain medical histories increases the risk of CTEPH in patients. A history of combined splenectomy is related to the occurrence of CTEPH, which may be due to the abnormal red blood cells not being filtered by the spleen or to the increase in reactive platelets causing the occurrence of chronic embolism [36]. Pengo et al. [6] have shown that the risk factors for the occurrence of CTEPH include a history of PTE, the greater perfusion defect in patients with acute PTE, and idiopathic PTE. The evidence is particularly strong between the risk factors associated with medical history and CTEPH.
Abnormal coagulation and fibrinolysis in CTEPH
Increased coagulation factor VIII levels in CTEPH
One study showed that elevated plasma levels of coagulation factor VIII are a risk factor for VTE [37]. Bonderman et al. [38] showed that the coagulation factor VIII levels in CTEPH patients (n = 122) were elevated and lasted 1 year after PEA compared to those in patients with PTE (n = 88) and healthy controls (n = 82). A coagulation factor VIII level > 230 IU/dl was significantly correlated with recurrent VTE and occurred in 41% of CTEPH patients. These findings suggest another potential mechanism for the development of CTEPH; however, not all patients have a significant increase in coagulation factor VIII levels. The causal relationship between the increase in coagulation factor VIII levels and CTEPH is not clear. These data suggest that elevated coagulation factor VIII levels may be one of the possible causes of disease progression in CTEPH patients.
Fibrinolysis abnormalities
Fibrinolysis is the initial stage of embolus decomposition, and fibrinolytic defects are considered to be the pathophysiological process of CTEPH. A decreased potential of plasma fibrin dissolution is a risk factor for VTE, and some research indicates that this risk factor is similar to CTEPH [39]. In patients with CTEPH, there may be a deficiency in or dysfunction of the fibrinolytic enzyme or a resistance to fibrinolysis [40].
Fibrinolytic system damage
The dissolution of thrombi depends on a normal fibrinolytic system, and fibrinolytic defects may be an important pathological link in the incomplete dissolution of thrombi in CTEPH patients. Physiological fibrinolysis is a very complex process that requires fibrinolytic enzymes, tissue plasminogen activators (t-PA), plasminogen activator inhibitor-1 (PAI-1), other regulatory proteins, and endothelial interactions at the site of vascular injury, where fibrin dissolves eventually; PAI-1 is the main inhibitor of T-PA and regulates the activity of the fibrinolytic system in vivo by combining with T-PA; PAI-1 can excessively stabilize thrombosis and promote the accumulation of collagen and other extracellular matrix proteins, which causes scar formation [41]. Vuylsteke et al. [42] found that the plasma levels of T-PA and PAI-1 were significantly increased in CTEPH patients, but there was no statistically significant difference between the T-PA and PAI-1 activity levels and those in the control group. Lang et al. [43] showed that there was no statistically significant difference in the basic levels of T-PA and PAI-1 in pulmonary endothelial cells in CTEPH patients compared with the control group. Therefore, the reduction in fibrinolysis caused by the activity or functional abnormalities of T-PA; and PAI-1 cannot fully explain the pathogenesis of CTEPH.
Thrombosis resistance to fibrin dissolution
Fibrin is the main component of acute thrombosis, and it is speculated that defective fibrin further affects the fibrinolytic process, which leads to CTEPH. Studies have shown that incomplete thrombosis can stimulate the remodeling of blood vessels into scar tissue. Morris et al. [40, 44] showed that fibrin extracted from CTEPH patients is resistant to fibrinolytic enzyme-mediated cleavage compared with that in the healthy control group. Morris et al. [45] speculated that this abnormal fibrinogen may impair fibrinolysis, promoting the development of CTEPH. However, not all CTEPH patients have abnormal fibrin. Since fibrin β chains are the most susceptible to resistance, Miniati et al. [46] compared the degradation rate of fibrin β chains by fibrinolytic enzymes in four groups of patients: normal control populations, CTEPH, other types of PH, and previous PTE history (not concurrent CTEPH); the researchers found that compared with normal control groups, the degradation rate of the fibrin β chain decreased in the other three groups of patients and that the CTEPH group and other types of PH patients showed more significant differences. These studies not only verify the conclusion of Morris that fibrin has resistance to fibrinolytic enzymes in CTEPH patients but also prove that fibrin resistance to fibrinolytic enzyme is not unique in CTEPH. In addition, Suntharalingam et al. [47] showed that fibrinogen AαThr312A1α allele mutations significantly increase the risk of CTEPH and that these alleles participate in regulating fibrin α-α crosslinking, which not only increases the risk of thromboembolism but also causes fibrinolytic resistance. Investigations by Standeven et al. [48] and other in vitro experimental studies have also confirmed the abovementioned point of view: human Aα312 bit Thr mutation to A1α, fibrin thickening, and elasticity enhancement. In summary, although damage to the fibrin dissolution system is an important pathogenetic mechanism of CTEPH, it can explain the etiology in only some CTEPH patients.
Inflammatory mechanisms
Under normal physiological conditions, endothelial cells mainly promote vasodilation and local fibrin dissolution by inhibiting the coagulation pathway and the activity of white blood cells and platelets. After thrombosis, white blood cells migrate into the region, initiating fibrinolytic systems and angiogenesis. Although the mechanism is not yet clear, it is currently recognized that platelet endothelial cell adhesion molecule 1 (PECAM-1) plays an important role. Kellermair et al. [49] found that the venous thrombosis was extensive and stable in PECAM-1 defective mice, and macrophage aggregation and neovascularization decreased, but the fibrosis composition increased obviously, which showed that a transient inflammatory response was beneficial to the dissolution of normal physiological thrombosis but that an excessive inflammatory response was not conducive to thrombosis.
Clinical studies [9] have found that staphylococcal infection, pacemaker infection, ventricular-atrial shunts, and other factors are related to the occurrence of CTEPH, that is, the infection mentioned above is not conducive to thrombosis. One study found that 3.7% of CTEPH patients had a previous history of intravascular implant infection; compared with other types of PH, those who had a history of previous intravascular implant devices were more likely have CTEPH (up to 76.4 times) (96% CI = 7.76~10.351) [9]. Through the study of seven patients with CTEPH combined with hydrocephalus and a parallel ventricular-atrial shunt, it was found that staphylococcal infection was detected in six cases of pulmonary endometrial exfoliation. Further animal experiments confirmed that the level of transforming growth factor β (TGF-β) in a mouse model of staphylococcal infections increased and led to delayed thrombosis. Bacterial infections can increase TGF-β and connective tissue growth factors that promote fibrosis, thus promoting collagen synthesis and thrombosis [50].
A number of studies have found that after other factors were corrected, the levels of peripheral blood interleukin 6 (IL-6), IL-8, IL-10, and γ interferon-induced protein 10 (IP-10), monocyte chemotactic protein-1 (MCP-1), monocyte interferon gamma inducing factor (MIG), and macrophage inflammatory protein-9 (MMP-9) were significantly increased in the CTEPH patients group compared to the levels in the healthy control group [51]. IP-10 can induce the proliferation of fibroblasts and may play important roles in thrombosis and the formation of chronic intravascular scars. Zabini et al. [51] found that compared with lung tissue in the healthy control group, the lung tissue harvested from PEA in CTEPH patients showed significantly higher levels of inflammatory markers (including IL-6, MCP-1, IP-10, Mip1α, and chemokine ligand-5). The level of C-reactive protein (CRP) in CTEPH patients was also higher than that in other PH patients [52].
Genetic susceptibility factors
The specific genes associated with CTEPH have not been identified in the present study.
The Leiden genetic mutation of coagulation factor V (FV Leiden), prothrombin gene, protein S, protein C, and thrombin-resistant mutations can increase the risk of VTE [53]. The roles of these mutations in the occurrence and development of CTEPH are still not clear. Wolf et al. [54] studied 46 cases of CTEPH and 64 patients with idiopathic pulmonary arterial hypertension (IPAH). The hereditary high coagulation state was evaluated; however, there were no significant increases in coagulation levels in patients with the mutations compared with the coagulation level in the healthy control group. Wong et al. [55] studied 45 patients with CTEPH and 200 other types of PH patients with detection of the abovementioned mutations; the FV Leiden mutation was increased only in the CTEPH Caucasians group (29% vs 7.8%, p = 0.001). These studies show that there is a lack of a clear causal relationship between genetic mutation and the development of CTEPH.
ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13), also known as vascular hemophilia factor lysis protease, regulates the size of vascular hemophilia factor and plays a fundamental role in hemostasis. A severe lack of ADAMTS13 leads to thrombotic thrombocytopenic purpura. Large numbers of rare and low-frequency coded nucleotide variants of ADAMTS13 were found in patients with DVT compared with the control group. In addition, these patients had a lower plasma level of ADAMTS13 [56]. Similar to other thrombotic diseases, the non-O blood groups are common in CTEPH, suggesting a mechanism by which von Willebrand factor levels are increased. The study of ABO blood type has shown that the levels of factor VIII and VWF are significantly reduced in the type O blood population [9]. A large proportion of the variation in VWF levels is genetically determined, with 30% due to ABO groups [57]. The ADAMTS13 gene is situated ~ 200 kb downstream of ABO and is genetically regulated with 20% of its variance attributable to common variants at the ADAMTS13 locus [58]. ADAMTS13 is not known to vary with ABO groups in healthy cohorts [59]. Recently, Toshner et al. [60] reported that CTEPH patients had decreased ADAMTS13 (adjusted β (95% CI) = − 23.4 (− 30.9 to − 15.1)%, p < 0.001) and increased VWF levels (β = + 75.5 (44.8 to 113)%, p < 0.001) compared to healthy controls. After reversal of pulmonary hypertension by pulmonary endarterectomy surgery, the ADAMTS13 levels were still low. The authors identified a genetic variant near the ADAMTS13 gene associated with ADAMTS13 protein that accounted for ~ 8% of the variation in levels. The ADAMTS13-VWF axis is dysregulated in CTEPH. This is unrelated to pulmonary hypertension, disease severity, or markers of systemic inflammation and implicates the ADAMTS13-VWF axis in CTEPH pathobiology.
Feng et al. [61] reported mutations in the bone morphogenetic protein II receptor (BMPR2) gene in patients diagnosed with CTEPH. However, earlier and larger studies did not support the role of BMPR2 mutations in the pathogenesis of CTEPH [62].
Vascular regeneration and thrombus dissolution
In CTEPH, angiogenesis relies on blood vessels from the systemic bronchial arteries. After the pulmonary artery is occluded, these vessels usually spread to the pulmonary arteries and open preexisting collaterals. During the healing process, the positive regulatory factors of angiogenesis dominate, and endothelial cells are activated, penetrating occlusive thrombosis [63]. Previous animal studies have shown that recanalization of the venous wall may occur within 24 h of thrombosis. In the early stages of thrombosis, the thrombus begins to shrink from the venous wall, resulting in the formation of intracellular pockets between the thrombus body and the inner membrane of the venous wall. The presence of vascular channels is associated with the expression of EGF and bFGF, which are believed to induce thrombosis. In tissue harvested from PEA, collagen secretory cells [64] are involved in the formation of the microenvironment, resulting in dysfunctional endothelial cells that do not support angiogenesis [63]. Previous animal studies have shown that VEGF increases thrombus dissolution in animal models [65]. Recent experiments suggest that stimulating the levels of hypoxia-inducible factor-1α in the venous wall will lead to vascular recanalization and thrombus dissolution. By differential display analysis and RT-PCR in CTEPH, we found gene expression defects of angiogenesis. These studies suggest that pulmonary vascular obstruction may be associated with defects in angiogenesis.
Platelets and in situ thrombosis
Platelets play a key role in coagulation and hemostasis, but their role in the development of CTEPH is unclear. A recent study compared the platelet-activating markers P-selectin and glycoprotein IIb/IIIa in a CTEPH group, a PAH group, and a healthy control group. Compared with the control group, platelets in CTEPH patients were activated, and CTEPH patients had a higher sensitivity to thrombin stimulation. However, platelets are also activated in patients with PAH; thus, it is still unclear whether platelet activation in CTEPH patients is secondary to PH [66]. Because of the strong correlation between CTEPH and a history of VTE, it is difficult to conceive of in situ thrombosis as the main pathogenetic mechanism in most CTEPH patients. It is suggested that primary PTE may be one of the causes of chronic large vessel obstruction and PH, and it is difficult to induce CTEPH in the recurrent PTE model, while the risk factors of traditional PTE are not directly related to CTEPH.
Vascular remodeling and other related factors
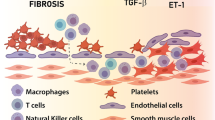
According to current knowledge, CTEPH is a “double” pulmonary vascular disease accompanied by thrombosis, which induces large vascular remodeling accompanied by distal pulmonary small vascular remodeling [67]. In patients with IPAH, pulmonary vascular lesions are mainly manifested in the thickening of the membrane smooth muscle cell layer in the pulmonary artery, the increase in extracellular mechanism fibers, and the proliferation of intimal endothelial cells and intimal smooth muscle cells (SMCs). Pathological examination results also suggest that some CTEPH patients also have similar pulmonary vascular remodeling with IPAH.
In the study by Lang et al. [68], endothelial cells and SMCs were isolated from the yellow and white endometrial tissue by PEA; the PAI-1 expression levels of the two cells were significantly higher, the endothelial cells in the thrombi were abnormally phenotypic and functional, and the proliferative capacity was improved, with resistance to apoptosis [69]. In vitro studies have shown that the fibroblast-like cells harvested from PEA specimens can induce endothelial cells to transform into stromal cells, and this change can be effectively inhibited by rapamycin [70]. Similar proliferative characteristics were observed in cells isolated and cultured from the proximal pulmonary artery during PEA, suggesting that, in addition to the metastasis or transdifferentiation of the extracellular model fibroblasts, the progenitor cells may be differentiated into SMC-type enhancement or dysfunctional differentiation in occluded vessels. Endothelial progenitor cells have been identified in tissues harvested from PEA in CTEPH patients. Firth et al. [71] and others reported the presence of pluripotent mesenchymal progenitor cells in the tissues of CTEPH patients. The unique microenvironment caused by a stable thrombus can promote the differentiation of progenitor cells into muscle fiber-like cells, while the erroneous differentiation of progenitor cells may strengthen the remodeling of the arterial intima; this field still requires further study.
Abnormal calcium homeostasis (increased calcium concentration and increased external calcium flow from the intracellular environment) also plays an important role in pulmonary vascular remodeling in patients with CTEPH [72]. Firth et al. [73] found that abnormalities in pulmonary artery SMCs and endothelial cell layers after exposure to fibrin and fibrinogen may be associated with changes in calcium ions in endothelial cells. In addition, similar SMCs can be isolated and cultured in the PEA tissue of CTEPH patients, while their voltage-dependent potassium ion channel expression is reduced, potassium current decreases, the voltage-gated calcium channel-mediated calcium flow increases, and intracellular free calcium ions increase.
Summary
In recent years, we have improved our understanding of CTEPH; however, the deep molecular mechanisms remain to be studied. There are many problems that are still unresolved; for example, why do only a small proportion of PTE patients develop CTEPH? Furthermore, genetic or acquired susceptibility is present in only some CTEPH patients. Damage to blood vessel regeneration after acute PTE is associated with the occurrence and development of CTEPH. Chronic inflammation and the occurrence and development of CTEPH play a certain role. In summary, for the vast majority of patients, a single factor cannot explain the occurrence and development of CTEPH; other factors, such as thrombosis risk factors, fibrinolytic abnormalities, chronic inflammation and other clinical conditions, and multiple mechanisms, participate together. It cannot be predicted whether acute PTE will develop into CTEPH. Moreover, the mechanisms involved in CTEPH development are not the same in different patients. Clinical symptoms are also changeable, especially during the “honeymoon period” that occurs in some CTEPH patients, which increases the difficulty of preventing CTEPH and predicting the occurrence of CTEPH. Gene chip, RNA sequencing [74] and proteomic [75] methods contribute to elucidating the pathogenesis of PAH to varying degrees, and research shows that these methods may also serve to study the mechanisms of the development of CTEPH and other types of PH [76].
References
Opitz C, Rosenkranz S, Ghofrani HA, Grünig E, Klose H, Olschewski H, Hoeper M (2016) ESC guidelines 2015 pulmonary hypertension: diagnosis and treatment. Dtsch Med Wochenschr 141(24):1764–1769
Lang IM, Pesavento R, Bonderman D, Yuan JX (2013) Risk factors and basic mechanisms of chronic thromboembolic pulmonary hypertension: a current understanding. Eur Respir J 41(2):462–468
Elwing JM, Vaidya A, Auger WR (2018) Chronic thromboembolic pulmonary hypertension: An Update. Clin Chest Med 39(3):605–620
Gall H, Hoeper MM, Richter MJ, Cacheris W, Hinzmann B, Mayer E (2017) An epidemiological analysis of the burden of chronic thromboembolic pulmonary hypertension in the USA, Europe and Japan. Eur Respir Rev 26(143):160121
Tapson VF, Platt DM, Xia F, Teal SA, de la Orden M, Divers CH, Satler CA, Joish VN, Channick RN (2016) Monitoring for pulmonary hypertension following pulmonary embolism: the INFORM study. Am J Med 129(9):978–985 e972
Pengo V, Lensing AW, Prins MH, Marchiori A, Davidson BL, Tiozzo F, Albanese P, Biasiolo A, Pegoraro C, Iliceto S, Prandoni P, Thromboembolic Pulmonary Hypertension Study Group (2004) Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. N Engl J Med 350(22):2257–2264
Yang S, Yang Y, Zhai Z, Kuang T, Gong J, Zhang S, Zhu J, Liang L, Shen YH, Wang C (2015) Incidence and risk factors of chronic thromboembolic pulmonary hypertension in patients after acute pulmonary embolism. J Thorac Dis 7(11):1927–1938
Escribano-Subias P, Blanco I, Lopez-Meseguer M et al (2012) Survival in pulmonary hypertension in Spain: insights from the Spanish registry. Eur Respir J 40(3):596–603
Fernandes T, Auger W, Fedullo P (2018) Epidemiology and risk factors for chronic thromboembolic pulmonary hypertension. Thromb Res 164:145–149
Woo K, Tse L, Tse C, Metreweli C, Vallance-Owen J (1988) The prevalence and pattern of pulmonary thromboembolism in the Chinese in Hong Kong. Int J Cardiol 20(3):373–380
Yang Y, Liang L, Zhai Z, He H, Xie W, Peng X, Wang C, on behalf of investigators for the National Cooperative Project for the Prevention and Treatment of PTE-DVT (2011) Pulmonary embolism incidence and fatality trends in Chinese hospitals from 1997 to 2008: a multicenter registration study. PLoS One 6(11):e26861
Pepke-Zaba J, Delcroix M, Lang I, Mayer E, Jansa P, Ambroz D, Treacy C, D'Armini AM, Morsolini M, Snijder R, Bresser P, Torbicki A, Kristensen B, Lewczuk J, Simkova I, Barberà JA, de Perrot M, Hoeper MM, Gaine S, Speich R, Gomez-Sanchez MA, Kovacs G, Hamid AM, Jaïs X, Simonneau G (2011) Chronic thromboembolic pulmonary hypertension (CTEPH): results from an international prospective registry. Circulation 124(18):1973–1981
Al-Naamani N, Espitia HG, Velazquez-Moreno H et al (2016) Chronic thromboembolic pulmonary hypertension: experience from a single center in Mexico. Lung 194(2):315–323
Riedel M, Stanek V, Widimsky J, Prerovsky I (1982) Longterm follow-up of patients with pulmonary thromboembolism. Late prognosis and evolution of hemodynamic and respiratory data. Chest 81(2):151–158
Lewczuk J, Piszko P, Jagas J, Porada A, Sobkowicz B, Wrabec K, Wójciak S (2001) Prognostic factors in medically treated patients with chronic pulmonary embolism. Chest 119(3):818–823
Auger WR, Kim NH, Trow TK (2010) Chronic thromboembolic pulmonary hypertension. Clin Chest Med 31(4):741–758
Egermayer P, Peacock AJ (2000) Is pulmonary embolism a common cause of chronic pulmonary hypertension? Limitations of the embolic hypothesis. Eur Respir J 15(3):440–448
Lang IM, Marsh JJ, Konopka RG, Olman MA, Binder BR, Moser KM, Schleef RR (1993) Factors contributing to increased vascular fibrinolytic activity in mongrel dogs. Circulation 87(6):1990–2000
Lang IM, Marsh J, Moser K, Schleef R (1992) Presence of active and latent type 1 plasminogen activator inhibitor associated with porcine platelets. Blood 80(9):2269–2274
Fedullo PF, Auger WR, Kerr KM, Rubin LJ (2001) Chronic thromboembolic pulmonary hypertension. N Engl J Med 345(20):1465–1472
Pesavento R, Filippi L, Palla A, Visonà A, Bova C, Marzolo M, Porro F, Villalta S, Ciammaichella M, Bucherini E, Nante G, Battistelli S, Muiesan ML, Beltramello G, Prisco D, Casazza F, Ageno W, Palareti G, Quintavalla R, Monti S, Mumoli N, Zanatta N, Cappelli R, Cattaneo M, Moretti V, Corà F, Bazzan M, Ghirarduzzi A, Frigo AC, Miniati M, Prandoni P, SCOPE Investigators (2017) Impact of residual pulmonary obstruction on the long-term outcome of patients with pulmonary embolism. Eur Respir J 49(5):1601980
Yamaki S, Ando M, Fukumoto Y, Higuchi Y, Kaneko K, Maeda K, Shimokawa H (2014) Histopathological examination by lung biopsy for the evaluation of operability and postoperative prognosis in patients with chronic thromboembolic pulmonary hypertension. Circ J 78(2):476–482
Lang IM, Madani M (2014) Update on chronic thromboembolic pulmonary hypertension. Circulation 130(6):508–518
Edward JA, Mandras S (2017) An update on the management of chronic thromboembolic pulmonary hypertension. Curr Probl Cardiol 42(1):7–38
Miniati M, Monti S, Bottai M, Scoscia E, Bauleo C, Tonelli L, Dainelli A, Giuntini C (2006) Survival and restoration of pulmonary perfusion in a long-term follow-up of patients after acute pulmonary embolism. Medicine (Baltimore) 85(5):253–262
Becattini C, Agnelli G, Pesavento R, Silingardi M, Poggio R, Taliani MR, Ageno W (2006) Incidence of chronic thromboembolic pulmonary hypertension after a first episode of pulmonary embolism. Chest 130(1):172–175
Klok FA, van Kralingen KW, van Dijk AP, Heyning FH, Vliegen HW, Huisman MV (2010) Prospective cardiopulmonary screening program to detect chronic thromboembolic pulmonary hypertension in patients after acute pulmonary embolism. Haematologica 95(6):970–975
Poli D, Grifoni E, Antonucci E, Arcangeli C, Prisco D, Abbate R, Miniati M (2010) Incidence of recurrent venous thromboembolism and of chronic thromboembolic pulmonary hypertension in patients after a first episode of pulmonary embolism. J Thromb Thrombolysis 30(3):294–299
Guerin L, Couturaud F, Parent F et al (2014) Prevalence of chronic thromboembolic pulmonary hypertension after acute pulmonary embolism. Prevalence of CTEPH after pulmonary embolism. Thromb Haemost 112(3):598–605
Otero R, Oribe M, Ballaz A, Jimenez D, Uresandi F, Nauffal D, Conget F, Rodriguez C, Elias T, Jara L, Cayuela A, Blanco I, Barberá J (2011) Echocardiographic assessment of pulmonary arterial pressure in the follow-up of patients with pulmonary embolism. Thromb Res 127(4):303–308
Dentali F, Donadini M, Gianni M, Bertolini A, Squizzato A, Venco A, Ageno W (2009) Incidence of chronic pulmonary hypertension in patients with previous pulmonary embolism. Thromb Res 124(3):256–258
Korkmaz A, Ozlu T, Ozsu S, Kazaz Z, Bulbul Y (2012) Long-term outcomes in acute pulmonary thromboembolism: the incidence of chronic thromboembolic pulmonary hypertension and associated risk factors. Clin Appl Thromb Hemost 18(3):281–288
Fedullo P, Kerr KM, Kim NH, Auger WR (2011) Chronic thromboembolic pulmonary hypertension. Am J Respir Crit Care Med 183(12):1605–1613
Bonderman D, Wilkens H, Wakounig S, Schäfers HJ, Jansa P, Lindner J, Simkova I, Martischnig AM, Dudczak J, Sadushi R, Skoro-Sajer N, Klepetko W, Lang IM (2009) Risk factors for chronic thromboembolic pulmonary hypertension. Eur Respir J 33(2):325–331
Bonderman D, Jakowitsch J, Adlbrecht C, Schemper M, Kyrle P, Schönauer V, Exner M, Klepetko W, Kneussl M, Maurer G, Lang I (2005) Medical conditions increasing the risk of chronic thromboembolic pulmonary hypertension. Thromb Haemost 93(3):512–516
Kim NH, Lang IM (2012) Risk factors for chronic thromboembolic pulmonary hypertension. Eur Respir Rev 21(123):27–31
Kyrle PA, Minar E, Hirschl M, Bialonczyk C, Stain M, Schneider B, Weltermann A, Speiser W, Lechner K, Eichinger S (2000) High plasma levels of factor VIII and the risk of recurrent venous thromboembolism. N Engl J Med 343(7):457–462
Bonderman D, Turecek PL, Jakowitsch J, Weltermann A, Adlbrecht C, Schneider B, Kneussl M, Rubin L, Kyrle P, Klepetko W, Maurer G, Lang I (2003) High prevalence of elevated clotting factor VIII in chronic thromboembolic pulmonary hypertension. Thromb Haemost 90(3):372–376
Lisman T, de Groot PG, Meijers JC, Rosendaal FR (2005) Reduced plasma fibrinolytic potential is a risk factor for venous thrombosis. Blood 105(3):1102–1105
Morris TA (2013) Why acute pulmonary embolism becomes chronic thromboembolic pulmonary hypertension: clinical and genetic insights. Curr Opin Pulm Med 19(5):422–429
Ghosh AK, Vaughan DE (2012) PAI-1 in tissue fibrosis. J Cell Physiol 227(2):493–507
Vuylsteke A, Sharples L, Charman G, Kneeshaw J, Tsui S, Dunning J, Wheaton E, Klein A, Arrowsmith J, Hall R, Jenkins D (2011) Circulatory arrest versus cerebral perfusion during pulmonary endarterectomy surgery (PEACOG): a randomised controlled trial. Lancet 378(9800):1379–1387
Lang IM, Marsh JJ, Olman MA, Moser KM, Schleef RR (1994) Parallel analysis of tissue-type plasminogen activator and type 1 plasminogen activator inhibitor in plasma and endothelial cells derived from patients with chronic pulmonary thromboemboli. Circulation 90(2):706–712
Morris TA, Marsh JJ, Chiles PG, Auger WR, Fedullo PF, Woods VL Jr (2006) Fibrin derived from patients with chronic thromboembolic pulmonary hypertension is resistant to lysis. Am J Respir Crit Care Med 173(11):1270–1275
Morris TA, Marsh JJ, Chiles PG, Magana MM, Liang NC, Soler X, DeSantis DJ, Ngo D, Woods VL (2009) High prevalence of dysfibrinogenemia among patients with chronic thromboembolic pulmonary hypertension. Blood 114(9):1929–1936
Miniati M, Fiorillo C, Becatti M, Monti S, Bottai M, Marini C, Grifoni E, Formichi B, Bauleo C, Arcangeli C, Poli D, Nassi PA, Abbate R, Prisco D (2010) Fibrin resistance to lysis in patients with pulmonary hypertension other than thromboembolic. Am J Respir Crit Care Med 181(9):992–996
Suntharalingam J, Goldsmith K, van Marion V, Long L, Treacy CM, Dudbridge F, Toshner MR, Pepke-Zaba J, Eikenboom JCJ, Morrell NW (2008) Fibrinogen Aalpha Thr312Ala polymorphism is associated with chronic thromboembolic pulmonary hypertension. Eur Respir J 31(4):736–741
Standeven KF, Grant PJ, Carter AM, Scheiner T, Weisel JW, Ariens RA (2003) Functional analysis of the fibrinogen Aalpha Thr312Ala polymorphism: effects on fibrin structure and function. Circulation 107(18):2326–2330
Kellermair J, Redwan B, Alias S, Jabkowski J, Panzenboeck A, Kellermair L, Winter MP, Weltermann A, Lang IM (2013) Platelet endothelial cell adhesion molecule 1 deficiency misguides venous thrombus resolution. Blood 122(19):3376–3384
Bonderman D, Jakowitsch J, Redwan B, Bergmeister H, Renner MK, Panzenböck H, Adlbrecht C, Georgopoulos A, Klepetko W, Kneussl M, Lang IM (2008) Role for staphylococci in misguided thrombus resolution of chronic thromboembolic pulmonary hypertension. Arterioscler Thromb Vasc Biol 28(4):678–684
Zabini D, Heinemann A, Foris V, Nagaraj C, Nierlich P, Bálint Z, Kwapiszewska G, Lang IM, Klepetko W, Olschewski H, Olschewski A (2014) Comprehensive analysis of inflammatory markers in chronic thromboembolic pulmonary hypertension patients. Eur Respir J 44(4):951–962
Quarck R, Nawrot T, Meyns B, Delcroix M (2009) C-reactive protein: a new predictor of adverse outcome in pulmonary arterial hypertension. J Am Coll Cardiol 53(14):1211–1218
Cohn DM, Roshani S, Middeldorp S (2007) Thrombophilia and venous thromboembolism: implications for testing. Semin Thromb Hemost 33(6):573–581
Wolf M, Boyer-Neumann C, Parent F, Eschwege V, Jaillet H, Meyer D, Simonneau G (2000) Thrombotic risk factors in pulmonary hypertension. Eur Respir J 15(2):395–399
Wong CL, Szydlo R, Gibbs S, Laffan M (2010) Hereditary and acquired thrombotic risk factors for chronic thromboembolic pulmonary hypertension. Blood Coagul Fibrinolysis 21(3):201–206
Lotta LA, Tuana G, Yu J, Martinelli I, Wang M, Yu F, Passamonti SM, Pappalardo E, Valsecchi C, Scherer SE, Hale W IV, Muzny DM, Randi G, Rosendaal FR, Gibbs RA, Peyvandi F (2013) Next-generation sequencing study finds an excess of rare, coding single-nucleotide variants of ADAMTS13 in patients with deep vein thrombosis. J Thromb Haemost 11(7):1228–1239
Orstavik KH, Magnus P, Reisner H, Berg K, Graham JB, Nance W (1985) Factor VIII and factor IX in a twin population. Evidence for a major effect of ABO locus on factor VIII level. Am J Hum Genet 37(1):89–101
Ma Q, Jacobi PM, Emmer BT, Kretz CA, Ozel AB, McGee B, Kimchi-Sarfaty C, Ginsburg D, Li JZ, Desch KC (2017) Genetic variants in ADAMTS13 as well as smoking are major determinants of plasma ADAMTS13 levels. Blood Adv 1(15):1037–1046
Chion CK, Doggen CJ, Crawley JT, Lane DA, Rosendaal FR (2007) ADAMTS13 and von Willebrand factor and the risk of myocardial infarction in men. Blood 109(5):1998–2000
Newnham M, South K, Bleda M, Auger WR, Barberà JA, Bogaard H, Bunclark K, Cannon JE, Delcroix M, Hadinnapola C, Howard LS, Jenkins D, Mayer E, Ng C, Rhodes CJ, Screaton N, Sheares K, Simpson MA, Southwood M, Su L, Taboada D, Traylor M, Trembath RC, Villar SS, Wilkins MR, Wharton J, Gräf S, Pepke-Zaba J, Laffan M, Lane DA, Morrell NW, Toshner M (2019) The ADAMTS13–VWF axis is dysregulated in chronic thromboembolic pulmonary hypertension. Eur Respir J 53(3):1801805
Feng YX, Liu D, Sun ML, Jiang X, Sun N, Mao YM, Jing ZC (2014) BMPR2 germline mutation in chronic thromboembolic pulmonary hypertension. Lung 192(4):625–627
Ulrich S, Szamalek-Hoegel J, Hersberger M, Fischler M, Garcia JS, Huber LC, Grünig E, Janssen B, Speich R (2010) Sequence variants in BMPR2 and genes involved in the serotonin and nitric oxide pathways in idiopathic pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension: relation to clinical parameters and comparison with left heart disease. Respiration 79(4):279–287
Distler JH, Hirth A, Kurowska-Stolarska M, Gay RE, Gay S, Distler O (2003) Angiogenic and angiostatic factors in the molecular control of angiogenesis. Q J Nucl Med 47(3):149–161
Waltham M, Burnand KG, Collins M, Smith A (2000) Vascular endothelial growth factor and basic fibroblast growth factor are found in resolving venous thrombi. J Vasc Surg 32(5):988–996
Henke PK, Wakefield TW, Kadell AM, Linn MJ, Varma MR, Sarkar M, Hawley A, Fowlkes JB, Strieter RM (2001) Interleukin-8 administration enhances venous thrombosis resolution in a rat model. J Surg Res 99(1):84–91
Yaoita N, Shirakawa R, Fukumoto Y, Sugimura K, Miyata S, Miura Y, Nochioka K, Miura M, Tatebe S, Aoki T, Yamamoto S, Satoh K, Kimura T, Shimokawa H, Horiuchi H (2014) Platelets are highly activated in patients of chronic thromboembolic pulmonary hypertension. Arterioscler Thromb Vasc Biol 34(11):2486–2494
Moser KM, Bloor CM (1993) Pulmonary vascular lesions occurring in patients with chronic major vessel thromboembolic pulmonary hypertension. Chest 103(3):685–692
Lang IM, Marsh JJ, Olman MA, Moser KM, Loskutoff DJ, Schleef RR (1994) Expression of type 1 plasminogen activator inhibitor in chronic pulmonary thromboemboli. Circulation 89(6):2715–2721
Sakao S, Tatsumi K (2013) Crosstalk between endothelial cell and thrombus in chronic thromboembolic pulmonary hypertension: perspective. Histol Histopathol 28(2):185–193
Sakao S, Hao H, Tanabe N, Kasahara Y, Kurosu K, Tatsumi K (2011) Endothelial-like cells in chronic thromboembolic pulmonary hypertension: crosstalk with myofibroblast-like cells. Respir Res 12:109
Firth AL, Yao W, Ogawa A, Madani MM, Lin GY, Yuan JX (2010) Multipotent mesenchymal progenitor cells are present in endarterectomized tissues from patients with chronic thromboembolic pulmonary hypertension. Am J Physiol Cell Physiol 298(5):C1217–C1225
Zabini D, Nagaraj C, Stacher E, Lang IM, Nierlich P, Klepetko W, Heinemann A, Olschewski H, Bálint Z, Olschewski A (2012) Angiostatic factors in the pulmonary endarterectomy material from chronic thromboembolic pulmonary hypertension patients cause endothelial dysfunction. PLoS One 7(8):e43793
Firth AL, Yau J, White A, Chiles PG, Marsh JJ, Morris TA, Yuan JXJ (2009) Chronic exposure to fibrin and fibrinogen differentially regulates intracellular Ca2+ in human pulmonary arterial smooth muscle and endothelial cells. Am J Physiol Lung Cell Mol Physiol 296(6):L979–L986
Rhodes CJ, Im H, Cao A, Hennigs JK, Wang L, Sa S, Chen PI, Nickel NP, Miyagawa K, Hopper RK, Tojais NF, Li CG, Gu M, Spiekerkoetter E, Xian Z, Chen R, Zhao M, Kaschwich M, del Rosario PA, Bernstein D, Zamanian RT, Wu JC, Snyder MP, Rabinovitch M (2015) RNA sequencing analysis detection of a novel pathway of endothelial dysfunction in pulmonary arterial hypertension. Am J Respir Crit Care Med 192(3):356–366
Abdul-Salam VB, Wharton J, Cupitt J, Berryman M, Edwards RJ, Wilkins MR (2010) Proteomic analysis of lung tissues from patients with pulmonary arterial hypertension. Circulation 122(20):2058–2067
Hemnes AR, Trammell AW, Archer SL, Rich S, Yu C, Nian H, Penner N, Funke M, Wheeler L, Robbins IM, Austin ED, Newman JH, West J (2015) Peripheral blood signature of vasodilator-responsive pulmonary arterial hypertension. Circulation. 131(4):401–409 discussion 409
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yan, L., Li, X., Liu, Z. et al. Research progress on the pathogenesis of CTEPH. Heart Fail Rev 24, 1031–1040 (2019). https://doi.org/10.1007/s10741-019-09802-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-019-09802-4