
Abstract
Sedentary life style and high calorie dietary habits are prominent leading cause of metabolic syndrome in modern world. Obesity plays a central role in occurrence of various diseases like hyperinsulinemia, hyperglycemia and hyperlipidemia, which lead to insulin resistance and metabolic derangements like cardiovascular diseases (CVDs) mediated by oxidative stress. The mortality rate due to CVDs is on the rise in developing countries. Insulin resistance (IR) leads to micro or macro angiopathy, peripheral arterial dysfunction, hampered blood flow, hypertension, as well as the cardiomyocyte and the endothelial cell dysfunctions, thus increasing risk factors for coronary artery blockage, stroke and heart failure suggesting that there is a strong association between IR and CVDs. The plausible linkages between these two pathophysiological conditions are altered levels of insulin signaling proteins such as IR-β, IRS-1, PI3K, Akt, Glut4 and PGC-1α that hamper insulin-mediated glucose uptake as well as other functions of insulin in the cardiomyocytes and the endothelial cells of the heart. Reduced AMPK, PFK-2 and elevated levels of NADP(H)-dependent oxidases produced by activated M1 macrophages of the adipose tissue and elevated levels of circulating angiotensin are also cause of CVD in diabetes mellitus condition. Insulin sensitizers, angiotensin blockers, superoxide scavengers are used as therapeutics in the amelioration of CVD. It evidently becomes important to unravel the mechanisms of the association between IR and CVDs in order to formulate novel efficient drugs to treat patients suffering from insulin resistance-mediated cardiovascular diseases. The possible associations between insulin resistance and cardiovascular diseases are reviewed here.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardiovascular diseases (CVDs) are often associated with metabolic diseases and are one of the major cause of early deaths in diabetic population. In diabetic patients, 65 % of the deaths are listed due to CVD [1]. In Framingham studies, it has been reported that diabetes is a precursor of cardiovascular morbidity, mortality and congestive heart failure [2, 3]. Several hypothesis such as hyperglycemia, oxidative stress, inflammation, obesity, sedentary life style, genetic predisposition etc. have emerged to connect the dots between diabetes and CVD. However, none of the hypothesis is able to completely define the underlying pathophysiology. In aggressive conventional therapy for diabetes the risk of CVD is reduced by about 42 %. Thus, a high-quality management of diabetes alone does not explain the high incidence of CVD in these patients. One of the overlooked links that is common in type 2 diabetic patients is the occurrence of insulin resistance (IR). The role of insulin as an atherogenic molecule as well as the significance of insulin signaling in the endothelial cells has been underappreciated even though these cells are the key players involved in the vascular function. Furthermore, the complexity of IR syndrome, arising in the major peripheral insulin-dependent tissues leading to the microvascular and macrovascular complications in diabetes due to systemic IR, is not clearly understood.
Pathophysiology of cardiovascular diseases in diabetes
Persistent hyperglycemia causes microvascular and macrovascular complications in both type 1 and type 2 diabetes. Microvascular complications include nephropathy, neuropathy and retinopathy, while macrovascular complications include coronary artery disease, peripheral arterial disease and stroke [4]. Some of the main etiological factors for CVD are as follows:
Oxidative stress, inflammatory response and endothelial dysfunction
Much of the enduring pathology of diabetes occurs as a consequence of persistent hyperglycemia leading to increased reactive oxygen species (ROS) production by mitochondria, which is the main source of oxidative stress involving complications of diabetic pathologies including CVD [1, 5, 6]. In type 1 diabetes, endothelial dysfunction is an essential determinant of inflammatory activities and considered as an early CVD marker. The inflammatory response is generated by innate immunity which includes augmentation of cytokine and chemokine release, enhanced leukocyte marginalization and increased superoxide release [7]. It is coupled with the impairment of the endothelial signal transduction and redox-regulated activation of transcription factors [8], and endothelial dysfunction in type 2 diabetes has also been shown to occur [9, 10]. It has been demonstrated that excess ROS production due to hyperglycemia induces epigenetic changes like: monomethylation of lysine from histone 3 which increases expression of p65 subunit of NFκ-B. These epigenetic reactions can be considered as mediators between diabetes, chronic inflammatory response and CVD [11, 12].
Dyslipidemia, obesity and hypertension
Risk of CVD persists in dyslipidemia, due to atherogenic profile which comprises of increased very low-density lipoprotein (VLDL) cholesterol, triglycerides and low-density lipoprotein (LDL) cholesterol levels whereas decreased high-density lipoprotein (HDL) cholesterol levels. This is also supported by studies showing that diabetic hyperlipidemia or hyperglycemia accelerates atherogenesis [13, 14], also obesity and diabetes are coupled with major increase in morbidity and mortality due to CVD [15, 16]. It is observed that in obesity, visceral fat deposition leads to inflammation which also plays an important role in diabetic complications. The association between hypertension and obesity is also known to cause higher rate of morbidity and mortality associated with CVD [17, 18]. It is reported that approximately 10–30 % type 1 diabetic and 60 % type 2 diabetics suffer from hypertension and thus high risk of CVD.
Hypoglycemia
Insulin and hypoglycemic drugs control glycemic load and may lead to frequent hypoglycemia-related cardiovascular mortality in diabetes patients [19]. Hypoglycemia is found to cause unusual electrical activity in the heart and is thus believed to aggravate sudden death [20, 21]. Inflammation in hypoglycemic condition occurs due to C-reactive protein (CRP), IL-6, vascular endothelial growth factor (VEGF), increased platelet and neutrophil activation [22].
Autonomic neuropathy
Cardiac autonomic neuropathy (CAN) is one of the common complications of type 1 and type 2 diabetes. It prevails in about 20 % and is reported to increase with age as well as duration of diabetes with annual increase of 2 % [23, 24]. EURODIAB study reported that poor glycemic control is strong risk factor for CAN which is the predictor of CVD morbidity and mortality in type 1 diabetes [25].
Apparently, all the above factors play an important role in diabetes related CVD risk. However, association of these factors with hyperinsulinemia leading to IR, hall marks of type 2 diabetes as a causal factor in cardiovascular diseases is not fully appreciated. An effort is made to delineate the relationship of insulin resistance and CVD in the pathogenesis of type 2 diabetes.
Molecular mechanism of insulin resistance
Insulin plays a central role in carbohydrate and lipid metabolism in peripheral system and also has other functions in heart and brain. Obesity is the major contributory factor to systemic interventions like hyperglycemia, hyperinsulinemia, hyperlipidemia etc. which leads to inefficiency or failure of insulin action that leads to systemic insulin resistance condition [26, 27] as shown in Fig. 1.

Etiological factors affecting insulin-dependent and insulin-independent pathways which cumulatively lead to alterations in metabolism and cause insulin resistance (IR). Factors such as obesity and sedentary life style leading to excess free fatty acids (FFA) availability and hyperinsulinemia are associated with IR. Pathophysiological processes involve insulin-independent pathway where oxidative stress leads to the activation of stress kinases such as p38 and JNK MAPKs stimulating secretion of proinflammatory cytokines like TNF-α and IL-6 under the transcriptional activation of NF-κB and induce protein kinase C (PKCs) at serine/threonine residues and GSK-3. Activation of PKC phosphorylates and subsequently activates IkB kinase to promote phosphorylation of insulin receptor substrate-1 (IRS-1) which inhibit the ability of IRS-1 to bind to SH2 domains of the p85 regulatory subunit of phosphatidylinositol 3-kinase (PI3K) and as a result impairs insulin signal transduction. Inhibition of PI3K causes an upregulation of phosphatase and tensin homolog (PTEN) and SH-2 containing inositol 5′-phosphatase (SHIP). On the other hand, oxidative stress also leads to mitochondrial dysfunction by downregulating the uncoupling protein-2 (UCP-2) decreasing ATP production and control glucose transport by the expression and translocation of glucose transporter 4 (GLUT4). Insulin-dependent pathway, involving etiological factors mainly excess FFA, hyperglycemia and hyperinsulinemia, leads to Ser phosphorylation of insulin receptors (IR-β) and its substrate (IRS-1) to desensitize tyrosine phosphorylation of PI3K and Akt. This reduces GLUT4 translocation mainly by the activation of serine/threonine phosphorylation caused by activated PKCs ultimately leading to insulin resistance
Muscle, adipose and liver are the most affected organs due to overload of lipid accumulation and thus, lead to peripheral insulin resistance [28]. Free fatty acids (FFA) circulate and deposit into the skeletal muscle myocytes and intramuscular lipid accumulation occurs, which further aggravates the insulin resistance condition by downregulating the expression as well as reducing total insulin receptor number [26].
Excess fat activates the Toll-like receptors (TLRs) on the resident macrophages of adipose tissue that secrete TNF-α. The later, activates NFκB-mediated cellular toxicity by activating various PKCs and downregulating tyrosine phosphorylation of insulin receptors. This compromises insulin signaling and promotes insulin resistance [29, 30]. Activation of various isoforms of PKCs, also activates Ser/Thr kinases which phosphorylates IRS-1 at Ser 307 that hampers the association of IRS-1 and PI3K, ultimately causing proteasomal degradation of IRS-1 and IRS-2. These events inhibit insulin signaling as well as the translocation of glucose transporter 4 (GLUT4) and insulin stimulated glucose uptake [29, 31]. Stimulated SREBP-1c also decreases IRS-2 levels in insulin resistance condition. Downstream in the signaling, altered ratio of PI3K subunits p110/p85 inhibits the dimerization of the enzyme and thus reduces its activity. Suppressors of cytokine signaling (SOCS) proteins, which are induced by inflammatory cytokines, bind to the insulin receptors and block their signaling. Insulin resistance can also be due to an increase in the activity as well as amount of the enzymes that normally reverse insulin action, including the phosphotyrosine phosphatases, e.g., PTP1b, and the PIP phosphatases, e.g., PTEN and SHIP.
All these events lead to a concomitant reduction in insulin stimulated glycogen synthesis and glucose uptake which leads to activation of the phosphoenolpyruvate carboxykinase, the rate limiting enzyme of gluconeogenesis. This increases hepatic glucose production which not only leads to hepatic insulin resistance but overall insulin resistance as well [31, 32].
Mechanism of oxidative stress in diabetes and insulin resistance
Glucose toxicity, being the hallmark of diabetes mellitus is responsible for ROS production leading to oxidative stress [32]. There are several mechanisms by which hyperglycemia can induce oxidative stress. These include the activation of polyol pathway, glucose auto oxidation, formation of advanced glycation end (AGEs) products, increased FFA, leptin levels and increased mitochondrial ROS generation [6, 33].
Activation of polyol pathway and formation of advanced glycation end (AGE) products
In hyperglycemia, up to 35 % of glucose is metabolized by polyol pathway. NADPH is needed for the production of reduced Glutathione, which is consumed by aldol-reductase (first enzyme of the pathway), thus preventing the regeneration of reduced Glutathione further aggravating oxidative stress. The second enzyme is sorbitol dehydrogenase that converts sorbitol to fructose producing NADH. NAD(P)H oxidases can use NADH to produce more superoxide anion [34].
Ketoaldehyde, protein reactive dicarbonyl sugar, is a product of glucose autoxidation and a member of AGE, which not only yields H2O2 but also the other highly reactive oxidants that become an important source of diabetes mellitus (DM)-induced oxidative stress [6]. Glyoxal and arabinose have been reported to be the major dicarbonyl intermediates that cause protein browning observed in DM and aging. Oxidative stress increases with age as well as severity of DM, which stimulates Maillard reaction where these dicarbonyl intermediates attach non-enzymatically to protein amino-structures generating ROS which can promote oxidation of glycation products leading to AGE formation, AGEs precursors can further bind to AGE receptors on the surface of endothelial cells and macrophages resulting in receptor-mediated production of oxygen free radicals and dysregulating their functions [35, 36].
FFA- and leptin-mediated oxidative stress during hyperglycemia
Free or non-esterified fatty acids (NEFA) are elevated in diabetic patients. Mitochondrial superoxide production increases when excess FFA enters into the citric acid cycle and generates acetyl-CoA to produce excess NADH. This also elevates isoprostanes, which are markers of lipid peroxidation. Leptin is an adipocyte secretory hormone that acts on the central nervous system to abate food intake. It also increases ROS levels when incubated with endothelial cells, vascular smooth muscle cells, monocytes, and macrophages. Plasma levels of leptin are increased in type 2 diabetics and are associated with CVD [37, 38].
Mechanism of hyperglycemia, hyperlipidemia and oxidative stress causing insulin resistance
Glucotoxicity and hyperinsulinemia induced IR is pathologically associated with hyper-Ser/Thr phosphorylation of IRS1 and IRS2 that impairs their interaction with the cytoplasmic domain of insulin receptor, which abolishes the propagation of normal insulin signaling [39]. Under normal conditions, this is an important counterbalancing mechanism that stops insulin’s action. However, in the case of DM, the hyperserine phosphorylation of the IRS proteins may lead to a chronic cellular desensitization to insulin [40].
It has been reported that under oxidative stress condition, insulin stimulated serine PKB phosphorylation and the translocation of GLUT4 from internal pool to the plasma membrane were dramatically reduced [41]. Introduction of prolonged oxidative stress to L6 myotubes and 3T3-L1 adipocytes mediates GLUT1 transcriptional activation and insulin-independent glucose uptake. This result in an increase in mitochondrial ROS production due to an increase in the basal glucose uptake and metabolism in various cell types including cardiomyocytes [42, 43]. It is known that cardiomyocytes expresses both GLUT4 and GLUT1 glucose transporters [44]. Binding affinity of C/EBP, i.e., the CCAAT enhancer binding protein to the GLUT4 promoter is affected during oxidative stress. This alteration in C/EBP function plays a role in the down-regulation of GLUT4 expression in the cells under oxidative stress [43].
Pathophysiology of DM is not only about an insulin–glucose axis, but fat derangements are also a major cause of type 2 diabetes. Central obesity is due to an overload of TG in abdominal adipocytes. Subcutaneous fat has a high rate of basal lipolysis. The enlarged visceral adipocytes pour out FFA which is mainly responsible for ectopic fat deposition [45]. This leads to ectopic TG accumulation in muscles, liver, heart and pancreatic β-cells, resulting in IR at the systemic level by interfering with both insulin secretion and insulin signaling [46].
Hyperinsulinemia is also known to enhance hepatic VLDL synthesis thus, leads to the increased plasma triglyceride and LDL cholesterol levels [47]. Resistance to the action of insulin on lipoprotein lipase in peripheral tissues further contributes to the elevated triglyceride and LDL cholesterol levels [48]. IR condition reduces the levels of HDL cholesterol despite enhanced HDL cholesterol synthesis. This decrease in plasma HDL cholesterol was entirely accounted by an increase in the rate of apolipoprotein A1/HDL cholesterol degradation, which exceeds the enhanced rate of its synthesis [49]. It further supports the view that dysregulation of fatty acid metabolism contributes to the pathophysiology of the IR syndrome which relates to the risk of cardiovascular disease [50].
Mitochondrial stress and IR
Superoxide anion production is promoted during hyperglycemia by the proton electrochemical gradient of the mitochondrial electron transport. In culture cells, it is observed that there is inhibition of mitochondrial superoxide formation followed by complete inhibition of PKCs and NF-κB activation. In normal conditions, heme oxygenase (HO)-1 has low expression but gets upregulated in response to oxidants such as heme, H2O2 and TNF-α, whereas its activity decreases in the case of hyperglycemia in diabetic rats having increased superoxide anion production. Thus, HO-1 is one of the major defense against oxidative stress which becomes vulnerable and contributes to mitochondrial oxidative stress in diabetes mellitus [51].
Association of IR and CVD
Hyperinsulinemia is a predictor of coronary artery disease (CAD) and has been confirmed by patient studies performed in Finland and Quebec [52, 53]. Other studies have also shown a relationship between carotid wall atherosclerotic lesions, angina, and insulin levels/resistance [54].
IR leading to hyperinsulinemia causes hypertension. It has been observed that the hypertensive patients have higher fasting and postprandial insulin levels than normal subjects [55]. Also, the relationship between insulin and hypertension is mainly seen in first-degree hypertensive patients, which does not occur with secondary hypertension [56, 57]. Accordingly, IR and hyperinsulinemia are not consequences of hypertension, instead, a genetic predisposition may contribute to both disorders. Activation of the sympathetic nervous system, renal sodium retention, altered transmembrane cation transport, growth-promoting effects of vascular smooth muscle cells, and vascular hyperreactivity are some of the mechanisms for developing hypertension in IR condition [58].
Microalbuminuria represents a significant risk factor for CVD in patients with (or without) clinical diabetes. Several studies have reported elevated systolic blood pressure in the development of microalbuminuria in type 2 diabetic patients. Thus understanding of the risks involved in the insulin-resistant patients becomes paramount as they are more prone toward elevated systolic blood pressures [59]. In patients both lean and obese hypertensive IR is also associated with enhanced salt sensitivity [60].
Obesity contributes significantly to impaired glucose tolerance, hyperinsulinemia, type 2 diabetes, dyslipidemia, and hypertension. All these factors play an important role in the pathophysiology of IR. Alteration in major metabolism of fat leads to obesity and IR-related complications such as atherosclerosis, hypertension and CVDs. IR thus is not simply a problem of deficient glucose uptake in response to insulin, but a multifaceted syndrome that significantly increases the risk for cardiovascular disease. The link between IR and the associated dyslipidemia, hypertension, hypercoagulability, and atherosclerosis are numerous and complex [61]. In Quebec study, 2000 middle aged men were monitored for 5 years. The study revealed that visceral fat, as compared to peripheral fat, is more resistant to the metabolic effects of insulin, more sensitive to lipolytic hormones and more prone to CVD [62]. Visceral obesity has been positively correlated with higher levels of plasminogen activator inhibitor-1 (PAI-1). It complexes with tissue-type plasminogen activator and eliminates its fibrinolytic activity [63]. Hence, CVD can be predicted by comparing low levels of plasminogen activator with PAI-1 levels. Type 2 diabetic patients have been observed to have higher levels of PAI-1 suggesting that hyperinsulinemia itself is a potent stimulator for PAI-1 production [64].
As stated above, patients with hypertension and IR are more prone to disturbances of the fibrinolytic system. Deficiency of clotting inhibitors such as endogenous antithrombotic factors (i.e., factors C and S and antithrombin III) has been associated with the insulin levels. Also, hyperfibrinogenemia is a powerful independent risk factor for CVD caused by elevated levels of fibrinogen, which have also been observed in the insulin-resistant state [65, 66].
Administration of inhibitors of Nitric oxide (NO) synthase abolishes peripheral vasodilatation in response to insulin, suggesting a crucial role for NO in the normal vasodilatory response to insulin. This response is lost in insulin-resistant/obese individuals suggesting resistance to the action of insulin to induce vascular NO production [66]. Further, abatement of insulin-mediated glucose uptake and decrease in insulin stimulated blood flow has been observed in insulin-resistant obese patients. In more specific vascular studies, it was observed that in insulin-resistant obese patients the ability of insulin to decrease aortic wave reflection was severely blunted, as determined from augmentation index. Also, this defect was a consequence of impaired insulin action, as it was not observed in the basal state suggesting that IR extends to large conduit vessels as well as to vessels regulating peripheral blood flow thereby increasing the risk toward cardiovascular diseases [60, 67, 68].
Inflammatory responses in obesity, IR and CVD
IR in hyperinsulinemia, hypertension, hyperlipidemia leads to type 2 DM, and an increased risk of atherosclerotic CVDs are the adverse effects of central obesity [69]. Apart from adults, obese children are also found to be on the alarming edge of IR, hypertension and abnormal lipid profiles [70]. Various bioactive compounds released by adipose tissue provoke IR, alterations in lipids, coagulation, fibrinolysis, inflammation that leads to endothelial dysfunction and atherosclerosis [71].
Adipose tissue is comprised of different cells such as adipose derived stem cells, adipocytes, endothelial cells and the immune cells (macrophages—M1 and M2). The M1 phenotype is involved in inflammatory processes and the M2 are associated with tissue remodeling. Excess of adipose tissue expansion and hypoxic conditions activate inflammatory responses that secrete various pro-inflammatory cytokines. Infiltrated pro-inflammatory leukocytes differentiate into M1 macrophages that clear the adipocytes that comprise large lipids and hence they resemble foam like cells (crown-like), structures characteristic of dysfunctional adipocytes [72]. Neuroimmune guidance cue netrin-1’s expression is high in adipose tissue of obese human and mice but not in lean. Netrin-1 is responsible for retention of macrophages into adipose tissue via Unc5b receptor and hence leads to increased IR in obesity [73].
Infiltration of macrophages into adipose tissue leads to expansion of the tissue causing chronic low-grade inflammation. Phenotypic switching of macrophages is governed by cells of innate and adaptive immunity which further leads to inflammation. The phenotypic switch involves recruitment of B cells and T cells by changes in the phenotype of T cells [74]. With concomitant increase in the subcutaneous fat tissue, the fat gets deposited in the visceral fat which releases more pro-inflammatory cytokines. Elevated central obesity is associated with worsened cardiovascular risk profiles, IR, hyperlipidemia, increasing the influx of FFA into cardiomyocytes. The common pathological pathways between obesity and CVDs are IR and low-grade inflammation. Presence of NEFA causes lipotoxicity and hence impairs endothelium-dependent vasodilation, increases oxidative stress and has cardio toxic effect [71]. Various NADPH oxidases (NOXs) present in macrophages are responsible for generation of ROS in adipose tissue which aids in cardiovascular diseases [75]. Increased expression levels of β3-adrenoreceptors make visceral adipose tissue (VAT) more sensitive to catecholamine-induced lipolysis that makes it eventually less sensitive to α2 effects and to anti-lipolytic activity of insulin. Excess influx of FFA into myocytes hampers the oxidative capabilities of substrate switching, leading to the deposition of the FFAs, and hence causing lipotoxicity. As a result cardiac dysfunction is promoted by ROS generation and ceramide production that further impairs insulin signaling, decreases sarcoplasmic reticular Ca2 + stores, and causes mitochondrial dysfunction. Due to FFA deposition, heart relies more on it for energy supply. Myocardial IR hampers insulin signaling proteins [72]. FFAs induce inflammation through TLR4 and the liver secretory protein fetuin-A (FetA), acts as an adaptor protein [76].
There is a strong link between leptin and cardiovascular functions and its remodeling. Hyperleptinemia, central leptin resistance, and leptin deficiency are all associated with impaired post-receptor leptin signaling and contractile response. Alterations in the pathways regulated by leptin in cardiomyocytes are associated with the pathology of these cells in obesity. Negative inotropic and hypertrophic responses are found due to the alterations in JAK/STAT, MAPK, NO, and β-adrenergic pathways [77].
Interplay of fat cells macrophages, endothelial cells in IR and CVD
As explained in the earlier section, adipose tissue harbors two types of macrophages of which M1-macrophages is predominant in obesity, secreting TNF-α and IL-6 thereby enhancing inflammation [78]. Both macrophages and adipocytes are capable of accumulating lipids and secreting cytokines. Adipocyte hypertrophy during obesity leads to release of more FFAs which can bind to TLR-4 resulting in NF-κB activation leading to augmentation of TNF-α levels [79]. In turn, macrophage-derived TNF-α activates adipocytes, thereby further inducing lipolysis and enhancing the expression of various genes [intracellular adhesion molecule-1 (ICAM-1), IL-6, macrophage chemo attractant protein-1 (MCP-1)]. FFA and TNF-α in turn also lead to the activation of serine threonine kinases and promoted IR condition [80].
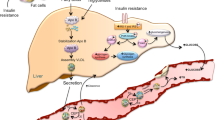
A key step in the initiation of CVD is the reduction of NO bioavailability [81, 82]. The bioavailability of NO is dependent on the balance between its production by eNOS and its inactivation by ROS. A feature action of insulin in the endothelial cells is the regulation of eNOS for nitric oxide production [83, 84]. Thus endothelial cells are crucial target during diabetes and whole body IR because of obesity and adipocyte inflammation which leads to a reduction in NO synthesis. Plasminogen activator inhibitor 1 (PAI-1) is a marker for risk of premature CVD [85, 86], and the link between elevated PAI-1 and IR has been studied extensively where endothelial cells respond to increased levels of insulin by synthesizing and secreting more PAI-1 [87]. Since there is a putative VLDL response element in the gene for PAI-1 in endothelial cells, VLDL (increased during diabetes) also increases PAI-1 synthesis and secretion [50]. The fat cell macrophages that are hyperactivated during obesity in addition to inflammation initiate the whole body IR. Hyperinsulinemia and IR consecutively affect normal functions of endothelial cells increasing the risks of CVD (Fig. 2).

Representation of interplay between fat cell macrophages, adipocytes, β-cells, hepatocytes and endothelial cells in insulin resistance (IR) and cardiovascular disease (CVD) complications. Excess of free fatty acids (FFA) and lipopolysaccharide (LPS) also activate TLR4 present on the fat cell macrophages (M1). This produces pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-6 and resistin through the activation of NF-κ-B which hamper insulin signaling proteins causing IR. The secreted FFAs are responsible for the ectopic fat deposition in muscle myocytes and hepatocytes thus causing peripheral IR due to increased lipolysis and decreased Glut4 translocation further elevating FFA and blood glucose (G) level. This in turn triggers insulin (I) secretion by the β-cells and results in hyperinsulinemia. The later also results in elevated PAI-1 promoter activity in the endothelial cells which is a marker for CVD. IR also results in decreased NO synthesis in endothelial cells resulting in decreased vasodilation, thus increasing the CVD complications. Furthermore, insulin binds to its receptor on hepatocytes and activates substrate to inhibit phosphorylation of tyrosine kinase (e.g., PI3K) and MAPK activity and controls fat and carbohydrate (CHO) metabolism thus decreasing HDL/LDL and promotes CVD problems including atherosclerosis. Thus cumulatively, an interplay of fat cell macrophages, adipocytes, β-cells, hepatocytes and endothelial cells as well as insulin resistance leads to cardiovascular diseases
Cross-talk of signaling pathways in development of IR in the heart
Discussion thus far makes a strong case that there is an association between IR and endothelial cell dysfunction with the cardiovascular diseases-including coronary artery disease, hypertension, heart failure, and stroke. Further in the regulation of various aspects of cardiovascular metabolism and function such as glucose and long-chain fatty acid (LCFA) metabolism, protein translation, and vascular tone, insulin plays a key role [88]. As the heart is an energy-consuming organ, it constantly requires supply of fuel and oxygen in order to maintain its intracellular ATP level. The heart gets the same from mitochondria and this ATP is essential for the uninterrupted myocardial contraction/relaxation cycle. Under physiological conditions, the heart produces ATP from the mitochondrial oxidation of different substrates, LCFAs (60–70 %) being predominant over glucose (20 %) and lactate (10 %). In pathological conditions such as starvation or chronic heart failure, ketone bodies become a major substrate and when glucose and insulin concentrations rise, glucose becomes the favored oxidized substrate of the heart [89].
Insulin signaling is a complex cascade wherein, the effector signaling proteins like IRS-(1/2)/PI3K/Akt have various downstream substrates, potentially activated by insulin thus, depicting varied biological roles of insulin.
Glucose uptake
Insulin favors the use of glucose in cardiomyocytes by activating cardiac 6-phosphofructo-2-kinase (PFK-2) isoform. Simultaneously, LCFA uptake in cardiomyocytes occurs through insulin stimulated activation of PI3K and then translocation of the LCFA transporter FAT/CD36 to the plasma membrane [88].
Protein translation
Insulin regulates protein synthesis in cardiomyocytes through the regulation of PKB/Akt/TSC2/mTOR and their downstream targets 4E-BP1, p70S6K/S6 and eEF2K/eEF2. Apart from these, insulin by inhibiting GSK-3 activates elF2B and stimulates the initiation of protein synthesis [90]. GSK-3 participates in the negative regulation of cardiac hypertrophy by phosphorylating and inactivating the nuclear factor of activated T (NFAT) cells responsible for the pro-hypertrophic gene expression [91].
Vasculature
Insulin functions as an important vasodilator by stimulating increased production of the potent vasodilator NO from vascular endothelium through the activation of eNOS by PI3K/PKB/Akt pathway [92]. PI3-kinase increases the trafficking and translocation of NO synthase and cation pump units as well as glucose transporters, which mediates an increase in NO, Na+ pump, K+ channel, and calcium (Ca2+) myofilament sensitivity [93]. These effects are blunted in IR states, predisposing to the development and the progression of cardiovascular diseases. Angiotensin II, a component of renin-angiotensin system (RAS) via stimulation of the angiotensin II, type I receptor (AT1R) activates JNK and MAP-kinase pathways, leading to increased serine phosphorylation of IRS-1 and 2 proteins ultimately inhibiting PI3K signaling and further causing deleterious effects [94].
Mitochondrial biogenesis in IR: a secondary cause of cardiovascular disorder
The myocardium maintains a high energy demand to keep the heart viable, and this energy is supplied by mitochondria. Cardiac muscle of IR individuals generally contains 30 % less mitochondria than that of insulin sensitive individuals [95]. High-fat diet tends to promote an increase in mitochondria in order to oxidize fat which is concomitant with the development of tissue-specific IR [96, 97]. Thus, increase in FFA, induced by high-fat diet can be correlated with mitochondrial biogenesis [98, 99]. Impaired mitochondrial oxidative phosphorylation (OXPHO) and mitochondrial biogenesis contributes to an inhibition of insulin metabolic signaling [100]. The mechanism by which mitochondrial dysfunction is directly involved, an impairment of IR signaling occurs by controlling the PGC-1 α, nuclear respiratory factors 1 (NRF1) and NRF2 genes [101], which therefore regulate mitochondrial ATP production [102–104]. It is also suggested that transcriptional activation of PGC1α promotes mitochondrial proliferation and its associated markers that are required for mitochondrial biogenesis in the myocardium [104, 105]. Studies on cardiac-specific deletion of NRF1 and ERRα suggest that PGC1α activates estrogen-related nuclear receptors-α and γ (ERRα and ERRγ) to induce genes participating in glucose and fatty acid uptakes. This results in upregulation of ATP transport via NRF1/2-mediated stimulation of mitochondrial transcription factor A (Tfam A) [106–108] and OXPHOS genes [109]. Thus, there is sufficient evidence that supports the role of mitochondrial biogenesis in CVD associated with IR.
IR related to CVDs and potential therapeutic approaches
Accumulation of fatty acid metabolites, DG and LCFA-CoA because of mitochondrial dysfunction by atypical PKCs activation results in IR [110, 111]. Mitochondrial function and insulin sensitivity can be improved by increased expression of UCP2/3 or decrease in ROS production by antioxidants [111]. Modulation of glucose/LCFA metabolism could be an approach for establishing a new equilibrium favoring glucose uptake and oxidation in opposition to LCFA oxidation. Adjustment of insulin signaling can also be done by using thiazolidinediones and metformin–insulin sensitizers, which are reported to reduce ROS production, increase expression of PGC-1α, and stimulate AMPK, thus improving mitochondrial function by reducing oxidative stress and stimulating mitochondrial biogenesis [111]. AMPK can also directly stimulate glucose uptake by phosphorylating and inactivating AS160 (converging point between insulin and AMPK-signaling pathways), enhancing PKB/Akt overactivation and decreasing serine phosphorylation of IRS-1 in cardiomyocytes [112]. Under ischemic conditions, the activated AMPK counteracts the PKB/Akt-mediated activation of p70S6K, phosphorylation of eEF2, and stimulation of protein synthesis in cardiomyocytes. In the cardiac system, expression of a kinase-dead phosphofructokinase 2 (PFK2) decreases glycolytic flux, induces hypertrophy and fibrosis, and reduces cardiomyocyte function thus explaining the importance of PFK2 in the regulation of cardiac function.
Under normal conditions, insulin stimulates the production of NO from endothelium, leading to vasodilation, increased blood flow, augments glucose disposal in skeletal muscle. Under IR condition, hyperinsulinemia overdrives unaffected MAPK-dependent pathways leading to secretion of the vasoconstrictor endothelin-1 (ET-1) from vascular endothelium. This imbalance between vasoconstrictor and vasodilator actions of insulin under IR condition is an important factor in the vascular pathophysiology of IR and endothelial dysfunction. Pharmacological blockage of ET-1 receptors (ET-A isoform) improves endothelial function in cardiovascular disorders [113]. Intraarterial vitamin C improves endothelial-dependent vasodilation in type 2 diabetes mellitus associated CVD [114]. Adiponectin directly stimulates the production of NO from vascular endothelium using a PI3-kinase-dependent signaling mechanism similar to that of insulin, thus opposing atherogenesis and improve endothelial function [115]. The development of IR along with cardiometabolic syndrome is associated with increased tissue renin–angiotensin system activity [111].
Angiotensin II via its type I receptors stimulates the production of ROS via NADPH oxidase, increases expression of ICAM-1 and increases ET-1 release from endothelium [111]. Pharmacological inhibitors such as angiotensin-converting enzyme (ACE) inhibitors reduce circulating angiotensin II levels, and angiotensin receptor blockers (ARBs) block the actions of angiotensin II ultimately helping in lowering of blood pressure, improving endothelial function and reduce circulating markers of inflammation, augmentation of insulin stimulated glucose uptake. ACE inhibitors (ramipril) and ARBs (losartan) support the existence of reciprocal relationships between endothelial dysfunction and insulin. Tempol, a superoxide scavenger, is able to ameliorate cardiac and vascular dysfunction, normalize angiotensin II–induced IR [116]. Fibrates are synthetic PPAR-α ligands that improve the circulating lipoprotein profile, resulting in improved endothelial function, reduced vascular inflammation, and reduction in cardiovascular events through increased adiponectin levels [117].
Conclusions
The complex interplay of IR and CVD has been studied extensively to elucidate the mechanistic pathway underlying the pathogenesis of the disease. The major causes of IR are hyperglycemia, oxidative stress and dyslipidemia which present as risk factors for CVD. The hampered insulin signaling abrogates insulin stimulated glucose uptake, endothelial functions, vasodilation and blood flow which in turn compromise functioning of cardiomyocytes and leads to hypertrophy, fibrosis and atherosclerosis. During severity of the disease anti-oxidants, anti-inflammatory and insulin sensitizers also enhance activities of various molecules such as NO, PI3K, Akt and GLUT4 receptors. Nonetheless, drugs regulating type 2 DM and hyperlipidemia have side effects associated with CVD and associated complications suggesting potential dual links between IR, type 2 DM and CVD at the molecular level. Thus treatment strategies for CVD would depend on the metabolic status of the individual. Given the serious consequences of the global CVD epidemic, understanding of the mechanisms that link IR with the development of CVD and comorbidities should be considered as a high priority in further research.
Abbreviations
- ACE:
-
Angiotensin-converting enzyme
- ARBs:
-
Angiotensin receptor blockers
- ADA:
-
American Diabetes Association
- AGEs:
-
Advanced glycation end-products
- AMPK:
-
AMP-activated protein kinase
- AT1R:
-
Angiotensin II type I receptor
- C/EBP:
-
CCAAT/enhancer binding protein
- CAN:
-
Cardiac autonomic neuropathy
- CRP:
-
C-reactive protein
- CVD:
-
Cardio vascular diseases
- DG:
-
Diacyl glycerol
- DM:
-
Diabetes mellitus
- eNOS:
-
Endothelial nitric oxide synthase
- ERR:
-
Estrogen-related nuclear receptors
- ET-1:
-
Endothelin-1
- FAT/CD36:
-
Fatty acid translocase
- FetA:
-
Fetuin-A
- FFA:
-
Free fatty acid
- Glut4:
-
Glucose transporter 4
- HDL:
-
High-density lipoprotein
- HO-1:
-
Heme oxygenase-1
- ICAM-1:
-
Intracellular adhesion molecule-1
- IL-6:
-
Interleukin-6
- IR:
-
Insulin resistance
- IR-β:
-
Insulin receptor β
- IRS-1:
-
Insulin receptor substrate-1
- JNK:
-
Janus kinase
- STAT:
-
Signal transducer and activator of transcription
- LCFA:
-
Long-chain fatty acid
- LDL:
-
Low-density lipoprotein
- LPL:
-
Lipoprotein lipase
- MAPK:
-
Mitogen-activated protein kinase
- MCP-1:
-
Macrophage chemo attractant protein-1
- mTOR:
-
Mammalian target of rapamycin
- NADP:
-
Nicotinamide adenine dinucleotide phosphate
- NEFA:
-
Non-esterified fatty acid
- NFAT:
-
Nuclear factor of activated T cells
- NFκ-B:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NO:
-
Nitric oxide
- NOXs:
-
NADPH oxidases
- NRF1:
-
Nuclear respiratory factor 1
- OXPHO:
-
Oxidative phosphorylation
- PAI-1:
-
Plasminogen activator inhibitor-1
- PFK2:
-
Phosphofructokinase 2
- PGC-1α:
-
PPAR-γ coactivator 1α
- PH:
-
Pleckstrin homology
- PI3K:
-
Phosphatidylinositol 3-kinase
- PKC:
-
Protein kinase C
- PKB/Akt:
-
Protein kinase B
- PPARs:
-
Peroxisome proliferator-activated receptors
- PTEN:
-
Phosphatase and tensin homolog
- PTP1B:
-
Protein tyrosine phosphatase 1B
- ROS:
-
Reactive oxygen species
- SHIP:
-
SH2-containing inositol 5′-phosphatase
- SOCS:
-
Suppressors of cytokine signaling
- SREBP:
-
Sterol regulatory element binding protein
- TAG:
-
Triacyl glycerol
- Tfam A:
-
Mitochondrial transcription factor A
- TLRs:
-
Toll-like receptors
- TNF-α:
-
Tumor necrosis factor-α
- UCP:
-
Uncoupling protein
- VAT:
-
Visceral adipose tissue
- VEGF:
-
Vascular endothelial growth factor
- VLDL:
-
Very low-density lipoprotein
References
Ceriello A (2005) Postprandial hyperglycemia and diabetes complications is it time to treat? Diabetes 54(1):1–7
Kannel WB, Hjortland M, Castelli WP (1974) Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol 34(1):29–34
Garcia M, McNamara P, Gordon T, Kannel W (1972) Cardiovascular complications in diabetics. Adv Metab Disord 2(Suppl 2):493–499
Fowler MJ (2008) Microvascular and macrovascular complications of diabetes. Clin Diabetes 26(2):77–82
Ceriello A (2003) New insights on oxidative stress and diabetic complications may lead to a “causal” antioxidant therapy. Diabetes Care 26(5):1589–1596
Farahmand F, Lou H, Singal PK (2003) Oxidative stress in cardiovascular complications of diabetes. In: Pierce GN, Nagano M, Zahradka P, Dhalla NS (eds) Atherosclerosis, hypertension and diabetes. Kluwer Academic Publications, Boston, pp 427–437
Gyurko R, Siqueira CC, Caldon N, Gao L, Kantarci A, Van Dyke TE (2006) Chronic hyperglycemia predisposes to exaggerated inflammatory response and leukocyte dysfunction in Akita mice. J Immunol 177(10):7250–7256
Lum H, Roebuck KA (2001) Oxidant stress and endothelial cell dysfunction. Am J Physiol Cell Physiol 280(4):C719–C741
Versari D, Daghini E, Virdis A, Ghiadoni L, Taddei S (2009) Endothelial dysfunction as a target for prevention of cardiovascular disease. Diabetes Care 32(suppl 2):S314–S321
Schram MT, Chaturvedi N, Schalkwijk C, Giorgino F, Ebeling P, Fuller JH, Stehouwer CD (2003) Vascular risk factors and markers of endothelial function as determinants of inflammatory markers in type 1 diabetes the EURODIAB Prospective complications study. Diabetes Care 26(7):2165–2173
El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, Cooper ME, Brownlee M (2008) Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med 205(10):2409–2417
Hink U, Li H, Mollnau H, Oelze M, Matheis E, Hartmann M, Skatchkov M, Thaiss F, Stahl RA, Warnholtz A (2001) Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res 88(2):e14–e22
Watts G, Playford D (1998) Dyslipoproteinaemia and hyperoxidative stress in the pathogenesis of endothelial dysfunction in non-insulin dependent diabetes mellitus: an hypothesis. Atherosclerosis 141(1):17–30
Renard CB, Kramer F, Johansson F, Lamharzi N, Tannock LR, von Herrath MG, Chait A, Bornfeldt KE (2004) Diabetes and diabetes-associated lipid abnormalities have distinct effects on initiation and progression of atherosclerotic lesions. J Clin Investig 114(5):659
Poirier P, Giles TD, Bray GA, Hong Y, Stern JS, Pi-Sunyer FX, Eckel RH (2006) Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss an update of the 1997 American Heart Association Scientific statement on obesity and heart disease from the obesity committee of the council on nutrition, physical activity, and metabolism. Circulation 113(6):898–918
Duncan BB, Schmidt MI, Pankow JS, Ballantyne CM, Couper D, Vigo A, Hoogeveen R, Folsom AR, Heiss G (2003) Low-grade systemic inflammation and the development of type 2 diabetes the atherosclerosis risk in communities study. Diabetes 52(7):1799–1805
Turner R, Holman R, Matthews D, Bassett P, Coster R, Stratton I, Cull C, Peto R, Frighi V, Kennedy I (1993) Hypertension in diabetes study (Hds). 1. Prevalence of hypertension in newly presenting type-2 diabetic-patients and the association with risk-factors for cardiovascular and diabetic complications. J Hypertens 11(3):309–317
ADA (1993) Treatment of hypertension in diabetes. Diabetes Care 16:1394–1401
Goto A, Arah OA, Goto M, Terauchi Y, Noda M (2013) Severe hypoglycaemia and cardiovascular disease: systematic review and meta-analysis with bias analysis. BMJ 347:F4533
Sommerfield AJ, Wilkinson IB, Webb DJ, Frier BM (2007) Vessel wall stiffness in type 1 diabetes and the central hemodynamic effects of acute hypoglycemia. Am J Physiol Endocrinol Metab 293(5):E1274–E1279
Frier BM, Schernthaner G, Heller SR (2011) Hypoglycemia and cardiovascular risks. Diabetes Care 34(Supplement 2):S132–S137
Desouza CV, Bolli GB, Fonseca V (2010) Hypoglycemia, diabetes, and cardiovascular events. Diabetes Care 33(6):1389–1394
Spallone V, Ziegler D, Freeman R, Bernardi L, Frontoni S, Pop-Busui R, Stevens M, Kempler P, Hilsted J, Tesfaye S (2011) Cardiovascular autonomic neuropathy in diabetes: clinical impact, assessment, diagnosis, and management. Diabetes Metab Res Rev 27(7):639–653
Witte D, Tesfaye S, Chaturvedi N, Eaton S, Kempler P, Fuller J, Group EPCS (2005) Risk factors for cardiac autonomic neuropathy in type 1 diabetes mellitus. Diabetologia 48(1):164–171
Astrup AS, Tarnow L, Rossing P, Hansen BV, Hilsted J, Parving H-H (2006) Cardiac autonomic neuropathy predicts cardiovascular morbidity and mortality in type 1 diabetic patients with diabetic nephropathy. Diabetes Care 29(2):334–339
DeFronzo RA, Tripathy D (2009) Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 32(suppl 2):S157–S163
Groop L, Bonadonna R, Del Prato S, Ratheiser K, Zyck K, DeFronzo R (1989) Effect of insulin on oxidative and non-oxidative pathways of glucose and FFA metabolism in NIDDM. Evidence for multiple sites of insulin resistance. J Clin Invest 84:205–213
Morino K, Petersen KF, Shulman GI (2006) Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes 55(Supplement 2):S9–S15
Langin D, Dicker A, Tavernier G, Hoffstedt J, Mairal A, Rydén M, Arner E, Sicard A, Jenkins CM, Viguerie N (2005) Adipocyte lipases and defect of lipolysis in human obesity. Diabetes 54(11):3190–3197
Kern PA (1997) Potential role of TNFα and lipoprotein lipase as candidate genes for obesity. J Nutr 127(9):1917S–1922S
Bugianesi E, McCullough AJ, Marchesini G (2005) Insulin resistance: a metabolic pathway to chronic liver disease. Hepatology 42(5):987–1000
Dröge W (2002) Free radicals in the physiological control of cell function. Physiol Rev 82(1):47–95
Brownlee M (2001) Biochemistry and molecular cell biology of diabetic complications. Nature 414(6865):813–820
Greene DA, Stevens MJ, Obrosova I, Feldman EL (1999) Glucose-induced oxidative stress and programmed cell death in diabetic neuropathy. Eur J Pharmacol 375(1):217–223
Wolff SP, Dean R (1987) Glucose autoxidation and protein modification. The potential role of ‘autoxidative glycosylation’ in diabetes. Biochem J 245:243–250
Yan S, Stern D, Schmidt A (1997) What’s the RAGE? The receptor for advanced glycation end products (RAGE) and the dark side of glucose. Eur J Clin Invest 27(3):179–181
Stojiljkovic MP, Lopes HF, Zhang D, Morrow JD, Goodfriend TL, Egan BM (2002) Increasing plasma fatty acids elevates F2-isoprostanes in humans: implications for the cardiovascular risk factor cluster. J Hypertens 20(6):1215–1221
S-i Yamagishi, Edelstein D, X-l Du, Kaneda Y, Guzmán M, Brownlee M (2001) Leptin induces mitochondrial superoxide production and monocyte chemoattractant protein-1 expression in aortic endothelial cells by increasing fatty acid oxidation via protein kinase A. J Biol Chem 276(27):25096–25100
Paz K, Hemi R, LeRoith D, Karasik A, Elhanany E, Kanety H, Zick Y (1997) A Molecular Basis for Insulin Resistance elevated serine/threonine phosphorylation of irs-1 and irs-2 inhibits their binding to the juxtamembrane region of the insulin receptor and impairs their ability to undergo insulin-induced tyrosine phosphorylation. J Biol Chem 272(47):29911–29918
Potashnik R, Bloch-Damti A, Bashan N, Rudich A (2003) IRS1 degradation and increased serine phosphorylation cannot predict the degree of metabolic insulin resistance induced by oxidative stress. Diabetologia 46(5):639–648
Ogihara T, Asano T, Katagiri H, Sakoda H, Anai M, Shojima N, Ono H, Fujishiro M, Kushiyama A, Fukushima Y (2004) Oxidative stress induces insulin resistance by activating the nuclear factor-κB pathway and disrupting normal subcellular distribution of phosphatidylinositol 3-kinase. Diabetologia 47(5):794–805
Khamaisi M, Potashnik R, Tirosh A, Demshchak E, Rudich A, Trischler H, Wessel K, Bashan N (1997) Lipoic acid reduces glycemia and increases muscle GLUT4 content in streptozotocin-diabetic rats. Metabolism 46(7):763–768
Pessler D, Rudich A, Bashan N (2001) Oxidative stress impairs nuclear proteins binding to the insulin responsive element in the GLUT4 promoter. Diabetologia 44(12):2156–2164
Castelló A, Rodríguez-Manzaneque JC, Camps M, Perez-Castillo A, Testar X, Palacin M, Santos A, Zorzano A (1994) Perinatal hypothyroidism impairs the normal transition of GLUT4 and GLUT1 glucose transporters from fetal to neonatal levels in heart and brown adipose tissue. Evidence for tissue-specific regulation of GLUT4 expression by thyroid hormone. J Biol Chem 269(8):5905–5912
Randle PJ, Kerbey AL, Espinal J (1988) Mechanisms decreasing glucose oxidation in diabetes and starvation: role of lipid fuels and hormones. Diabetes Metab Rev 4(7):623–638
Shulman GI (2000) Cellular mechanisms of insulin resistance. J Clin Investig 106(2):171
Stalder M, Pometta D, Suenram A (1981) Relationship between plasma insulin levels and high density lipoprotein cholesterol levels in healthy men. Diabetologia 21(6):544–548
Sadur UN, Yost TJ, Eckel RH (1984) Insulin responsiveness of adipose tissue lipoprotein lipase is delayed but preserved in obesity*. J Clin Endocrinol Metab 59(6):1176–1182
Golay A, Zech L, Shi M-Z, Chiou Y-A, Reaven G, Chen Y-D (1987) High density lipoprotein (HDL) metabolism in noninsulin-dependent diabetes mellitus: measurement of HDL turnover using tritiated HDL*. J Clin Endocrinol Metab 65(3):512–518
Eriksson P, Nilsson L, Karpe F, Hamsten A (1998) Very-low-density lipoprotein response element in the promoter region of the human plasminogen activator inhibitor-1 gene implicated in the impaired fibrinolysis of hypertriglyceridemia. Arterioscler Thromb Vasc Biol 18(1):20–26
Nishikawa T, Edelstein D, Du XL, S-i Yamagishi, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes H-P (2000) Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404(6779):787–790
Pyörälä K (1979) Relationship of glucose tolerance and plasma insulin to the incidence of coronary heart disease: results from two population studies in Finland. Diabetes Care 2(2):131–141
Després J-P, Lamarche B, Mauriège P, Cantin B, Dagenais GR, Moorjani S, Lupien P-J (1996) Hyperinsulinemia as an independent risk factor for ischemic heart disease. N Engl J Med 334(15):952–958
Suzuki M, Shinozaki K, Kanazawa A, Hara Y, Hattori Y, Tsushima M, Harano Y (1996) Insulin resistance as an independent risk factor for carotid wall thickening. Hypertension 28(4):593–598
Shen D-C, Shieh S-M, Fuh M-T, Wu D-A, Chen Y-D, Reaven G (1988) Resistance to insulin-stimulated-glucose uptake in patients with hypertension*. J Clin Endocrinol Metab 66(3):580–583
Reaven GM, Chang H (1991) Relationship between blood pressure, plasma insulin ana triglyceride concentration, and insulin action in spontaneous hypertensive and Wistar-Kyoto rats. Am J Hypertens 4(1 Pt 1):34–38
Sechi LA, Melis A, Tedde R (1992) Insulin hypersecretion: a distinctive feature between essential and secondary hypertension. Metabolism 41(11):1261–1266
Reaven G (1996) Hypertension and associated metabolic abnormalities—the role of insulin resistance and the sympathoadrenal system. N Engl J Med 334:374–381
Mitchell TH, Nolan B, Henry M, Cronin C, Baker H, Greely G (1997) Microalbuminuria in patients with non-insulin-dependent diabetes mellitus relates to nocturnal systolic blood pressure. Am J Med 102(6):531–535
Laine H, Yki-Jarvinen H, Kirvela O, Tolvanen T, Raitakari M, Solin O, Haaparanta M, Knuuti J, Nuutila P (1998) Insulin resistance of glucose uptake in skeletal muscle cannot be ameliorated by enhancing endothelium-dependent blood flow in obesity. J Clin Investig 101(5):1156
Ginsberg HN (2000) Insulin resistance and cardiovascular disease. J Clin Investig 106(4):453
Seidell JC, Pérusse L, Després J-P, Bouchard C (2001) Waist and hip circumferences have independent and opposite effects on cardiovascular disease risk factors: the Quebec Family Study. Am J Clin Nutr 74(3):315–321
Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH (1997) Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med 336(14):973–979
Brown NJ, Kim K-S, Chen Y-Q, Blevins LS, Nadeau JH, Meranze SG, Vaughan DE (2000) Synergistic effect of adrenal steroids and angiotensin II on plasminogen activator inhibitor-1 production 1. J Clin Endocrinol Metab 85(1):336–344
Sowers JR, Sowers PS, Peuler JD (1994) Role of insulin resistance and hyperinsulinemia in development of hypertension and atherosclerosis. J Lab Clin Med 123(5):647–652
Chen Y-Q, Su M, Walia RR, Hao Q, Covington JW, Vaughan DE (1998) Sp1 sites mediate activation of the plasminogen activator inhibitor-1 promoter by glucose in vascular smooth muscle cells. J Biol Chem 273(14):8225–8231
Westerbacka J, Vehkavaara S, Bergholm R, Wilkinson I, Cockcroft J, Yki-Järvinen H (1999) Marked resistance of the ability of insulin to decrease arterial stiffness characterizes human obesity. Diabetes 48(4):821–827
Steinberg HO, Chaker H, Leaming R, Johnson A, Brechtel G, Baron AD (1996) Obesity/insulin resistance is associated with endothelial dysfunction. Implications for the syndrome of insulin resistance. J Clin Investig 97(11):2601
Després J-P (2006) Abdominal obesity: the most prevalent cause of the metabolic syndrome and related cardiometabolic risk. Eur Heart J Suppl 8(suppl B):B4–B12
Steinberger J, Daniels SR (2003) Obesity, insulin resistance, diabetes, and cardiovascular risk in children an American Heart Association scientific statement from the atherosclerosis, hypertension, and obesity in the Young Committee (Council on Cardiovascular Disease in the Young) and the Diabetes Committee (Council on Nutrition, Physical Activity, and Metabolism). Circulation 107(10):1448–1453
Van Gaal LF, Mertens IL, Christophe E (2006) Mechanisms linking obesity with cardiovascular disease. Nature 444(7121):875–880
Turer AT, Hill JA, Elmquist JK, Scherer PE (2012) Adipose tissue biology and cardiomyopathy translational implications. Circ Res 111(12):1565–1577
Ramkhelawon B, Hennessy EJ, Ménager M, Ray TD, Sheedy FJ, Hutchison S, Wanschel A, Oldebeken S, Geoffrion M, Spiro W (2014) Netrin-1 promotes adipose tissue macrophage retention and insulin resistance in obesity. Nat Med 20(4):377–384
Sell H, Habich C, Eckel J (2012) Adaptive immunity in obesity and insulin resistance. Nat Rev Endocrinol 8(12):709–716
De Marchi E, Faldassari B, Bononi A, Wieckowski M, Pinton P (2013) Oxidative stress in cardiovascular diseases and obesity: role of p66Shc and protein kinase C. Oxid Med Cell Longev 2013:564961. doi:10.1155/2013/564961
Borén J, Taskinen MR, Olofsson SO, Levin M (2013) Ectopic lipid storage and insulin resistance: a harmful relationship. J Intern Med 274(1):25–40
Yang R, Barouch LA (2007) Leptin signaling and obesity cardiovascular consequences. Circ Res 101(6):545–559
Lumeng CN, Bodzin JL, Saltiel AR (2007) Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Investig 117(1):175
Ruan H, Hacohen N, Golub TR, Van Parijs L, Lodish HF (2002) Tumor necrosis factor-α suppresses adipocyte-specific genes and activates expression of preadipocyte genes in 3T3-L1 adipocytes nuclear factor-κB activation by TNF-α is obligatory. Diabetes 51(5):1319–1336
Permana PA, Menge C, Reaven PD (2006) Macrophage-secreted factors induce adipocyte inflammation and insulin resistance. Biochem Biophys Res Commun 341(2):507–514
Schächinger V, Britten MB, Zeiher AM (2000) Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation 101(16):1899–1906
Bugiardini R, Manfrini O, Pizzi C, Fontana F, Morgagni G (2004) Endothelial function predicts future development of coronary artery disease a study of women with chest pain and normal coronary angiograms. Circulation 109(21):2518–2523
Han S, Liang C-P, DeVries-Seimon T, Ranalletta M, Welch CL, Collins-Fletcher K, Accili D, Tabas I, Tall AR (2006) Macrophage insulin receptor deficiency increases ER stress-induced apoptosis and necrotic core formation in advanced atherosclerotic lesions. Cell Metab 3(4):257–266
Kuboki K, Jiang ZY, Takahara N, Ha SW, Igarashi M, Yamauchi T, Feener EP, Herbert TP, Rhodes CJ, King GL (2000) Regulation of endothelial constitutive nitric oxide synthase gene expression in endothelial cells and in vivo a specific vascular action of insulin. Circulation 101(6):676–681
Hamsten A, Wiman B, de Faire U, Blombäck M (1985) Increased plasma levels of a rapid inhibitor of tissue plasminogen activator in young survivors of myocardial infarction. N Engl J Med 313(25):1557–1563
Wiman B, Andersson T, Hallqvist J, Reuterwall C, Ahlbom A (2000) Plasma levels of tissue plasminogen activator/plasminogen activator inhibitor-1 complex and von Willebrand factor are significant risk markers for recurrent myocardial infarction in the Stockholm Heart Epidemiology Program (SHEEP) study. Arterioscler Thromb Vasc Biol 20(8):2019–2023
Calles-Escandon J, Mirza SA, Sobel BE, Schneider DJ (1998) Induction of hyperinsulinemia combined with hyperglycemia and hypertriglyceridemia increases plasminogen activator inhibitor 1 in blood in normal human subjects. Diabetes 47(2):290–293
Bertrand L, Horman S, Beauloye C, Vanoverschelde J-L (2008) Insulin signalling in the heart. Cardiovasc Res 79(2):238–248
Randle P, Garland P, Hales C, Newsholme E (1963) The glucose fatty-acid cycle its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 281(7285):785–789
Proud C (2007) Signalling to translation: how signal transduction pathways control the protein synthetic machinery. Biochem J 403:217–234
Heineke J, Molkentin JD (2006) Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol 7(8):589–600
Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM (1999) Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399(6736):601–605
McFarlane SI, Banerji M, Sowers JR (2001) Insulin resistance and cardiovascular disease. J Clin Endocrinol Metab 86(2):713–718
Mehta PK, Griendling KK (2007) Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol 292(1):C82–C97
Lowell BB, Shulman GI (2005) Mitochondrial dysfunction and type 2 diabetes. Science 307(5708):384–387
Kim YJ, Park T (2008) Genes are differentially expressed in the epididymal fat of rats rendered obese by a high-fat diet. Nutr Res 28(6):414–422
Dong F, Li Q, Sreejayan N, Nunn JM, Ren J (2007) Metallothionein prevents high-fat diet-induced cardiac contractile dysfunction role of peroxisome proliferator-activated receptor γ coactivator 1α and mitochondrial biogenesis. Diabetes 56(9):2201–2212
Garcia-Roves P, Huss JM, Han D-H, Hancock CR, Iglesias-Gutierrez E, Chen M, Holloszy JO (2007) Raising plasma fatty acid concentration induces increased biogenesis of mitochondria in skeletal muscle. Proc Natl Acad Sci 104(25):10709–10713
Hancock CR, Han D-H, Chen M, Terada S, Yasuda T, Wright DC, Holloszy JO (2008) High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc Natl Acad Sci 105(22):7815–7820
Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI (2004) Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med 350(7):664–671
Huo L, Scarpulla RC (2001) Mitochondrial DNA instability and peri-implantation lethality associated with targeted disruption of nuclear respiratory factor 1 in mice. Mol Cell Biol 21(2):644–654
Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R (2003) Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci 100(14):8466–8471
Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E (2003) PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34(3):267–273
Russell LK, Mansfield CM, Lehman JJ, Kovacs A, Courtois M, Saffitz JE, Medeiros DM, Valencik ML, McDonald JA, Kelly DP (2004) Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor γ coactivator-1α promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ Res 94(4):525–533
Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP (2000) Peroxisome proliferator–activated receptor γ coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Investig 106(7):847
Huss JM, Kelly DP (2004) Nuclear receptor signaling and cardiac energetics. Circ Res 95(6):568–578
Kelly DP, Scarpulla RC (2004) Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev 18(4):357–368
Garnier A, Fortin D, Delomenie C, Momken I, Veksler V, Ventura-Clapier R (2003) Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J Physiol 551(2):491–501
Ritz P, Berrut G (2005) Mitochondrial function, energy expenditure, aging and insulin resistance. Diabetes Metab 31:5S67–65S73
Savage DB, Petersen KF, Shulman GI (2005) Mechanisms of insulin resistance in humans and possible links with inflammation. Hypertension 45(5):828–833
J-a Kim, Wei Y, Sowers JR (2008) Role of mitochondrial dysfunction in insulin resistance. Circ Res 102(4):401–414
Zick Y (2005) Ser/Thr phosphorylation of IRS proteins: a molecular basis for insulin resistance. Sci STKE 2005(268):pe4. doi:10.1126/stke.2682005pe4
Mather KJ, Lteif A, Steinberg HO, Baron AD (2004) Interactions between endothelin and nitric oxide in the regulation of vascular tone in obesity and diabetes. Diabetes 53(8):2060–2066
Ting HH, Timimi FK, Boles KS, Creager SJ, Ganz P, Creager MA (1996) Vitamin C improves endothelium-dependent vasodilation in patients with non-insulin-dependent diabetes mellitus. J Clin Investig 97(1):22
Chen H, Montagnani M, Funahashi T, Shimomura I, Quon MJ (2003) Adiponectin stimulates production of nitric oxide in vascular endothelial cells. J Biol Chem 278(45):45021–45026
Whaley-Connell A, Govindarajan G, Habibi J, Hayden MR, Cooper SA, Wei Y, Ma L, Qazi M, Link D, Karuparthi PR (2007) Angiotensin II-mediated oxidative stress promotes myocardial tissue remodeling in the transgenic (mRen2) 27 Ren2 rat. Am J Physiol Endocrinol Metab 293(1):E355–E363
Koh KK, Quon MJ, Han SH, Chung W-J, Ahn JY, Seo Y-H, Choi IS, Shin EK (2005) Additive beneficial effects of fenofibrate combined with atorvastatin in the treatment of combined hyperlipidemia. J Am Coll Cardiol 45(10):1649–1653
Acknowledgments
Prof. Sarita Gupta was a visiting scientist in Institute of Cardiovascular Sciences. Nathalia Bernardes and Danielle da Silva Dias were exchange students, under the Canada-Brazil Training program. Dr. Pawan Singal is the holder of the Dr. Naranjan S. Dhalla Chair in Cardiovascular Research supported by St. Boniface Hospital and Research Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Rights and permissions
About this article

Cite this article
Patel, T.P., Rawal, K., Bagchi, A.K. et al. Insulin resistance: an additional risk factor in the pathogenesis of cardiovascular disease in type 2 diabetes. Heart Fail Rev 21, 11–23 (2016). https://doi.org/10.1007/s10741-015-9515-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-015-9515-6