
Abstract
The pathophysiology of acute heart failure syndromes (AHFS), defined as a change or worsening in heart failure symptoms and signs, is complex. The variety of adverse neurohormonal adaptations includes increased levels of plasma renin, aldosterone and angiotensin II, all responsible for cardio-renal dysfunction. In fact, such alterations result in an array of clinical changes that include abnormal haemodynamics, altered ventricular filling pressures, pathological neurohormonal responses, leading to fluid overload, congestion and ultimately heart failure symptoms. Clinical pictures can be various: in spite of a usual improvement in dyspnoea, little weight change and significant morbidity are generally observed during hospitalization. Short-term outcomes are characterized by a high 60-day re-hospitalization and high mortality rates; apparently, both can be predicted from pre-discharge characteristics. The most frequently used treatments for AHF care include diuretics, inotropic agents, and vasodilator/vasoactive agents; however, the final therapeutic strategy is often individualized. Diuretics are currently the most used agents, but resistance to diuretic therapy is common. In addition, several studies have demonstrated that aggressive diuresis can contribute to reduced renal function, and high doses of diuretics have been associated with increased morbidity and mortality. Many patients with AHFS also suffer from acute or from chronic renal dysfunction (cardio-renal syndromes type 1 and 2, respectively), which further complicate the outcomes and treatment strategies. A personalized patient evaluation of the combined heart and kidney functions is advised to implement the best possible multidisciplinary diagnostic and therapeutic approach.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute heart failure (AHF) is defined as a ‘rapid onset or change in the signs and symptoms of HF, resulting in the need for urgent therapy’, according to the recent European Society of Cardiology (ESC) guidelines for the diagnosis and treatment of heart failure (HF) [1].
The decompensated state begins with a haemodynamic derangement [2], which leads to pulmonary congestion, with or without fluid overload and peripheral oedema, in the presence of reduced or preserved left ventricular systolic function. These are almost universal haemodynamic findings in patients with AHF. However, despite their importance as a cause of symptoms and immediate hospitalization, pulmonary congestion and fluid overload remain poorly related to long-term outcomes [3]. Additional mechanisms activated in AHF should therefore be taken into account, such as myocardial damage and, most notably, renal dysfunction.
Monitoring of such haemodynamic complications of AHF is a cornerstone in the management of the critically ill patient. It may help to identify different patients’ risk profiles and response to therapy. However, the efficacy of monitoring depends upon its capacity to guide treatments that improve outcome.
This review will focus on the haemodynamic complications in AHFS and will address the relevant pathophysiological issues and therapeutic strategies with particular emphasis on the impact of renal dysfunction on outcomes.
Clinical presentation of AHF and pathophysiology
Several clinical characteristics and presentations of AHF syndromes (AHFS) have been previously described [1]:
-
Worsening or decompensated chronic HF: characterized by progressive worsening of known chronic HF on treatment, with evidence of systemic and pulmonary congestion.
-
Pulmonary oedema: characterized by severe respiratory distress, tachypnoea and orthopnoea with rales over the lung fields.
-
Hypertensive HF: characterized by signs and symptoms of HF accompanied by high BP and usually relatively preserved LV systolic function.
-
Cardiogenic shock: defined on the basis of signs of tissue hypoperfusion after adequate correction of preload and arrhythmias, typically characterized by reduced systolic blood pressure and absent or low urine output.
-
Isolated right HF: which presents as a low output syndrome with increased jugular venous pressure, with or without hepatomegaly, and low LV filling pressures, in the absence of pulmonary congestion.
Finally, according to 2008 ESC guidelines on the management of HF, another type of AHF syndrome can be distinguished, which is HF developing in the setting of an acute coronary syndrome.
This classification is based mainly upon major pathophysiologic characteristics of the different syndromes, which are complex and still incompletely understood, in particular for the precise events that trigger the transition from the compensated to the decompensated state of HF.
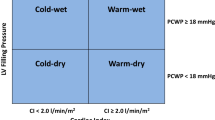
Congestion, as noted above, is one of the key findings of AHFS and may be due to two main initial mechanisms: a LV systolic dysfunction with increased ventricular filling pressures, peripheral hypoperfusion, renal sodium and water retention, and/or an excessive increase in LV afterload with concomitant LV diastolic dysfunction. This is considered a factor of paramount importance in the genesis of renal dysfunction.
In advanced chronic heart failure (CHF), the reduction in resting cardiac output is accompanied by marked redistribution of the forward flow to regional circulations [4, 5]. This redistribution of limited cardiac output favours vital organs (i.e. heart and brain) over the liver, limbs, skeletal muscles and kidney. These physiological adaptations associated with iatrogenic interventions sustain the compensated state of CHF making some organs, however, highly sensitive to insults. Renal sodium and water retention is caused by both reduced cardiac output with renal hypoperfusion, and increased central venous and renal venous pressure [6, 7], as well as by neurohormonal activation. Activation of the renin–angiotensin–aldosterone system (RAAS) together with an altered glomerular-tubular feedback leads to increased sodium and water retention in the kidney [8, 9]. Increased (non-osmotically induced) vasopressin secretion causes increased water reabsorption in the distal nephron and collecting ducts with consequent hyponatraemia [10]. Neurohormonal activation in HF has been linked to the unfavourable progression of the disease, ventricular remodelling and myocyte damage on one side [11] and to a high risk for renal dysfunction on the other side.
In response to increased cardiac filling pressures, there is an increased synthesis of atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP). These peptides promote arterial and venous vasodilation, increased water and sodium excretion, and they limit the effects of the activation of the RAAS and sympathetic nervous system [12].
In some cases, AHF may occur without salt and water retention and weight gain, and the underlying mechanism is rather a redistribution of fluids to the lungs with pulmonary, but not systemic, congestion [13]. This is likely to occur mostly in patients with HF and preserved LVEF and with clinical presentations of pulmonary oedema or hypertensive AHF. This is a condition of diastolic dysfunction often occurring in patients with chronic kidney disease (CKD).
Recent studies suggest the role of an “acute endothelitis” as responsible, albeit in part, for the sudden transition from compensated to decompensated HF. The “key mechanism” of acute “vascular” heart failure is an abrupt increase in vascular stiffness/resistance, probably secondary to neurohormonal/inflammatory activation, that contributes to progressive fluid retention and centralization of blood volume through vascular, renal and neurohormonal mechanisms. In this condition, high ventricular filling pressures may negatively impact cardiac function, by causing subendocardial ischaemia, left ventricular remodelling, impairment of cardiac venous drainage from coronary veins and a lower threshold for arrhythmias. The resulting decrease in cardiac output may further impair renal perfusion and function causing additional fluid retention. According to this “vasculocentric view,” an initial inflammatory stimulus may lead to vicious cycles and progressive vascular, cardiac and renal impairment. In particular, an inflammatory insult of biochemical (i.e. respiratory and urinary infections, non-compliance with medications) or biomechanical nature (i.e. fluid retention due to non-compliance with medications or diet) causes or worsens systemic endothelitis, with enhanced endothelial oxidative stress and activation in the venous system at the time of clinical decompensation.
Systemic endothelitis may offset preferential distribution of limited cardiac output to vital organs, and most importantly, to the kidneys. Kidney hypoperfusion may trigger the vascular, neural and humoral mechanism leading to sodium and water retention typical of cardio-renal syndrome type 1. The activation and stretching of the endothelium in the kidney may itself worsen “systemic endothelitis,” with further induction of vasoactive mediators and upregulation of pro-inflammatory genes and secondary capillary leak-mediated fluid retention. Concurrently, congestion may worsen cardiac function, leading to additional fluid retention, thereby creating a vicious circle [14].
Progressive fluid overload and centralization of blood volume may ultimately lead to clinical decompensation, which occurs several days to weeks after the beginning of the inflammatory stimuli.
In this context, giving the central role of kidney hypoperfusion in the fluid retention, the concept of “vasomotor nephropathy” should be stressed.
The so-called vasculo-renal syndrome is defined as transient renal dysfunction related to a perfusion mismatch of the afferent/efferent glomerular arteries due to vascular (reduction in endothelial NO bioavailability and/or an imbalance in prostaglandin production may cause vasoconstriction), haemodynamic (i.e. intravascular pressures/reduction in renal blood flow), neural (i.e. sympathetic) and humoral (i.e. renin–angiotensin–aldosterone system) mechanisms, leading to progressive sodium and water retention and ultimately AHF [8–10]. From a vascular standpoint, a reduction in endothelial NO bioavailability and/or an imbalance in prostaglandin production may cause afferent vasoconstriction and reduce renal blood flow and sodium excretion.
From a haemodynamic standpoint, perception of inadequate filling of the arterial circulation triggers direct vascular, renal and humoral mechanism that reduce sodium excretion. Enhanced renal sodium and water reabsorption predominantly fills the venous circulation, increasing atrial pressures. Normally, an increase in atrial pressure suppresses arginine-vasopressin release and enhances water diuresis, decreases renal sympathetic tone and augments natriuretic peptide secretion. In patients with HF, these atrial–renal reflexes are altered by neurohormonal activation and extension of venous congestion to the renal veins further impairs the glomerular filtration rate (Fig. 1) [11].

Mechanism of sodium and water retention in heart failure; compensatory mechanisms and altered renal responses leading to progressive and ultimately acute decompensation
Impact of renal dysfunction on heart failure outcomes
The majority of patients admitted with AHF have renal dysfunction. Data from Acute Decompensated Heart Failure National Registry (ADHERE) database (118,465 hospitalization) report normal renal function (estimated glomerular filtration rate (eGFR) ≥ 90 mL/min/1.73 m2) in 9% of patients at admission, mild renal dysfunction (eGFR 60–89 mL/min/1.73 m2) in 27.4%, moderate (eGFR 30–59 mL/min/1.73 m2) and severe renal dysfunction (eGFR 15–29 mL/min/1.73 m2), in 43.5 and 13.1% of patients, respectively. In-hospital mortality increases from 1.9% in patients with normal renal function to 7.6 and 6.5% in patients with severe kidney dysfunction and end stage kidney disease, respectively [15]. Renal dysfunction is an independent predictor of 6-month mortality: data from ESCAPE trial (Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheterization Effectiveness) also confirm this relationship between renal function and mortality. In this study, designed to understand the impact and pathophysiology of renal dysfunction in patients hospitalized with advanced HF, renal parameters were correlated with haemodynamic measurements (baseline renal insufficiency defined as eGFR < 60 mL/min/1.73 m2) and worsening renal function (WRF) (delta serum creatinine Δ[SCr] > or = 0.3 mg/dL) during the treatment of decompensated HF. Baseline and discharge renal insufficiency were associated with an increased risk of death and/or rehospitalization [16].
In patients undergoing intensive treatment for heart failure, WRF [increase in SCr ≥ 25% [17] or ≥ 0.3 mg/dL [18] is common and clinically significant (20–25% of admissions for AHF) and WRF predicts adverse outcomes in AHF both in-hospital and post-discharge [19]. The relation among risk factors (e.g. older age, hypertension and chronic kidney disease) and worsened outcome may reflect complex cardio-renal interactions; however, clinical characteristics available at hospital admission can be used to identify patients at increased risk for developing WRF.
Predominant haemodynamic factor associated with WRF in patients hospitalized with AHFS can be summarized as follows:
-
a.
Low cardiac output, high pulmonary capillary wedge pressure (pulmonary venous hypertension often resulting in pulmonary interstitial and alveolar oedema) and increased systemic venous pressure (high right atrial pressure) may contribute to the development of the cardio-renal syndrome [9]
-
b.
Central venous pressure: venous congestion is the most important haemodynamic factor driving WRF in patients with advanced heart failure admitted for ADHF. Mullens et al. studied a total of 145 patients treated with intensive medical therapy guided by pulmonary artery catheter to determine whether venous congestion, rather than impairment of cardiac output, is primarily associated with the development of WRF. Patients who developed WRF had a greater central venous pressure on admission and after intensive medical therapy [7].
-
c.
Intra-abdominal pressure (IAP): elevated IAP ≥ 8 mm Hg) is associated with intra-abdominal organ dysfunction. In 40 patients admitted for intensive management of ADHF, IAP was measured using transvesical technique at the time of admission and before the removal of pulmonary artery catheter. The study sought to determine whether changes in IAP with aggressive diuretic or vasodilator therapy are associated with improvement in renal function. Elevated IAP is prevalent in patients with ADHF and is associated with impaired renal function. In the setting of intensive medical therapy for ADHF, changes in IAP were better correlated with changes in renal function than any haemodynamic variable [20].
Monitoring and management of haemodynamic complications
AHFS are among the most frequent causes of hospitalizations in United States and Europe, but despite available therapies, these patients have high readmission and mortality rates [21]. The hospital course is associated with improvement in dyspnoea, little weight change and significant morbidity. Data from ADHERE indicate that 23% of hospitalized ADHF patients will be readmitted with the same diagnosis within the next 6 months [22]. It would be of great importance monitoring hospitalized patient both to improve haemodynamics and to reduce pulmonary or peripheral congestion. It would also be important to improve haemodynamics without determining arrhythmias, hypotension or renal dysfunction.
There are many challenges to successfully managing haemodynamic complications of AHFS.
Clinical evaluation remains the mainstay of patient management; however, it is subjective and based on the doctor’s skills, especially with regard to the evaluation of congestion signs. This aspect is of outstanding importance, in particular for the early identification of the transition from compensated to decompensated status in the setting of a worsening HF. In fact, clinical congestion may be present late, as the “tip of the iceberg” of the haemodynamic derangements preceding symptoms thus the need for an early identification of congestion. Studies with continuous monitoring of right ventricular or pulmonary artery pressure have shown that an increase in intracardiac pressures precedes an episode of acute decompensation, independently of whether the patient has a low or a normal LVEF [23, 24].
Significant changes in body weight are also often used to identify fluid overload, and an increase in body weight has often been reported to precede HF hospitalizations [2]. However, as previously noted, fluid redistribution to the lungs may cause AHF in the absence of peripheral oedema [13], and body weight is less sensitive than monitoring of intracardiac pressures.
Measurement of blood pressure plays a central role in the assessment and treatment of the patients with AHF [2]. Most patients admitted for acute HF present with normal or high blood pressure. Systolic blood pressure has been consistently shown to be one of the most important, independent predictor of both in-hospital and post-discharge outcomes in patients with AHF [25].
Many mechanisms can be involved in AHF patients including left ventricular (LV) diastolic and/or systolic dysfunction and/or right ventricular (RV) dysfunction. Although echocardiography often helps to assess the mechanism of heart dysfunction, it cannot be considered as a monitoring tool. In some cases, pulmonary artery catheter (PAC) can help to assess and monitor cardiovascular status and to evaluate responses to treatments. The recent publication of guidelines for diagnosis and treatment of cardiac failure provides an opportunity to better position the use of PAC particularly in haemodynamically unstable patients who are not responding as expected to traditional treatments [1].
In patients admitted with AHF, BNP plasma levels increase during the unstable phase due to volume overload and decrease with a reduction in both filling pressures and body hydration status during diuretic and vasoactive therapy [26, 27]. Patients who respond quickly to intravenous diuretics and experience a decline in BNP values to <250 pg/mL (reaching euvolemia and clinical stability) may be safely discharged. Furthermore, lowering of BNP below target after clinical stability by additional treatment guided by body hydration status using bioelectric impedance vectorial analysis (BIVA) could be achieved. This pre-discharge value for BNP is predictive of a low rate of adverse events. [28].
-
Bioelectric Impedance Vectorial Analysis (BIVA): this test is a reliable method to assess fluid status and fluid distribution in heart failure patients. The analysis can be repeatedly conducted in a non-invasive way using the patient as a control of himself. Fluid removal by diuretics or ultrafiltration can be guided by repeated assessment of fluid status by BIVA. The combination of BIVA with BNP (wet and dry values) and NGAL, a biomarker of acute kidney injury, can help preventing the occurrence of iatrogenically induced renal dysfunction or cardio-renal syndrome type 1 [29].
-
Although loop diuretics are a mainstay of treatment of ADHF, diuretic use has been shown to cause decline in glomerular filtration rate, further activation of the deleterious neurohormones associated with HF, and reaccumulation of Na + [30]. Diuretic resistance occurs in many HF patients due to adaptive changes occurring within the kidney in response to Na + accumulation, thus decreasing the efficacy of diuretic therapy and necessitating changes in diuretic regimens or combination diuretic therapy. This renal dysfunction may result from further neurohormonal and haemodynamic abnormalities, which may be aggravated by high-dose loop diuretic. Furthermore, the balance of plasma refill rates is the most important determinant during diuretic treatment. Inducing an aggressive diuresis in hospitalized patients with congestion can result in decreased plasma volume and low renal perfusion pressure related to WRF during or after discharge [31]. The same concept applies when fluid removal is obtained by extracorporeal ultrafiltration. In this circumstance, not only BIVA and BNP can become helpful to guide ultrafiltration kinetics, but special information can be gathered by a direct blood volume measurement in the extracorporeal circuit. This measure may help to understand the optimal velocity of fluid removal i.e. the speed at which blood volume remains unchanged.
All intervention strategies for AHFS now focus on early identification of clinical and instrumental indicators, which allow in-hospital treatment and the pre-discharge stratification of patients at high risk of rehospitalization. It is also useful to understand the importance of renal perfusion pressure, coronary perfusion pressure and the importance of transitional care. Recent data demonstrate deterioration in signs and symptoms, neurohormonal profile and renal function during the first few weeks after discharge. Novel diagnostic systems for continuous monitoring of intracardiac pressures and of intrathoracic impedance may allow early detection of congestion, thereby offering investigational opportunities to clarify the pathophysiology of AHF [32, 33]. In addition, there have been no substantial improvements in the treatment of patient with AHFS and impaired renal function during the past three decades. Treatment options are limited after therapeutic agents for AHF have failed in improving outcomes and the ability of mechanical fluid removal approaches to preserve renal function or improve cardio-renal syndrome remains unclear. Because of this, agents that could have little or no effect on vascular resistance within the kidney are currently being investigated.
Conclusions
Episodes of decompensation leading to hospitalization are characterized by deterioration in patient’s haemodynamic status, with water and salt retention. This deterioration occurs in spite of standard therapy, including beta-blockers, ACE inhibitors or ARB, and often aldosterone-blocking agents. To make progress in this field, it is important to first understand the pathophysiological bases of the syndrome and to define the severity of the disease. Short-term outcomes can be predicted from admission and pre-discharge characteristics, such as, coexistent cardiac and renal dysfunction, worsening renal function during the treatment of heart failure and diuretic resistance. Monitoring for potential cardiac injury and renal function is important to prevent iatrogenic insults. Any degree of renal dysfunction has negative prognostic implications and complicates the treatment of AHFS limited by side effects and increased mortality.
We need to learn to risk-stratify our patients upstream using multiple parameters, such as, BNP changes, renal dysfunction and intolerance to neurohormonal antagonism. A practical definition of cardio-renal syndromes is also useful to prevent negative heart–kidney interactions, to early diagnose a dangerous organ crosstalk and to set the stage for targeted management strategies. The secret to these approaches is the multidisciplinary management of the complicate heart failure patient with renal dysfunction.
References
Dickstein K, Cohen-Solal A, Filippatos G et al. (2008) ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the Task Force for the diagnosis and treatment of acute and chronic heart failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM). Eur Heart J 29:2388–2442
Gheorghiade M, Pang PS (2009) Acute heart failure syndromes. J Am Coll Cardiol 53:557–573
Harjola VP, Follath F, Nieminen MS et al (2010) Characteristics, outcomes, and predictors of mortality at 3 months and 1 year in patients hospitalized for acute heart failure. Eur J Heart Fail 12:239–248
Zelis R, Nellis SH, Longhurst J et al (1975) Abnormalities in regional circulations accompanying congestive heart failure. Progr Cardiovasc Dis 18:181–202
Leithe ME, Margorien RD, Hermiller JB et al (1984) Relationship between central hemodynamics and regional blood flow in normal subjects and in patients with congestive heart failure. Circulation 69:57–64
Reilly AW, French RM, Lau FYK et al (1954) Blood volume in cardiac decompensation. W Calif Med 81:256–258
Mullens W, Abrahams Z, Francis GS et al (2009) Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J Am Coll Cardiol 53:589–596
Firth JD, Raine AE, Ledingham JG (1988) Raised venous pressure: a direct cause of renal sodium retention in oedema. Lancet 1:1033–1035
Damman K, Navis G, Smilde TD et al (2007) Decreased cardiac output, venous congestion and the association with renal impairment in patients with cardiac dysfunction. Eur J Heart Fail 9:872–878
Blake WD, Wegria R, Keating RP et al (1949) Effect of increased renal venous pressure on renal function. Am J Physiol 157:1–13
Schrier RW (2006) Role of diminished renal function in cardiovascular mortality: marker or pathogenetic factor? J Am Coll Cardiol 47:1–8
Maisel A, Mueller C, Adams K Jr et al (2008) State of the art: using natriuretic peptide levels in clinical practice. Eur J Heart Fail 10:824–839
Cotter G, Metra M, Milo-Cotter O et al (2008) Fluid overload in acute heart failure—redistribution and other mechanisms beyond fluid accumulation. Eur J Heart Fail 10:165–169
Colombo PC, Onat D, Sabbah HN (2008) Acute heart failure as ‘acute endothelitis’—interaction of fluid overload and endothelial dysfunction. Eur J Heart Fail 10:170–175
Heywood JT, Fonarow GC, Costanzo MR et al (2007) High prevalence of renal dysfunction and its impact on outcome in 118, 465 patients hospitalized with acute decompensated heart failure: a report from the ADHERE database. J Card Fail 13(6):422–430
Nohria A, Hasselblad V, Stebbins A et al (2008) Cardiorenal interactions: insights from the ESCAPE trial. J Am Coll Cardiol 51(13):1268–1274
Weinfeld MS, Chertow GM, Stevenson LW (1999) Aggravated renal dysfunction during intensive therapy for advanced chronic heart failure. Am Heart J 138:285–290
Forman DE, Butler J, Wang Y et al (2004) Incidence, predictors at admission, and impact of worsening renal function among patients hospitalized with heart failure. J Am Coll Cardiol 43(1):61–67
Damman K, Navis G, Voors AA et al (2007) Worsening renal function and prognosis in heart failure: systematic review and meta-analysis. J Card Fail 13(8):599–608
Mullens W, Abrahams Z, Skouri HN et al (2008) Elevated intra-abdominal pressure in acute decompensated heart failure: a potential contributor to worsening renal function? J Am Coll Cardiol 51(3):300–306
Thom T, Haase N, Rosamond W et al (2006) Heart disease and stroke statistics—2006 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 113(6):e85–e151
Adams KF Jr, Fonarow GC, Emerman CL et al (2005) Characteristics and outcomes of patients hospitalized for heart failure in the United States: rationale, design, and preliminary observations from the first 100, 000 cases in the Acute Decompensated Heart Failure National Registry (ADHERE). Am Heart J 149(2):209–216
Gheorghiade M, De Luca L, Fonarow GC et al (2005) Pathophysiologic targets in the early phase of acute heart failure syndromes. Am J Cardiol 96:11G–17G
Metra M, Dei Cas L, Bristow MR (2008) The pathophysiology of acute heart failure—it is a lot about fluid accumulation. Am Heart J 155:1–5
Harjola VP, Follath F, Nieminen MS et al (2010) Characteristics, outcomes, and predictors of mortality at 3 months and 1 year in patients hospitalized for acute heart failure. Eur J Heart Fail 12(3):239–248
Kazanegra R, Chen V, Garcia A et al (2001) A rapid test for B-type natriuretic peptide correlates with falling wedge pressures in patients treated for decompensated heart failure: a pilot study. J Card Fail 7:21–29
Paterna S, Di Pasquale P, Parrinello G et al (2005) Changes in brain natriuretic peptide levels and bioelectrical impedance measurements after treatment with high-dose furosemide and hypertonic saline solution versus high-dose furosemide alone in refractory congestive heart failure. J Am Coll Cardiol 45:1997–2003
Valle R, Aspromonte N, Giovinazzo P et al (2008) B-type natriuretic Peptide-guided treatment for predicting outcome in patients hospitalized in sub-intensive care unit with acute heart failure. J Card Fail 14:219–224
Valle R, Aspromonte N (2010) Use of brain natriuretic Peptide and bioimpedance to guide therapy in heart failure patients. Contrib Nephrol 164:209–216
Francis GS, Siegel RM, Goldsmith SR et al (1985) Acute vasoconstrictor response to intravenous furosemide in patients with chronic congestive heart failure. Activation of the neurohumoral axis. Ann Intern Med 103(1):1–6
Hasselblad V, Gattis Stough W, Shah MR et al (2007) Relation between dose of loop diuretics and outcomes in a heart failure population: results of the ESCAPE trial. Eur J Heart Fail 9:1064–1069
Yu CM, Wang L, Chau E et al (2005) Intrathoracic impedance monitoring inpatients with heart failure: correlation with fluid status and feasibility of early warning preceding hospitalization. Circulation 112:841–848
Adamson PB, Magalski A, Braunschweig F et al (2003) Ongoing right ventricular hemodynamics in heart failure. Clinical value of measurements derived from an implantable monitoring system. J Am Coll Cardiol 41:565–571
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Aspromonte, N., Cruz, D.N., Valle, R. et al. Management and monitoring of haemodynamic complications in acute heart failure. Heart Fail Rev 16, 575–581 (2011). https://doi.org/10.1007/s10741-011-9229-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-011-9229-3