
Abstract
Sobetirome, also known as GC-1 and QRX-431, is a member of a class of compounds known as selective thyromimetics (Scanlan et al., Curr Opin Drug Discov Dev 4:614–622). These compounds are synthetic structural analogs of thyroid hormone that have tissue-specific thyroid hormone actions. Many of the compounds in this class, including sobetirome, also are subtype-selective thyroid hormone receptor (TR) agonists. Sobetirome selectively binds to and activates TRβ over TRα and this receptor selectivity led to the hypothesis that sobetirome would lower cholesterol through activation of liver TRβ without stimulating cardiac function through TRα activation in the heart. The tissue selective thyromimetic properties of sobetirome have been demonstrated in numerous animal models, which led to its clinical development as a novel cholesterol-lowering agent. This review will describe the discovery and development journey of sobetirome as a case history.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
As seems to be the case with all hormones, an excess quantity of circulating thyroid hormone, commonly referred to as hyperthyroidism, results in both positive and negative outcomes [1]. On the negative side, increased heart rate, body temperature, osteoporosis, muscle fatigue, and undesirable mood and mental state changes all can accompany hyperthyroidism. However, dramatic reductions in circulating cholesterol and body weight also can arise from hyperthyroidism and such changes would be considered therapeutically beneficial for individuals with dyslipidemia, obesity, and metabolic syndrome. Can these beneficial hyperthyroid effects by achieved safely by simply dosing with the endogenous thyroid hormone thyroxine (T4)? The answer to this question is unequivocally “no” [2]. There is simply no therapeutic index with T4 whereby the desired effects on cholesterol and/or body weight reduction can be separated from the tachycardia and other undesired effects. With this limitation in mind, the next obvious question is, can a synthetic thyroid hormone agonist be discovered that stimulates cholesterol lowering and weight loss at doses where cardiac function and other undesired hyperthyroid side effects are absent? [3] The answer to this question is liberally “yes” and conservatively “maybe,” and the case history of sobetirome, a selective thyromimetic currently in clinical development, will be presented such that the reader may judge for him/herself.
Discovery of the TRβ-selective agonist GC-1
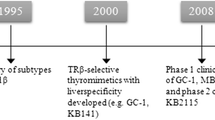
In about 1995 my laboratory, then in the Department of Pharmaceutical Chemistry at the University of California-San Francisco (UCSF), began a project aimed at creating new and interesting ligands for the thyroid hormone receptor (TR). There are actually two thyroid hormone receptors, TRα and TRβ, expressed as four different isoforms at different ratios in different tissues. Details of the molecular and structural biology of the TRs have been reviewed extensively elsewhere and will not be covered here [4]. In 1995, the different biological roles of TRα and TRβ in mammals were poorly understood. The different TR subtype- and isoform-specific knockout mice were still being developed and analyzed and subtype- and isoform specific ligands had not yet been described. The crystal structure of the TR ligand binding domain (LBD) in complex with the active form of thyroid hormone, 3,5,3′-triiodothyronine (T3, Fig. 1), and additional analogs had just been solved by my UCSF colleagues Robert Fletterick and John Baxter [5]. After studying this structure our primary goal was to design the first thyroid hormone antagonist. We eventually succeeded in this endeavor, but that is another story [6, 7].

Chemical structures of thyroid hormones and synthetic thyromimetics
There are many different tools, techniques, approaches, strategies, programs, algorithms, rules, etc. available today to the practicing medicinal chemist advertised as being useful, or even essential, for the drug discovery process. However, one of the most often overlooked, yet most important, aspects of drug discovery is synthetic accessibility of the target compounds. A class of compounds that is straightforward to synthesize is usually advantageous to a class of compounds obtained via a difficult synthesis; more compounds can be made, tested, and optimized with an efficient synthetic route. In our initial attempts to make thyroid hormone analogs, it became clear that there were structural elements to this class that were difficult to contend with from a synthetic standpoint. The biaryl ether linkage joining the inner and outer aryl rings of T3 is a particularly difficult bond-forming reaction for which there were no robust synthetic methods available at the time. In addition, the three iodine atoms of T3, which are essential for its high affinity interaction with TR, are labile in a number of different reaction conditions, thus limiting the scope of chemistry that could be used. With these considerations in mind, and guided by our insights from the TR–T3 crystal structure, as well as previously reported structure-activity relations on thyroid hormone analogs [8], we designed and synthesized a novel thyroid hormone analog that we named GC-1 (Fig. 1) [9]. GC-1 differs structurally from T3 in that the three iodine atoms are replaced by hydrocarbon residues, the biaryl ether linkage is replaced by a methylene linkage, and the amino acid side chain is replaced by an oxyacetic acid side chain. The synthesis of GC-1 was straightforward and efficient, and we considered GC-1 to be a good synthetic entry point for making additional analogs to test our ideas about TR ligand binding and receptor activation.
When we measured the TR binding of GC-1 we received our first surprise and indication that this was an unusual compound [9]. GC-1 was found to bind to TRs with exceptionally high affinity—on the same order as the affinity of TR for its endogenous hormone ligand T3. This was unanticipated because in other analogs where hydrocarbon for iodine replacement had been done, such as 3,5-dimethyl-3′-isopropylthyronine (DIMIT, Fig. 1), a 10–100-fold decrease in binding affinity occurred [10, 11]. A second surprise was the finding that GC-1 was TR subtype selective; GC-1 bound to TRβ1 with the same affinity as T3, but bound to TRα1 with approximately 10-fold lower affinity. This same TRβ selectivity was also observed in a variety of other in vitro TR-mediated transactivation assays.
Our most unequivocal evidence for TRβ selective activation by GC-1 came from Xenopus laevis tadpole metamorphosis experiments [12]. Metamorphosis of tadpoles into frogs is largely governed by thyroid hormone and X. laevis TRα and TRβ play distinct and tissue-specific roles in this developmental program. In the pre-metamorphic tadpole TRα is expressed at high levels in tissues targeted for proliferation such as limb buds, brain, and skin, and TRα activation by T3 plays a critical role in driving the proliferation of these tissues during metamorphosis. Near the peak of metamorphosis, TRβ expression is induced in tissues destined for death such as the tail and gill and TRβ activation by T3 drives the resorption of these larval tissues. The timing of different T3-activated events in this process is very precise; TRα-mediated tissue proliferation occurs before TRβ-mediated tissue resorption. These TR-mediated processes can be activated by addition of exogenous T3 or thyromimetic analogs to the bath water, because the compounds are readily absorbed by the tadpoles through their skin and gills. This makes for a convenient and very powerful in vivo bioassay for evaluating TR-subtype selectivity of thyromimetics. When GC-1 was tested in this assay, we observed what one would predict from a TRβ selective agonist; GC-1 potently stimulated tail and gill resorption, but showed little activity compared to T3 for inducing limb bud proliferation.
In an effort to understand the molecular basis of this TRβ selectivity, we performed structure–activity relationship (SAR) studies where hybrid analogs of GC-1 and DIMIT were prepared and their binding affinity to TRα and TRβ was compared [13]. This SAR analysis pointed to the oxyacetic acid side chain of GC-1 as the substructural element that was responsible for the majority of the TRβ-selective ligand binding. Structural studies of the TRβ–GC-1 complex confirmed this assessment [14]. Compared to the TRβ–T3 complex, the TRβ–GC-1 complex showed a structural rearrangement around the oxyacetic acid side chain that appears to result in additional hydrogen bonding interactions between ligand, receptor, and bound water molecules. A 10-fold difference in binding affinity to TRβ over TRα would result from a small, ~1.4 kcal/mol difference in binding energy, and this difference could arise from a single additional hydrogen bond in the TRβ versus TRα complex. It is important to note that such small differences in ligand-receptor binding energy are hard, if not impossible, to design or predict. In fact, the oxyacetic acid side chain, which is the source of the GC-1 TRβ selectivity, was used purely for ease of synthesis, on the basis that an inner ring fragment containing the oxyacetic acid moiety was commercially available for use as a starting material [15]. Such serendipity in medicinal chemistry is a common occurrence and leads to the advice that I have heard from other medicinal chemists, that it usually does not pay to think too much about compound design. This is always said in jest, but is unfortunately also very close to the truth, and speaks to our fundamentally crude understanding of how organic compounds behave in complex biological systems.
Proof of therapeutic concept in vivo
With a TRβ-selective agonist in hand, we began to wonder what to do with it, and established a variety of collaborations that ultimately showed the utility of GC-1 as a pharmacological tool for studying the selective activation of TRβ in vivo. Important thyroid hormone-driven processes where GC-1 proved useful as a tool include adaptive thermogenesis [16], brain development [17–19], hypothalamic–pituitary–thyroid (HPT) axis regulation [20], bone development [21], angiogenesis [22], and cell proliferation [23]. We also began to wonder if a TRβ-selective agonist would have therapeutic uses. The liver, where regulation of lipid homeostasis occurs, is generally regarded as a TRβ dominant organ whereas T3 effects on the heart, where the most dangerous acute effects of hyperthyroidism occur, are thought to result largely from TRα activation [4]. These tissue selective actions are exemplified by the clinical extremes in thyroid status. Hyperthyroid individuals generally have low-serum low-density lipoprotein (LDL) cholesterol and increased heart rate, while in cases of hypothyroidism, high-serum LDL cholesterol and bradycardia are usually displayed. The endogenous thyroid hormone T3 is not selective and does not distinguish between the liver TRβ and the heart TRα, but a TRβ selective agonist such as GC-1 should make this distinction. We reasoned that the expected pharmacological outcome of GC-1 treatment would be efficient LDL lowering through activation of TRβ in the liver with decreased effects on cardiac function compared to T3.
This hypothesis was first tested in a hypothyroid mouse model [24]. Normal mice have a high basal heart rate making it difficult to accurately measure drug-induced tachycardia, and this problem is remedied by making the mice hypothyroid which induces bradycardia by about 25%. Hypothyroid mice treated with a hormone replacement dose of T3 showed a 25% increase in heart rate, effectively a normalizing tachycardia, whereas hypothyroid mice treated with the same dose of GC-1 showed no increase in heart rate, and retained the 25% bradycardia from the induced hypothyroidism. Hypothyroidism in mice also increases serum cholesterol approximately twofold above normal levels and both T3 and GC-1 treatment decreased cholesterol to normal, euthyroid levels.
GC-1 was evaluated next in the hypercholesterolemic rat model [24, 25]. Rodents do not have the same circulating lipoprotein inventory as humans. Unlike primates, most of the circulating cholesterol in rats is high-density lipoprotein (HDL). By feeding rats a high cholesterol diet, however, the circulating LDL/HDL ratio can be artificially adjusted to a ratio closer to that of humans. In hypercholesterolemic rats, both T3 and GC-1 were found to dramatically decrease plasma cholesterol by about 75% in a dose-dependent fashion. The T3-treated rats also showed a dose-dependent increase in heart rate and decrease in circulating thyroid stimulating hormone (TSH) levels, and as expected, there was no therapeutic window separating cholesterol lowering from tachycardia and TSH suppression. GC-1, on the other hand, showed no significant tachycardia at any tested dose, and displayed about a 20-fold potency window separating cholesterol lowering from TSH suppression.
Drug-induced changes to cardiac and thyroid function were among the first mechanism-based deleterious side effects that concerned us, and we were happy to see that therapeutic windows, or safety margins, existed in these rodent models separating therapeutically beneficial lipid lowering from these side effects. We also were concerned about chronic thyroid hormone mechanism-based side effects such as osteoporosis and muscle wasting, and tested GC-1 in rat models of these diseases. The osteoporosis test involved treating rats chronically (60 days) with either T3 or GC-1, followed by measuring bone mass at several different skeletal sites [26]. T3-treated rats showed significantly reduced bone mineral density (BMD), whereas GC-1 treatment did not affect BMD at any of the sites examined. For the effects on skeletal muscle, we used a derivative of GC-1 called GC-24 (Fig. 1) that is structurally and pharmacologically very similar to GC-1 [27]. For this study the type 1 to type II fiber shift in rats was used as a marker of skeletal muscle weakness and fatigue that results from hyperthyroidism [28]. Here, T3 treatment significantly drove the type 1 to type II fiber shift, whereas GC-24 treatment was found to induce no significant changes to muscle fiber type composition. The combination of these studies suggested that there could be an acceptable safety margin separating the therapeutic effect of GC-1 from both the acute and chronic side effects expected from a mechanism related to hyperthyroidism.
In an effort to test our compound in a preclinical model closer to humans in terms of cardiac function and lipid homeostasis, GC-1 was evaluated side by side with T3 in adult cynomolgus monkeys [25]. Cardiac function and lipid inventory in these animals closely resemble those of humans. In addition to LDL cholesterol, lipoprotein (a) (Lp(a))—also considered an atherosclerosis risk factor—can be measured in serum from these animals. Monkeys treated with T3 showed 30–40% reductions in both cholesterol and Lp(a), but this was accompanied by a 30% increase in heart rate. In contrast, monkeys treated with an equi-molar dose of GC-1 showed a more therapeutically acceptable outcome; a 35–40% decrease in cholesterol and Lp(a) with no significant effect on heart rate. Interestingly, a small but significant reduction in body weight (~4%) was observed in both the T3- and GC-1-treated groups. This weight loss could result from a drug-induced stimulation in basal metabolic rate, a thyroid hormone-regulated process. A study in rats showed that GC-1 stimulated energy expenditure and weight loss like T3, although unlike T3 which promoted both fat and adipose mass reduction, the GC-1-induced weight loss was selective for fat mass and lean muscle mass was not significantly affected [29].
An obvious question is how does GC-1 compare to the statins, which are the current first-line treatment for high cholesterol. The first point to make is that the statins are proven therapeutic agents in humans with well-established efficacy and safety profiles; GC-1 and other thyromimetics [30, 31] are experimental therapeutics at present, and their safety and efficacy in humans are not fully understood. However, GC-1 is a substantially more potent cholesterol-lowering agent than statins; GC-1 lowers cholesterol more effectively than atorvastatin in both rat and monkey at about 1,000-fold lower doses [32]. In addition, GC-1 and the statins work by completely different mechanisms. The statins are inhibitors of HMG CoA-reductase, a key enzyme in the biosynthesis of cholesterol. Thus, the therapeutic effect of statins presumably results from limiting the body’s ability to make new cholesterol from diet-derived carbon building blocks. Thyroid hormone and GC-1, however, lower cholesterol by a completely different mechanism. These agents appear to exert their effects on cholesterol breakdown and clearance in the liver by up-regulating expression of hepatic LDL-receptors and stimulating reverse cholesterol transport (RCT), a process that involves the conversion of HDL-associated cholesterol into bile [33]. Because statins and thyromimetics such as GC-1 work at opposite ends of the cholesterol spectrum, it is conceivable that the two drugs would constitute an effective combination therapy capable of inducing more effective cholesterol lowering with low doses of both drugs.
Proof of therapeutic concept in humans
The bulk of the work described above takes us from 1996 to 2004, at which time we realized that the next step was to test GC-1 in humans. In the United States, however, this requires approval from the Food & Drug Administration (FDA) in the form of an approved Investigational New Drug application (IND), and the resources and experience needed for proper IND-enabling studies on preclinical drug candidates are generally hard to find in academic institutions. Instead, we were able to form a partnership in 2005 with QuatRx Pharmaceuticals, a company with substantial expertise and experience in developing drugs in endocrine, metabolic, and cardiovascular therapeutic areas. Upon obtaining the license to GC-1 in 2005, QuatRx changed the name of the compound to QRX-431 and began IND-enabling studies [34]. These studies were completed in 2006 and soon thereafter Phase 1 clinical studies were initiated [35]. These studies were completed in 2008 and the results were reported at the 2008 Endocrine Society meeting [36]. In addition, a summary of the results is available on the QuatRx website [37]. Upon completion of these studies, QuatRx obtained the United States Adopted Name (USAN) of sobetirome for QRX-431.
The results of the Phase 1 sobetirome trials are encouraging in that the drug was found to be generally well tolerated at all doses studied and proof of the therapeutic concept of cholesterol lowering was clearly demonstrated. In a multiple-dose sobetirome study of 24 subjects over 2 weeks of dosing, LDL cholesterol was reduced up to 41% at daily doses of 100 μg in subjects with non-enriched LDL levels.
Conclusions and perspectives
When this project began in my lab in 1996, I was mostly concerned with chemistry and structural biology issues related to in vitro ligand binding and receptor activation; I had no idea that a compound from my lab would be used in animal studies, let alone human clinical trials. The journey to this point has been nothing short of fascinating, even if at times stressful, and I look forward to finding out whether sobetirome and/or other thyromimetics in the class will find a use in medicine. Although my description of this journey from bench to clinic may make it seem to some that a great deal of work has been accomplished, major challenges and significant regulatory hurdles still lie ahead. An expert in clinical development would probably take the position that this journey is just beginning, as there is a great deal yet to be learned about how the action of these agents in humans. Will the tissue-specific hyperthyroidism mechanism of action for cholesterol lowering used by sobetirome and other compounds in the class have the clean safety profile expected of a chronically administered therapeutic agent? Will there be unanticipated problems—or possibly benefits—that arise from long-term exposure to these agents? Are there other metabolic diseases in addition to dyslipidemia, such as obesity, diabetes, and metabolic syndrome that could be treated safely and effectively by these agents? Are there thyroid-related disorders and illnesses that would be better treated by these thyromimetics than by T4 replacement therapy? These are all important questions confronting the clinical development of sobetirome and other thyromimetics that will only be answered through future clinical trials.
References
Greenspan FS (1997) The thyroid gland. In: Greenspan FS, Strewler GJ (eds) Basic and clinical endocrinology, 5th edn. Appleton & Lange, Stamford, pp 192–262
The Coronary Drug Project Group (1972) The coronary drug project. J Am Med Assoc 222:996–1008
Scanlan TS, Yoshihara HA, Nguyen N-H et al (2001) Selective thyromimetics: tissue-selective thyroid hormone analogs. Curr Opin Drug Discov Dev 4:614–622
Yen PM (2001) Physiological and molecular basis of thyroid hormone action. Physiol Rev 81:1097–1142
Wagner RL, Apriletti JW, McGrath ME et al (1995) A structural role for hormone in the thyroid hormone receptor. Nature 378:690–697. doi:10.1038/378690a0
Nguyen N-H, Apriletti JW, Cunha Lima ST et al (2002) Rational design and synthesis of a novel thyroid hormone antagonist that blocks co-activator recruitment. J Med Chem 45:3310–3320. doi:10.1021/jm0201013
Lim W, Nguyen N-H, Yang HY et al (2002) A thyroid hormone antagonist that inhibits thyroid hormone action in vivo. J Biol Chem 277:35664–35670. doi:10.1074/jbc.M205608200
Yokoyama N, Walker GN, Main AJ et al (1995) Synthesis and structure-activity relationships of oxamic acid and acetic acid derivatives related to l-thyronine. J Med Chem 38:695–707. doi:10.1021/jm00004a015
Chiellini G, Apriletti JW, Yoshihara HA et al (1998) A high-affinity subtype-selective agonist ligand for the thyroid hormone receptor. Chem Biol 5:299–306. doi:10.1016/S1074-5521(98)90168-5
Jorgensen EC (1978) Thyroid hormones and analogs. I. Synthesis, physical properties and theoretical calculations. In: Li CH (ed) Hormonal proteins and peptides. Academic Press, New York, pp 57–105
Jorgensen EC (1978) Thyroid hormones and analogs. II. Structure-activity relationships. In: Li CH (ed) Hormonal proteins and peptides. Academic Press, New York, pp 107–204
Furlow JD, Yang HY, Hsu M et al (2004) Induction of larval tissue resorption in Xenopus laevis tadpoles by the thyroid hormone receptor agonist GC-1. J Biol Chem 279:26555–26562. doi:10.1074/jbc.M402847200
Yoshihara HA, Apriletti JW, Baxter JD et al (2003) Structural determinants of selective thyromimetics. J Med Chem 46:3152–3161. doi:10.1021/jm0301181
Wagner RL, Huber BR, Shiau AK et al (2001) Hormone selectivity in thyroid hormone receptors. Mol Endocrinol 15:398–410. doi:10.1210/me.15.3.398
Chiellini G, Nguyen N-H, Yoshihara HAI et al (2000) Improved synthesis of the iodine-free thyromimetic GC-1. Bioorg Med Chem Lett 10:2607–2611. doi:10.1016/S0960-894X(00)00531-X
Ribeiro MO, Carvalho SD, Schultz JJ et al (2001) Thyroid hormone-sympathetic interaction and adaptive thermogenesis are thyroid hormone receptor isoform-specific. J Clin Investig 108:97–105
Morte B, Manzano J, Scanlan TS et al (2002) Deletion of the thyroid hormone receptor α1 prevents the structural alterations of the cerebellum induced by hypothyroidism. Proc Natl Acad Sci USA 99:3985–3989. doi:10.1073/pnas.062413299
Manzano J, Morte B, Scanlan TS et al (2003) Differential effects of triiodothyronine and the thyroid hormone receptor β-specific agonist GC-1 on thyroid hormone target genes in the brain. Endocrinology 144:5480–5487. doi:10.1210/en.2003-0633
Morte B, Manzano J, Scanlan TS et al (2004) Aberrant maturation of astrocytes in thyroid hormone receptor α1 knockout mice reveals an interplay between thyroid hormone receptor isoforms. Endocrinology 145:1386–1391. doi:10.1210/en.2003-1123
Dupre SM, Guissouma J, Flamant F et al (2004) Both thyroid hormone receptor (TR)β1 and TRβ2 isoforms contribute to the regulation of hypothalamic thyrotropin-releasing hormone. Endocrinology 145:2337–2345. doi:10.1210/en.2003-1209
Freitas FRS, Capelo LP, O’Shea PJ et al (2005) The thyroid hormone receptor β-specific agonist GC-1 selectively affects bone development in hypothyroid rats. J Bone Miner Res 20:294–304. doi:10.1359/JBMR.041116
Mousa SA, O’Connor LJ, Bergh JJ et al (2005) The proangiogenic action of thyroid hormone analogue GC-1 is initiated at an integrin. J Cardiovasc Pharmacol 46:356–360. doi:10.1097/01.fjc.0000175438.94906.a0
Columbano A, Pibiri M, Deidda M et al (2006) The thyroid hormone receptor-β agonist GC-1 induces cell proliferation in rat liver and pancreas. Endocrinology 147:3211–3218. doi:10.1210/en.2005-1561
Trost SU, Swanson E, Gloss B et al (2000) The thyroid hormone receptor-β-selective agonist GC-1 differentially affects plasma lipids and cardiac activity. Endocrinology 141:3057–3064. doi:10.1210/en.141.9.3057
Grover GJ, Egan DM, Sleph PG et al (2004) Effects of the thyroid hormone receptor agonist GC-1 on metabolic rate and cholesterol in rats and primates: selective actions relative to 3,5,3′-triiodo-l-thyronine. Endocrinology 145:1656–1661. doi:10.1210/en.2003-0973
Freitas FRS, Moriscot AS, Jorgetti V et al (2003) Spared bone mass in rats treated with thyroid hormone receptor TRβ-selective compound GC-1. Am J Physiol Endocrinol Metab 285:E1135–E1141
Borngraeber S, Budny M-J, Chiellini G et al (2003) Ligand selectivity by seeking hydrophobicity in thyroid hormone receptor. Proc Natl Acad Sci USA 100:15358–15363. doi:10.1073/pnas.2136689100
Miyabara EH, Aoki MS, Soares AG et al (2005) Thyroid hormone receptor-β-selective agonist GC-24 spares skeletal muscle type I to II fiber shift. Cell Tissue Res 321:233–241. doi:10.1007/s00441-005-1119-3
Villicev CM, Freitas FRS, Aoki MS et al (2007) Thyroid hormone receptor β-specific agonist GC-1 increases energy expenditure and prevents fat-mass accumulation in rats. J Endocrinol 193:21–29. doi:10.1677/joe.1.07066
Berkenstam A, Kristensen J, Mellstrom K et al (2008) The thyroid hormone mimetic compound KB2115 lowers plasma LDL cholesterol and stimulates bile acid synthesis without cardiac effects in humans. Proc Natl Acad Sci USA 105:663–667. doi:10.1073/pnas.0705286104
Erion MD, Cable EE, Ito BR et al (2007) Targeting thyroid hormone receptor-β agonists to the liver reduces cholesterol and triglycerides and improves the therapeutic index. Proc Natl Acad Sci USA 104:15490–15495. doi:10.1073/pnas.0702759104
Baxter JD, Webb P, Grover G et al (2004) Selective activation of thyroid hormone signaling pathways by GC-1: a new approach to controlling cholesterol and body weight. Trends Endocrinol Metab 15:154–157. doi:10.1016/j.tem.2004.03.008
Johansson L, Rudling M, Scanlan TS et al (2005) Selective thyroid receptor modulation by GC-1 reduces serum lipids and stimulates steps of reverse cholesterol transport in euthyroid mice. Proc Natl Acad Sci USA 102:10297–10302. doi:10.1073/pnas.0504379102
QuatRx Pharmaceuticals (2005) QuatRx Pharmaceuticals licenses novel lipid-lowering drug. In: QuatRx Pharmaceuticals website. http://www.quatrx.com/news/03_15_05.htm. Accessed 15 Mar 2005
QuatRx Pharmaceuticals (2006) QuatRx Pharmaceuticals initiates phase I trial for treatment to reduce both LDL cholesterol and lipoprotein (a). In: QuatRx Pharmaceuticals website. http://www.quatrx.com/news/09_28_06.htm. Accessed 28 Sept 2006
Lin VH, Klepp HM, Hanley RM (2008) Sobetirome is a TRβ- and liver-selective thyromimetic that can effect substantial LDL-C lowering without significant changes in heart rate or the thyroid axis in euthyroid men. In: The Endocrine Society annual meeting ENDO 08 (http://www.endo-society.org/endo/), San Francisco. http://www.abstracts2view.com/endo/view.php?nu=ENDO08L_OR36-3. Accessed 17 June 2008
QuatRx Pharmaceuticals (2008) Phase I studies show promise of Quatrx’s novel compound, sobetirome, for lowering LDL cholesterol levels. In: QuatRx Pharmaceuticals website. http://www.quatrx.com/news/01_29_08.htm. Accessed 29 Jan 2008
Acknowledgments
I wish to thank John Baxter and Robert Fletterick for engaging me in and teaching me about the wonderful field of thyroid hormone action. I wish to further thank my former students and postdocs, particularly Hikari Yoshihara and Grazia Chiellini, for bringing GC-1 to life. A hearty “thank you” also goes to all of my collaborators around the world, whose work described here illuminated the utility of GC-1 as a research tool and potential therapeutic agent. I am very grateful to the team at QuatRx Pharmaceuticals who brought sobetirome to life, and who educated and included me in a fascinating journey of drug development. Finally, I am indebted to the National Institute of Diabetes & Digestive & Kidney Diseases of the National Institutes of Health for generous and continuous financial support of this research program.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Scanlan, T.S. Sobetirome: a case history of bench-to-clinic drug discovery and development. Heart Fail Rev 15, 177–182 (2010). https://doi.org/10.1007/s10741-008-9122-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-008-9122-x