
Abstract
The formation of advanced glycation end-products(AGEs) is an important cause of metabolic memory in diabetic patients and a key factor in the formation of atherosclerosis(AS) plaques in patients with diabetes mellitus. Related studies showed that AGEs could disrupt hemodynamic steady-state and destroy vascular wall integrity through the endothelial barrier damage, foam cell(FC) formation, apoptosis, calcium deposition and other aspects. At the same time, AGEs could initiate oxidative stress and inflammatory response cascade via receptor-depended and non-receptor-dependent pathways, promoting plaques to develop from a steady state to a vulnerable state and eventually tend to rupture and thrombosis. Numerous studies have confirmed that these pathological processes mentioned above could lead to acute coronary heart disease(CHD) and other acute cardiovascular and cerebrovascular events. However, the specific role of AGEs in the progression and regression of AS plaques has not yet been fully elucidated. In this paper, the formation, source, metabolism, physical and chemical properties of AGEs and their role in the migration of FCs and plaque calcification are briefly described, we hope to provide new ideas for the researchers that struggling in this field.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
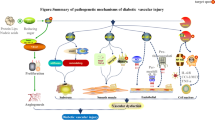
Advanced glycation end-products(AGEs), the most important metabolite of diabetic glycotoxin, are involved in the formation and evolution of cardiovascular complications of diabetes. More and more studies indicated that AGEs had multiple pathological effects in a receptor-depended or non-receptor-depended manner, such as disrupting hemodynamic steady-state, destroying vascular wall integrity, etc. These effects eventually promoted the vicious progression to vulnerable plaques,thrombosis formation and the occurrence of acute coronary syndrome [1]. However, the specific role of AGEs in atherosclerosis(AS) plaques remains poorly understood. Considering that AGEs have important clinical significance, identifying their mechanisms is essential for designing treatment strategies of AS (Fig. 1).

The diagram of role of AGEs in progression and regression of AS plaques. CML inhibited the outward migration of FCs via receptor CD36 pathway, further increased the apoptosis of FCs in plaque. Produced ABs provided a suitable microenvironment for calcification in plaque. CML promoted calcification formation through receptor RAGE pathway. The progression of AS is facilitated by both pathways above. Targeted blocking the correlation signals on these two signal cascades to design optimization of intervention strategies, which may promote AS plaque regression. It may bring new breakthroughs in the prevention and treatment of AS cardiovascular and cerebrovascular diseases
Characteristics of AS
AS is the main cause of coronary heart disease(CHD), cerebral infarction, peripheral vascular disease. Lipid metabolic disorders are the basis of AS lesions. Involved arterial lesions began from the intima, generally first presented the lipid and complex carbohydrate accumulation, bleeding and thrombosis formation. Then fibrous tissue hypertrophied and calcium deposited, accompanied by the gradual transformation of and calcification in the middle of the arteries, leading to arterial wall thickening and hardening, vascular stenosis. The lesions often involved large and medium muscle arteries. The tissue or organ supplied by these arteries would be ischemic or necrotic once the lesions progressed enough to block the arterial cavity. It was named as AS, which was due to the lipid accumulated in the arterial intima presented as a yellow porridge. AS is caused by a combination of multiple factors and the pathogenesis of which is complex, which has not been fully elucidated yet. Previous studies showed that the AS calcification in large and middle arteries, especially in the coronary artery, may lead to acute myocardial infarction, congestive heart failure, arterial dissection, limb ischemic gangrene and percutaneous coronary intervention failure. These clinical events are all due to mechanical changes in the blood vessel wall which are caused by the abnormal calcium deposition in plaques [2]. At present, some studies have found that plaque calcification is one of the advanced manifestations of AS, while the migration of FCs is closely related to the regression of AS plaque. Therefore, relationship between calcification of plaque, the outward migration of FCs and progression and regression of plaque were explored in this part.
Plaque progression and intimal calcification
Vascular calcification concept and classification
We have gradually clarified that vascular calcification(VC) is similar to the process of bone development and is a cell-mediated, highly regulative and reversible pro-active process. It is a common adverse event in the progression of AS disease and is an important predictor for death from cardiovascular and cerebrovascular diseases [3]. According to pathological anatomy, VC is divided into: (1) venous calcification; (2) arterial calcification, including endometrial calcification, calcification of the media, valve calcification and calcification defenses and many other types. According to calcification diameter, VC can be divided into micro-calcification, spotted calcification, fragmented calcification and diffuse calcification. The first two can be collectively referred to as micro-calcification, the latter two can be collectively referred to as macro-calcification [4, 5].
Pathological mechanism of VC
VC was a pathological process that bone-specific hydroxyapatite crystals actively deposited in the vascular wall, which is caused by high calcium and phosphorus environment and local or systemic mineralization inducer up-regulation, suppressor down-regulation. During this process, vessel wall mineralization defense mechanisms was exhausted. Vascular smooth muscle cells(VSMCs) and other mesenchymal cells lost the original phenotype and obtained osteoblastic phenotype and released mineralized lipid vesicles (there are at least two forms of matrix vesicles and apoptotic bodies in the arterial wall). The vesicles provided a suitable nucleation microenvironment for calcification crystallization, while the vascular wall elastin provided a stent structure for the hydroxyapatite deposition. Matrix vesicles released from macrophages and VSMCs during apoptosis played a pivotal role in formation of micro-calcification, while osteogenic differentiation of VSMCs contributed to progression of advanced calcification. Thus, after the balance between osteoblast-like cell-mediated calcium deposition and osteoclast-like cell-mediated calcium uptake was broken, vascular wall intima, medial membrane or aortic valve may form ectopic calcification [6].
Role of VC in AS
Recent studies have shown that at the early stages of AS such as grease period, it appears that bone-associated proteins had been expressed. And histological evidence of calcification could be detected once the lipid core is formed [7]. AS plaque calcification is the endometrial calcification. The macro-calcification will reduce the mechanical stress of adjacent plaques, leading to the formation of stable plaques. Therefore, it is related to stable angina. While the micro-calcification will increase the stress at the edge of the plaque, leading to the formation of unstable plaque. Unstable plaque refers to those plaques that are more prone to rupture under the mechanical stress caused by vasoconstriction and “wheel effect” caused by micro-calcification in the plaque. Previous studies confirmed that micro-calcification is related to unstable angina or myocardial infarction [8].
It is proved that there is close relationship between coronary calcification exists and the presence of AS lesions in the coronary arterial system [9], and AS plaque calcification is a risk sign for cardiovascular events. Therefore, the study of AS calcification is of great significance on the study of plaque stability and prevention and treatment of AS.
Plaque regression and FCs migration
FCs migration concept
FCs are those monocytes or tissue cells that have phagocytosed lipids and contain many lipid droplets in their cytoplasm. They are characteristic pathological cells that appear in AS plaques and are mainly derived from macrophages and VSMCs. FCs formation is an early event in the formation of AS. In the state of dyslipidemia, there exists a positive feedback mechanism of phagocytosis of ox-LDL by CD36, the scavenger receptor of macrophage. At the same time, cholesterol reverse transport mechanism is impaired in diabetic condition. Both of the mechanisms mentioned above result in the accumulation of large amounts of lipid in macrophages and eventually forming FCs. With the in-depth understanding of the pathogenesis of AS, researchers gradually cleared that the inward migration of macrophages and the outward migration of FCs in arterial wall lesions are always a dynamic process [10, 11].
Role of FCs migration in plaque regression
Assuming that AS lesions are placed in a suitable internal and external environment, it is possible to reverse the course of the disease, change the plaque structure and even make it completely subsided. This is confirmed by the available basic research and clinical evidence [12]. In the early stages of AS plaque regression, the number of FCs significantly decreased or even disappeared. FCs were replaced by many non-foamed mononuclear macrophages around the necrotic nucleus, and then the necrotic nucleus volume was significantly reduced. Williams KJ had transplanted experimental AS plaque to wild-type mice, the results showed that the rapidly disappearing FCs mainly flowed to the lymph nodes and still retain the foamy shape. It is considered that outward migration dysfunction of FCs is one of the key factors in the formation of AS plaque, but this dysfunction is reversible [13, 14]. In addition to the outward migration of FCs within the plaque, the improvement of the internal and external environment of the plaque, the reduction of the lipid core in the plaque, and the elimination of ox-LDL to inhibit oxidative stress are all beneficial to the regression of AS plaque [10].
Although great advances have been made in experimental studies of AS plaque regression, the actual clinical interventions are limited in terms of effectiveness, safety and viability. Therefore, the study of the role of FCs migration in the regression of AS plaques is of great significance.
AGEs
Source of AGEs
AGEs are a class of heterogeneous biological macromolecules. They are produced by glucose, protein, lipid and even nucleic acid through a series of non-enzymatic glycosylation reactions (known as Maillard reaction) [15, 16]. AGEs pool in human body is formed by two major sources, including endogenous and exogenous. Exogenous AGEs are from the daily diet, especially after high temperature treatment of food (such as fried, baked, roasted, etc.). Endogenous AGEs are mainly derived from Maillard reaction in body, people with hyperglycemia in diabetes are particularly likely to form endogenous AGEs. Previous article showed that after intaking of high AGEs diet, diabetes animal model is more likely to form AS, diabetic nephropathy and other vascular complications. The study by Ikeda T [17] confirmed that serum pentosidine levels were associated with branch atheromatous disease.
Metabolism of the AGEs
AGEs are irreversible after formation, and these deposited proteins are not easily decomposed by proteases. AGEs have the ability to further produce reactive oxygen and react with cell surface specific structures. It is by kidney that AGEs are removed through binding with the macrophage-specific receptor, then being degraded by macrophage into a small soluble peptide and released [18]. Therefore, the effective clearance of AGEs relies on normal renal function, reducing renal function will result in the accumulation of AGEs in the body. AGEs content is very low under physiological conditions, but the body’s aging or diabetes hyperglycemia will accelerate the process of glycosylation, followed by human body continuing to spontaneously generate AGEs. Although most of the peptides can undergo post-transcriptional glycosylation/oxidative modification, but the formation of AGEs mainly depends on three factors: the degree of hyperglycemia, the rate of regeneration of the glycosylated/oxidized substrate, and the oxidative properties of the tissue micro-environment. In patients with diabetes,the formation of AGEs speeded up under these three factors, increasing the risk to cardiovascular system [1].
Classification and characteristics of AGEs
So far, more than 20 kinds of AGEs have been identified, including carboxymethyl-lysine (CML), carboxyethyl-lysine (CEL), pentosidine and so on. They are involved in atherosclerosis, normal aging process, arthritis, cancer and progression of age-related neurodegenerative diseases like Alzheimer’s disease [19]. CML is the product of fructose lysine oxidative cleavage, which is the most widely used marker for food AGEs analysis and animal related research [20]. Studies have shown that CML is the main structure that AGEs antibody recognize.
Most of AGEs has unique physical and chemical properties: ①brown; ②have a unique fluorescent properties; ③have cross-linking properties; ④are not easy to be degraded by enzymes. But not all of AGEs have such attributes, such as CML is no color, no fluorescence and no cross-linking properties [21].
Relationship between AGEs and plaque calcification
AGEs exert their biological functions through receptor and non-receptor pathways, the former is the main pathogenesis. Wang and his group [2] successfully induced AS calcification model of apoE−/− mice with STZ-CML-HFD triple treatment, the experimental results showed that: AGEs mediated AS plaque calcification by affecting osteoblastic differentiation of VSMCs.
Previous studies [22,23,24] have shown that AGEs activate a series of signal transduction pathways to participate in calcification in AS plaques. The relationship between AGEs and their receptors and plaque calcification was described through 4 aspects in this section.
Role of AGEs and their receptors in inflammatory and oxidative stress-mediated plaque calcification
Inflammation plays a major role in plaque calcification. It is associated with endothelial cells dysfunction, VSMCs migration, increased metalloprotease activity, extracellular matrix degradation, oxidative stress, elastin and collagen degradation. Various studies have shown that, there was a significant correlation between arterial stiffness and inflammatory markers such as leukocyte count, adhesion molecules, fibrinogen, C-reactive protein, etc. in patients with metabolic syndrome, diabetes, coronary heart disease and other diseases. Studies have shown that induction of vascular inflammation can promote plaque rupture, while inhibition of pro-inflammatory response can promote the transition from micro-calcification to macro-calcification, which can stabilize the plaque [25]. There is strong evidence that inflammation plays an important, at least partially reversible role in the development of AS [26].
Plaque calcification is associated with inflammation and plaque instability in a dual fashion. The micro-calcified area is characterized by abundant inflammatory cells, high expression of RAGE, CML and S100 calcium binding protein A12, less expression of Galectin-3 by VSMCs. In contrast, most of the VSMCs in the macro-calcified region highly expressed Galectin-3, α-SMA and osteoblast differentiation marker alkaline phosphatase, whereas inflammatory cells expressed less RAGE and inflammatory markers. It has been confirmed [25] that in addition to the opposite effect on inflammation, Galectin-3 and RAGE regulated plaque calcification in different ways. They dominated the formation of macro-calcification and micro-calcification, which in turn regulate the development of plaques towards stable or unstable phenotypes, respectively.
Oxidative stress is associated with various organ dysfunctions, especially with cardiovascular disease. It has been demonstrated [27] that plaque calcification proceeds in a time-dependent manner, parallel to osteogenic differentiation of VSMCs. Accumulation of oxidative stress is also identified in calcified areas. AGE/RAGE signaling has been implicated in oxidative stress associated with diabetes-mediated vascular calcification [28]. Time-course studies showed that oxidative stress was associated with plaque calcification.
Previous experimental studies have also confirmed the correlation between oxidative stress and the occurrence of plaque calcification [29,30,31,32,33], Liberman M found ROS production and increased NAD (P) H oxidase expression around plaque calcification. These data suggested that ROS, especially hydrogen peroxide, enhanced the progression of plaque calcification [31].
Effect of AGEs and their receptors on plaque calcification in osteogenesis and osteoclast balance
In all vertebrates, bone tissue is continuously regenerated through osteoclasts reabsorbing bone tissue and osteoblasts reconstituting it. This process is regulated by several chemical signals and involves multiple types of cells [34]. AGEs have been shown to induce osteogenic trans-differentiation of VSMCs. The vitro experiments by Molinuevo MS [35] showed that AGEs and SR (anti-osteoporosis agents), alone or in combination, stimulate L-type calcium channels, resulting in reactive oxygen increasing and activation of ERK and NFκB, ultimately contributing to osteogenic metastasis of VSMCs.
AGEs did not increase the number of newly formed osteoclasts, but AGEs-modified β2-microglobulin and BSA both increased the number of absorbed pits produced by osteoclasts, which was observed in vivo and vitro experiments by Miyata T [36]. This suggested that AGE enhanced osteoclast-induced bone resorption. Moreover, calcitonin and ipriflavone, both of which are inhibitors of bone resorption, effectively inhibited the enhanced bone resorption by AGEs. These results suggested that AGEs are beneficial to osteoclastic bone resorption via activating osteoclasts or altering the microenvironment. Studies by Menini [25] and Li G [37] found that RAGE and galectin-3 played different roles in VSMCs trans-differentiation into osteoblasts, respectively.
In summary, AGEs and their receptors promote plaque calcification and pathological bone remodeling by breaking the osteogenesis and osteoclastic balance.
Effect of AGEs and their receptors on plaque calcification by elastin and collagen
Previous studies have shown that with the remodeling of the arterial wall, the significant change in the composition of the extracellular matrix and the vascular cell phenotype, especially the production of elastin fragments, increased protease activity and activated transforming growth factor-β signaling. In addition, the deposition of collagen and proteoglycan provided the suitable conditions for plaque calcification.
Takino J [38] treated mice with RAGE inhibitor FPS-ZM1, exercise training reduced aortic stiffness, decreased collagen levels, increased elastin levels, and reduced pulse wave velocity (PWV). And it was able to prevent related endothelial dysfunction, such as plaque calcification. Above evidence indicated that AGEs and their receptors may alter the structure of extracellular matrix proteins such as collagen and elastin by activating intracellular signal transduction [39], leading to the change of cell phenotype and thus accelerate plaque calcification.
AGEs and their receptors affect plaque calcification by fetuin
Fetal globulin-A is a negative acute phase inhibitor that inhibits Ca-P precipitation and VC and plays a biomarker role in calcification prediction [40]. Marcus Baumann [39] found a positive correlation between CML and fetuin A, suggesting that CML may reflect the state of vascular inflammation. This result is further enhanced by the observation of CML in AS human subcutaneous carotid arteries. The study by Janda K [41] showed that serum levels of AGEs were positively correlated with BMI, serum C-reactive protein (hsCRP), fetuin A, plasminogen activator inhibitor-1 (PAI-1) and 2,2-diphenyl-1-picrylhydrazyl (DPPH) scavenging, but only fetuin A was an independent predictor of multiple regression. These results suggested that AGEs and their receptors may affect plaque calcification by fetuin.
There is growing evidence that apoptosis plays an important role in VC. Proudfoot D confirmed that inhibition of apoptosis by caspase inhibitor ZVAD.fmk could reduce about 40% of calcification in nodules, while the promotion of apoptosis could increase the chance of calcification by ten times. Studies by Duan XH demonstrated that endoplasmic reticulum stress (ERS) -mediated apoptosis was activated during VC [42]. More in-depth studies had shown that FC apoptotic bodies(ABs) and VSMC necrotic debris could be locally enriched in concentrated calcium and phosphate ions, which could provide a suitable nucleation microenvironment for the formation and rearrangement of hydroxyapatite calcium crystals. Consistent with these existing studies, Wang [43] showed that CML may first induce intracellular glucose and lipid metabolism disorders, then initiated the apoptotic cascade of macrophage-derived FCs at the key multi-signal site of endoplasmic reticulum. Annexin V-FITC/PI double staining fluorescence qualitative and quantitative analysis of flow cytometry showed that CML could induce RAW264.7 macrophage apoptosis through a dose-dependent approach in high fat environment.
So, how does CML start diabetes AS? Wang showed that CML can significantly promote RAGE expression in RAW264.7 macrophages and A7R5 aortic SMCs. NADPH oxidase is the main source of active oxygen in VSMCs, and active oxygen is an important physiological and pathological regulatory medium for intracellular signal cascade. Studies showed that AGEs could be activated by active oxygen-mediated activation of p38MAPK signaling to induce calcification cascade [44]. Previous experimental results suggested that the CML / RAGE axis may first initiate apoptosis of macrophages in AS lesions, then induced the transmission of CML/RAGE→ROS → p38MAPK → cbfα1 → ALP calcification cascade in the coexistence environment of high-fat and apoptosis [2].
AGEs and FCs migration
As mentioned above, the accumulation of CML promoted vascular cell apoptosis. Apoptotic cells (mainly FCs) increased VC through various forms, promoted the formation of AS plaques, and thus promoted the evolution of the disease. Previous article [45] also showed that the early manifestation of AS plaque regression was a large number of FCs in plaque migrating outside the blood vessels. Therefore, we hypothesized that the migration dysfunction of FCs leaded to a large accumulation of FCs in the intima, which further promoted the progression of AS through apoptosis.
Cell migration involved two processes, namely cell elongation and localized adhesion plaque formation of cells. These two processes were dynamically controlled by cytoskeletal actin and local focal adhesion kinase [46]. Our previous study [47] showed that CML promoted FCs actin polymerization, which affected RAW264.7-derived FCs migration.
As a member of the scavenger receptor B family, CD36 is a transmembrane glycoprotein receptor, which is expressed in a variety of cells including monocytes and macrophages. CD36 mediates the uptake of ox-LDL and the formation of FCs by identifying cut fatty acids or oxidized lecithin. Recent studies also showed that CD36 of macrophages are a direct signaling molecule that can initiate related signal cascades, such as regulate the skeletal dynamics of macrophages as a regulator of cell migration. AGEs are a major risk factor in the development of AS in diabetes mellitus, while scavenger receptor CD36 is a receptor for AGE. Therefore, AGEs may be involved in the pathogenesis of diabetic AS FCs migration by interacting with CD36. Previous in vivo and in vitro migration experiments suggested that ox-LDL inhibited macrophage migration in CD36-dependent manner. The underlying mechanism was that the binding of ox-LDL to the cell surface CD36 induced cell actin polymerization, hence affecting cell mobilization and other intracellular signaling pathways. These pathways include the accumulation of FCs, the production of intracellular reactive oxygen species, Src family kinase and FAK activation [48]. Existing study showed that CML promoted the expression of CD36 in FCs and enhanced the binding capacity of CML and CD36 in RAW264.7-derived FCs. These results support CML inhibition of FCs migration through CD36.
Our previous studies have shown that CML could increase cellular cholesterol [43]. The cholesterol efflux out of the FCs played an important role in the treatment of AS, in which apoA-1 is one of the important apolipoprotein involved in the process. The study by Hong Sun [49] confirmed that CML increased cholesterol synthesis and cholesterol uptake, and reduced cholesterol efflux, which ultimately caused lipid accumulation in HK-2 cells. Park demonstrated that elevated levels of local free cholesterol in cells could lead to localized actin polymerization [48], and the NADPH-derived ROS production was closely related to the formation of bioactive lipids. Previous experimental results showed that CML/CD36 mediated FCs to produce free cholesterol and reactive oxygen, CD36 signal promoted cell spread and inhibited cell migration through the production of ROS. Furthermore, it is at the upstream of ROS production that CML/CD36 promoting free cholesterol production.
Studies proved that activation of FAK signaling contributed to actin polymerization and could activate Arp2/3 complex at the same time, affecting cell migration [50]. To investigate the specific pathways that CML/CD36 affects the migration of RAW264.7-derived FCs by producing free cholesterol and ROS, we used western blot to detect the expression of related signaling pathways, respectively, CD36, gp91phox, p-FAK and Arp2/3. Western blot and semi-quantitative results indicated that CML/CD36-mediated migration of RAW264.7-derived FCs was strongly associated with ROS production, FAK phosphorylation, Arp2 / 3 complex activation and F-actin aggregation.
In summary, AGEs inhibited FCs in the plaques migrating to the lymph nodes, and accelerated the evolution of AS. The mechanism may be related to the polymerization of FCs actin, the increase of cell cholesterol and reactive oxygen, the activation of p-FAK, Arp2 / 3 complex and F-actin polymerization. Previous studies confirmed that anti-CD36 promoted macrophage-derived FCs reduction in aortic AS plaque, and the reduced partial FCs may migrate out to adjacent lymph nodes. When the outward migration of FCs was inhibited, AS lesions would be malignant evolution. Therefore, it is of great significance to study the abnormal mechanism of FCs outward migration from arterial wall to promote the regression of AS plaque.
Conclusion
In conclusion, AGEs promote the progress of AS by inhibiting FCs outward migration from AS plaques and inducing calcification cascades in hyperlipidemic environments. Targeted blocking the correlation signals on these two signal cascades, such as using anti-CD36 receptor antibody and anti-RAGE neutralizing antibody to design optimization of intervention strategies, which may promote AS plaque regression. It may bring new breakthroughs in the prevention and treatment of AS cardiovascular and cerebrovascular diseases.
Abbreviations
- AGEs:
-
advanced glycation end-products
- AS:
-
atherosclerosis
- FC:
-
foam cell
- CHD:
-
coronary heart disease
- VC:
-
vascular calcification
- VSMC:
-
vascular smooth muscle cell
- AB:
-
apoptotic body
- CML:
-
Nε-Carboxymethyllysine
References
Singh, V.P., Bali, A., Singh, N., Jaggi, A.S.: Advanced glycation end products and diabetic complications[J]. Korean J Physiol Pharmacol. 18(1), 1–14 (2014)
Wang, Z., Jiang, Y., Liu, N., Ren, L., Zhu, Y., An, Y., Chen, D.: Advanced glycation end-product Nε-carboxymethyl-lysine accelerates progression of atherosclerotic calcification in diabetes. Atherosclerosis. 221(2), 387–396 (2012)
Criqui, M.H., Denenberg, J.O., Ix, J.H., et al.: Calcium density of coronary artery plaque and risk of incident cardiovascular events[J]. JAMA. 3(3), 271–278 (2014)
Pugliese, G., Iacobini, C., Blasetti, F.C., et al.: The dark and bright side of atherosclerotic calcification [J]. Atherosclerosis. 238(2), 220–230 (2015)
Otsuka, F., Sakakura, K., Yahagi, K., Joner, M., Virmani, R.: Has our understanding of calcification in human coronary atherosclerosis progressed? Arterioscler. Thromb. Vasc. Biol. 34(4), 724–736 (2014)
Giacheli, C.M.: Inducers and inhibitors of biomineralization: lessons from pathological calcification [J]. Orthod. Craniofacial Res. 8(4), P229–231–P229–229 (2005)
Bobryshev, Y.V.: Transdifferentiation of smooth muscle cells into chondrocytes in atherosclerotic arteries in situ: implications for diffuse intimal calcification. J. Pathol. 205, 641–650 (2005)
Ruiz JL, Hutcheson JD, Aikawa E. Cardiovascular calcification: current controversies and novel concepts. 2015 24(4), 207–212
Goel, R., Garg, P., Achenbach, S., Gupta, A., Song, J.J., Wong, N.D., Shaw, L.J., Narula, J.: Coronary artery calcification and coronary atherosclerotic disease. Cardiol. Clin. 30(1), 19–47 (2012 Feb)
Kalanuria, A.A., Nyquist, P., Ling, G.: The prevention and regression of atherosclerotic plaques:emerging treatments. Vasc. Health Risk Manag. 8, 549–561 (2012)
Curtiss, L.K.: Reversing atherosclerosis? [J]. N. Engl. J. Med. 360, 1144–1146 (2009)
Feig, J.E., Vengrenyuk, Y., Reiser, V., et al.: Regression of atherosclerosis is characterized by broad changes in the plaque macrophage transcriptome. PLoS One. 7, e39790 (2012)
Williams, K.J., Feig, J.E., Fisher, E.A.: Rapid regression of atherosclerosis: insights from the clinical and experimental literature. Nat. Rev. Cardiol. 5, 91–102 (2008)
Williams, K.J., Feig, J.E., Fisher, E.A.: Cellular and molecular mechanisms for rapid regression of atherosclerosis: from bench top to potentially achievable clinical goal. Curr. Opin. Lipidol. 18(4), 443–450 (2007 Aug)
Vistoli, G., De Maddis, D., Cipak, A., et al.: Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): an overview of their mechanisms of formation. Free Radic. Res. 47(Suppl 1), 3–27 (2013 Aug)
Shen, C., Li, Q., Zhang, Y.C., Ma, G., Feng, Y., Zhu, Q., Dai, Q., Chen, Z., Yao, Y., Chen, L., Jiang, Y., Liu, N.: Advanced glycation end products increase EPC apoptosis and decrease nitric oxide release via MAPK pathways[J]. Biomed Pharmacother. 64(1), 35–43 (2010 Jan)
Ikeda, T., Maruyama, K., et al.: Higher serum pentosidine, an advanced glycation end product, in branch atheromatous disease among small vessels occlusion. J. Neurosurg. Sci. (2016 Feb 19)
Xu, H., Wang, Z., Wang, Y., et al.: Biodistribution and elimination study of fluorine-18 labeled Nε-carboxymethyl-lysine following intragastric and intravenous administration [J]. PLoS One. 8(3), 1–10 (2013)
Ajith, T.A., Vinodkumar, P.: Advanced glycation end products: association with the pathogenesis of diseases and the current therapeutic advances. Curr. Clin. Pharmacol. 11(2), 118–127 (2016)
Semba, R.D., Arab, L., Sun, K., Nicklett, E.J., Ferrucci, L.: Fat mass is inversely associated with serum carboxymethyl-lysine,an advanced glycation end product,in adults. J. Nutr. 141, 1726–1730 (2011)
Prasad, A., Bekker, P., Tsimikas, S.: Advanced glycation end products and diabetic cardiovascular disease [J]. Cardiol. Rev. 4, 177–183 (2012 Jul-Aug)
Wang, Y., Zhang, Z.Y., Chen, X.Q., Wang, X., Cao, H., Liu, S.W.: Advanced glycation end products promote human aortic smooth muscle cell calcification in vitro via activating NF-κB and down-regulating IGF1R expression. Acta Pharmacol. Sin. 34(4), 480–486 (2013 Apr)
Ren, X., Shao, H., Wei, Q., Sun, Z., Liu, N.: Advanced glycation end-products enhance calcification in vascular smooth muscle cells. J Int Med Res. 37(3), 847–854 (2009 May-Jun)
Brodeur, M.R., Bouvet, C.R., Bouchard, S., Moreau, S., Leblond, J., deBlois, D., Moreau, P.: Reduction of advanced-glycation end products levels and inhibition of RAGE signaling decreases rat vascular calcification induced by diabetes. PLoS One. 9(1), e85922 (2014 Jan 21)
Menini, S., Iacobini, C., Ricci, C., Blasetti Fantauzzi, C., Salvi, L., Pesce, C.M., Relucenti, M., Familiari, G., Taurino, M., Pugliese, G.: The galectin-3/RAGE dyad modulates vascular osteogenesis in atherosclerosis. Cardiovasc. Res. 100(3), 472–480 (2013)
Mozos, I., Malainer, C., Horbańczuk, J., Gug, C., Stoian, D., Luca, C.T., Atanasov, A.G.: Inflammatory markers for arterial stiffness in cardiovascular diseases. Front. Immunol. 8, 1058 (2017)
Yamada, S., Taniguchi, M., Tokumoto, M., Toyonaga, J., Fujisaki, K., Suehiro, T., Noguchi, H., Iida, M., Tsuruya, K., Kitazono, T.: The antioxidant tempol ameliorates arterial medial calcification in uremic rats: important role of oxidative stress in the pathogenesis of vascular calcification in chronic kidney disease. J. Bone Miner. Res. 27(2), 474–485 (2012 Feb)
Kay, A.M., Simpson, C.L., Stewart Jr., J.A.: The role of AGE/RAGE signaling in diabetes-mediated vascular calcification. J Diabetes Res. 2016, 6809703 (2016)
Sutra, T., Morena, M., Bargnoux, A.S., Caporiccio, B., Canaud, B., Cristol, J.P.: Superoxide production: a procalcifying cell signalling event in osteoblastic differentiation of VSMCs exposed to calcification media. Free Radic. Res. 42, 789–797 (2008)
Kennedy, J.A., Hua, X., Mishra, K., Murphy, G.A., Rosenkranz, A.C., Horowitz, J.D.: Inhibition of calcifying nodule formation in cultured porcine aortic valve cells by nitric oxide donors. Eur. J. Pharmacol. 602, 28–35 (2009)
Liberman, M., Bassi, E., Martinatti, M.K., Lario, F.C., Wosniak, J., Pomerantzeff, P.M.A., Laurindo, F.R.M.: Oxidant generation predominates around calcifying foci and enhances progression of aortic valve calcification. Arterioscler. Thromb. Vasc. Biol. 28, 463–470 (2008)
You, H., Yang, H., Zhu, Q., Li, M., Xue, J., Gu, Y., Lin, S., Ding, F.: Advanced oxidation protein products induce vascular calcification by promoting osteoblastic trans-differentiation of VSMCs via oxidative stress and ERK pathway. Ren. Fail. 31, 313–319 (2009)
Byon, C.H., Javed, A., Dai, Q., Kappes, J.C., Clemens, T.L., Darley-Usmar, V.M., McDonald, J.M., Chen, Y.: Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J. Biol. Chem. 283, 15319–15327 (2008)
Benito, M., Chen-Charpentier, A., et al.: A mathematical model of bone remodeling with delays. J. Comput. Appl. Math. 291, 76–84 (2016)
Molinuevo, M.S., Fernández, J.M., Cortizo, A.M., McCarthy, A.D., Schurman, L., Sedlinsky, C.: Advanced glycation end products and strontium ranelate promote osteogenic differentiation of VSMCs in vitro: preventive role of vitamin D. Mol. Cell. Endocrinol. 450, 94–104 (2017)
Miyata, T., Notoya, K., et al.: Advanced glycation end products enhance osteoclast-induced bone resorption in cultured mouse unfractionated bone cells and in rats implanted subcutaneously with devitalized bone particles. J. Am. Soc. Nephrol. 8(2), 260–270 (1997 Feb)
Li, G., Xu, J., Li, Z.: Receptor for advanced glycation end products inhibits proliferation in osteoblast through suppression of Wnt, PI3K and ERK signaling[J]. Biochem. Biophys. Res. Commun. 423(4), 684–689 (2012)
Takino, J., Nagamine, K., Hori, T., Sakasai-Sakai, A., Takeuchi, M.: Contribution of the toxic advanced glycation end-products-receptor axis in nonalcoholic steatohepatitis-related hepatocellular carcinoma. World J. Hepatol. 7(23), 2459–2469 (2015 Oct)
Marcus, B., Tom, R., et al.: Association between carotid diameter and the advanced glycation end product Nε-Carboxymethyllysine (CML). Cardiovasc. Diabetol. 8, 45 (2009)
Demiryurek, B.E., Gundogdu, A.A., Fetuin-A, S.: Levels in patients with bilateral basal ganglia calcification. Neurosci. Lett. 666, 148–152 (2017)
Janda, K., Krzanowski, M., Gajda, M., et al.: Vascular effects of advanced glycation end-products: content of immunohistochemically detected AGEs in radial artery samples as a predictor for arterial calcification and cardiovascular risk in asymptomatic patients with chronic kidney disease [J]. Dis. Markers. 153978, 2015 (2015)
Duan, X.H., Chang, J.R., Zhang, J., Zhang, B.H., Li, Y.L., Teng, X., Zhu, Y., du, J., Tang, C.S., Qi, Y.F.: Activating transcription factor 4 is involved in endoplasmic reticulum stress-mediated apoptosis contributing to vascular calcification. Apoptosis. 18(9), 1132–1144 (2013 Sep)
Wang, Z., Yan, J., Li, L., Liu, N., Liang, Y., Yuan, W., Chen, X.: Effects of Nε-carboxymethyl-lysine on ERS-mediated apoptosis in diabetic atherosclerosis. Int. J. Cardiol. 172(3), e478–e483 (2014)
Wang, Z., Li, L., Du, R., et al.: CML/RAGE signal induces calcification cascade in diabetes. Diabetol. Metab. Syndr. 83(1–12), 8 (2016)
Ramsey, S.A., Vengrenyuk, Y., Menon, P., Podolsky, I., Feig, J.E., Aderem, A., Fisher, E.A., Gold, E.S.: Epigenome-guided analysis of the transcriptome of plaque macrophages during atherosclerosis regression reveals activation of the Wnt signaling pathway. PLoS Genet. 10(12), e1004828 (2014 Dec 4)
Cipres, A., O'Malley, D.P., Li, K., et al.: Sceptrin, a marine natural compound, inhibits cell motility in a variety of cancer cell lines[J]. ACS Chem. Biol. 5(2), 195–202 (2010 Feb)
Xu, S., Li, L., Yan, J., Ye, F., Shao, C., Sun, Z., Bao, Z., Dai, Z., Zhu, J., Jing, L., Wang, Z.: CML/CD36 accelerates atherosclerotic progression via inhibiting foam cell migration. Biomed Pharmacother. 97, 1020–1031 (2018 Jan)
Park YM, Febbraio M, Silverstein RL. CD36 modulates migration of mouse and human macrophages in response to oxidized LDL and may contribute to macrophage trapping in the arterial intima [J]. J. Clin. Invest., 2009, 119(1):136–145
Sun, H., Yuan, Y., Sun, Z.: Update on mechanisms of renal tubule injury caused by advanced glycation end products. Biomed. Res. Int. 2016, 5475120 (2016)
Xanthis, A., Hatzitolios, A., Fidani, S., Befani, C., Giannakoulas, G., Koliakos, G.: Receptor of advanced glycation end products(RAGE) positively regulates CD36 expression and reactive oxygen species production in human monocytes in diabetes[J]. Angiology. 60(6), 772–779 (2009)
Availability of data and materials
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 81770450, 81370408, 81670105); Jiangsu Provincial Health and Family Planning Commission project(QNRC2016836); the Open Program of Key Laboratory of Nuclear Medicine, Ministry of Health and Jiangsu Key Laboratory of Molecular Nuclear Medicine(KF201504); Innovation plan for postgraduate research in Jiangsu Province(KYCX17_1801).
Author information
Authors and Affiliations
Contributions
All authors contributed to conception and design and wrote the review; All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflicts of interest
The authors declare that they have no competing interests.
Ethical approval
Not applicable.
Rights and permissions
About this article

Cite this article
Wang, Zq., Jing, Ll., Yan, Jc. et al. Role of AGEs in the progression and regression of atherosclerotic plaques. Glycoconj J 35, 443–450 (2018). https://doi.org/10.1007/s10719-018-9831-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10719-018-9831-x