
Abstract
Glycosphingolipids and glycoproteins play pivotal roles in the complex series of events governing cell adhesion and signal transduction. Aberrant glycosilation, typical of tumor cells, represents a key event in the induction of invasion and metastasis. Sialidases remove sialic acid residues from sialoconjugates and, in mammals, these enzymes have been proved to be involved in several cellular phenomena, including cell proliferation and differentiation, membrane function, and malignant transformation. Herein we show that only the lysosomal sialidase Neu1 and the plasma membrane-associated sialidase Neu3 are expressed in CFU-E erythroid precursors and K562 erythroleukemic cells. Tumour cells show much higher expression levels than CFU-E cells and, during differentiation, the content of the two enzymes progressively decreases. The sialoglycoconjugate pattern is different in the two cell types. In fact, the differentiating erythroid precursors show an increase of the typical erythrocyte sphingolipids, whereas K562 cells treated with butyrate show a marked increase of GD1a, GM2, PE, and ceramide. Finally, during differentiation the sialoglycoprotein content of erythroid cells shows a marked increase, and in K562 cells the process induces the synthesis of some sialoglycoprotein typical of the erythroid membrane. Overall, these results point out the great differences in sialoglycoconjugate and sialidase patterns exhibited by normal and tumour cells.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Sialoglycoconjugates represent a large class of biomolecules, characterized by the presence of sialic acid as terminal residue(s) of the oligosaccharide portion [1]. They include sialooligosaccharides, sialoglycoproteins, and sialoglycolipids or gangliosides, the latter being inserted into the external leaflet of the cellular membranes. Sialoglycoproteins and gangliosides are involved in several crucial biological processes including the control of cell survival, proliferation, differentiation, and adhesion [2, 3]. In particular, it is established that many malignant transformations are associated with abnormal sialylation, resulting in the expression of altered carbohydrate determinants on the cell surface [4]. This aberrant glycosilation profile is considered a key feature associated with invasiveness and metastatic potential of cancer cells [5–7]. The cellular sialic acid content is metabolically regulated mainly by sialyltransferases and sialidases [8–10]. Sialyltransferases belong to the enzyme class of glycotransferases and add sialic acid residues to sialoglycoproteins and sialoglycolipids during their biosynthesis [11], while sialidases or neuraminidases are glycohydrolitic enzymes that remove sialic acid residues during sialoglycoconjugate degradation [3]. Sialyltransferase expression level in malignant cells has been determined by several authors [12–14]. Based on these results, it is quite unlikely that sialyltransferases alone are responsible for the increase of the sialoglyconjugates content in tumor cells. Indeed, further studies have shown that also sialidases play a key role in this process, and their expression levels significantly change during tumorigenesis [15]. Four genetically distinct forms of mammalian sialidase have been characterized, with a predominant cellular localization (lysosomal, cytosolic and membrane-associated) and substrate specificity [16, 17]. Lysosomal sialidase (Neu1) has a catabolic role in desialylating glycoproteins and glycolipids in the lysosome [18]. Plasma membrane-associated sialidase (Neu3) is localized mainly on the cell surface [19], and its association with membrane microdomains has been demonstrated [20, 21]. Neu3 acts preferentially on ganglioside substrates, and therefore modulates the sialic acid content on the cell surface [22]. For example, it was shown that Neu3 is involved in cell growth control and differentiation of neuroblastoma cells by triggering selective ganglioside desialylation [23–25].
The substrate specificities and kinetic properties of the cytosolic sialidase (Neu2) have been fully characterized using the highly purified recombinant enzyme [26]. Neu2 showed a broad substrate specificity and desialylates both glycoproteins and gangliosides in vitro. Rodent Neu2 enzymes are involved in the differentiation of myoblasts to myotubes of rat L6 [27] and mouse C2C12 cells [28]. Unfortunately, the precise nature of their natural substrate(s) in the process of myotube formation and maintenance remains unclear.
More recently, the fourth member of the mammalian sialidase gene family (Neu4) has been identified [29, 30]. Although the subcellular localization of this enzyme is still under investigation, a recent report described an association of the human gene product with lysosomes [31].
In the attempt to elucidate the relationships among sialidases, sialoconjugates, and the complex series of events leading to malignant transformation, we compared the expression levels of the four enzymes and the sialoconjugate cell content in erythroid and erythroleukemic cells. Moreover, since cell proliferation (uncontrolled in tumour pathologies) and other tumoral characteristics can be also modulated through differentiation, we decided to make a comparative analysis of sialidases expression and sialoconjugates content during erythroid cell differentiation.
The human leukemic K562 cell line, originally established from a patient with chronic myeloid leukemia [32], is characterized by the Philadelphia chromosome and exhibits erythroid cell markers, including the presence of glycophorin-A [33], spectrin [34], and erythroglycan [35] and can be induced to differentiate and synthesize haemoglobin by sodium butyrate [36]. For these reasons K562 is considered a suitable model for erythroleukemia. In our comparative study we used as a healthy control the second stage erythroid progenitors (CFU-E, colony forming unit-erythroid cells) corresponding to the differentiation stage preceding hemoglobin production. At this stage the hemopoietic progenitors generate, in vitro, small erythroid colonies and finally give rise to morphologically recognizable erythroid precursors. During the differentiation process, cells became progressively sensitive to erythropoietin, due to the appearance of erythropoietin receptors on their plasma membrane [37].
Herein we report that sialidase Neu1 and Neu3 are expressed at higher levels in K562 compared to CFU-E cells, but they are progressively down-regulated when cell differentiation is induced.
On the other hand, we found that sialidase Neu2 is absent in K562 cells but low levels of its mRNA are detectable at the end of CFU-E differentiation. Furthermore, the two cell types present a different sialoglycoconjugate pattern. In fact, while CFU-E cells show all the typical erythrocyte sphingolipids, K562 cells have a distinct pattern, abundant in GD1a, GM2, and phosphatidylethanolamine (PE). The comparative analysis of the sialo-glycoproteins in the two cell types shows two distinct patterns as well. On the other hand, when K562 cells were induced to differentiate, they started to present some sialoglycoproteins typical of the erythroid membrane.
2 Materials and methods
Commercial chemicals were the purest available, common solvents were distilled before use and deionized water, obtained by a MilliQ system (Millipore), was distilled in a glass apparatus. High performance silica gel precoated thin-layer plates (HPTLC Kieselgel 60, 20 × 10 cm) were purchased from Merck GmbH; 4-methylumbellyferyl-N-acetylneuraminic acid (MU-NeuAc) and 4-methylumbellyferone (MU), N-acetylneuraminic acid (Neu Ac); bovine serum albumin (BSA); fetal bovine serum; αMEM; cyclosporine A; erythropoietin; sodium butyrate; ECACC (European Collection of Cell Culture), RPMI 1640 medium; pepstatin A, apoprotinin, leupeptin; penicillin and streptomycin from Sigma Chemical Co (St. Louis, MO, USA). TRIzol, SuperScript III First-Strand Synthesis System for RT-PCR from Invitrogen Life Technology (Carlsbad, CA, USA) ; dNTPs, iTaq DNA Polymerase, SYBR Green (iQ SYBR Green Supermix) from BioRad Laboratories (Richmond, VA, USA); PVDF membrane, horseradish peroxidase-conjugated IgG from Amersham Pharmacia Bioscience (Buckinghamshire, England). Sphingosine was prepared from cerebroside [38]. DIG Glycan Differentiation Kit for Glycan Analysis (Roche Diagnostics, Indianapolis, IN, USA) [1-3H]sphingosine (radiochemical purity over 98%; specific radioactivity 2.08 Ci/mmol) was prepared by specific chemical oxidation of the primary hydroxyl group of sphingosine followed by reduction with sodium boro[3H]hydride [39]. Ganglioside GD1a was purified from the total ganglioside mixture extracted from bovine brains [40]. Radioactive GD1a containing erythro C18-sphingosine, isotopically tritium labeled at position 3, [3-3H(Sph18)]GD1a, was prepared by the dichloro-dicyano-benzoquinone/sodium boro[3H] hydride method followed by reversed phase HPLC purification (homogeneity over 99%; specific radioactivity of 1.2 Ci/mmol) [41, 42].
Radioactive sphingolipids were extracted from cells fed with [1-3H]sphingosine, purified, characterized as previously described [22], and used as chromatographic standards. Standard molecular biology techniques were carried out as described by Sambrook and Russell [43].
2.1 Cell cultures
Human erythroid cells were amplified by the two-phase liquid culture method from BFU (burst-forming unit) present in peripheral blood of healthy donors, according to Fibach and Rachmilewitz [44]. In the first phase, mononuclear cells were isolated by Lymphoprep and cultured for a week in α-MEM medium supplemented with 10% FBS, 1 μg/ml cyclosporine A, and 10% conditioned medium collected from bladder carcinoma 5,637 culture. After 7 days, the non-adherent cells were harvested and recultured in phase II medium composed of α-MEM, 30% fetal bovine serum (FBS), 1% deionized bovine serum albumin (BSA), 10−5 M 2-mercaptoethanol, and 1 U/ml erythropoietin to induce erythroid differentiation. Cell samples were harvested and analyzed on days 0, 3, 6, 9, 13 of phase II. Human erythroleukemia K562 cells were purchased from ECACC and cultured in RPMI 1640 medium supplemented with 10% FBS, 2 mM glutamine, 100 IU/ml penicillin, 100 μg/ml streptomycin. Erythroid differentiation of K562 cells was triggered incubating 2 × 105 cells/ml in culture medium containing 1 mM sodium butyrate [36]. All cultures were performed at 37°C in humidified incubator with 5% CO2.
2.2 RNA extraction and real-time PCR
Total RNA was isolated using TRIzol (Invitrogen) and its manufacturer’s guidelines. Using random hexamers, 2.5 μg of RNA were reverse-transcribed with the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen). cDNA representing 25 ng of total RNA was used as template for real-time PCR. PCR mixture included 0.2 μM primers, 50 mM KCl, 20 mM Tris/HCl, pH 8.4, 0.8 mM dNTPs, 0.7U iTaq DNA Polymerase, 3 mM MgCl2, and SYBR Green (iQ SYBR Green Supermix, BioRad), in a final volume of 25 μl. Amplification and real-time data acquisition were performed in iCycler (BioRad) using the following cycle-conditions: initial denaturation at 95°C for 5 min , followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. The gene coding for the ribosomal protein S14 was chosen as reference gene.
The following primers were used:
Gene | Forward primer | Reverse primer |
NEU1 | 5′-CCTGGATATTGGCACTGAA-3′ | 5′-CATCGCTGAGGAGACAGAAG-3′ |
NEU2 | 5′-AGAAGGATGAGCACGCAGA-3′ | 5′-GGATGGCAATGAAGAAGAGG-3′ |
NEU3 | 5′-TGAGGATTGGGCAGTTGG-3′ | 5′-CCCGCACACAGATGAAGAA-3′ |
NEU4 | 5′-ACCGCCGAGAGTGTTTTG-3′ | 5′-CGGGCATTGCAGTAGAGG-3′ |
S14 | 5′-GGCAGACCGAGATGAATCCTCA-3′ | 5′-CAGGTCCAGGGGTCTTGGT CC-3′ |
2.3 Sialidase activity assay
Erythroid cells and K562 cells were harvested by centrifugation and resuspended in PBS containing 1 μg/ml pepstatin A, 10 μg/ml apoprotinin, and 10 μg/ml leupeptin. Total cell extracts were prepared by sonication and centrifuged at 800 × g for 10 min to eliminate unbroken cells and nuclear components. The crude extract was subsequently centrifuged at 200,000 × g for 20 min on TL100 Ultracentrifuge (Beckman) in order to separate membranes from cytosolic proteins. Aliquots of particulate fraction and supernatant were used for protein (Coomassie Protein Assay Reagent; Pierce) and enzymatic assay. For Neu1 sialidase, 20 μg of membrane proteins were incubated with 0.3 mM (4-MU-NeuAc), in 50 mM sodium-acetate buffer pH 4.6, in a final volume of 100 μl according to Miyagi [45]. Neu2 sialidase activity present in the cell cytosol was assayed using 30 μg of soluble protein and 0.3 mM 4MU-NeuAc in 50 mM sodium-acetate buffer, pH 5.5 according to Miyagi [26]. The activity of Neu3 sialidase was assessed toward 0.2 mM ganglioside [3H]GD1a (carrying 2.0 × 105 dpm) using 20 μg of membrane protein in 50 mM sodium-acetate buffer pH 4.6, in the presence of 0.04% Triton X-100 [22]. Enzyme activity was expressed as units (U), representing the enzyme amount that liberates 1 μM of product min/mg protein, under optimal conditions.
2.4 Metabolic labeling of cell sphyngolipids with [1-3H]Sphingosine and lipid extraction
[1-3H]Sphingosine in methanol was dried under a nitrogen stream and dissolved in culture medium containing 10% FBS, to obtain a final concentration of 3 × 10−8 M (corresponding to 0.4 μCi); 1 × 107 erythroid cells at 0, 6, 9, and 13 days of differentiation and 2 × 106 K562 cells were incubated in this medium for a 2-h pulse followed by a 24-h chase. At the end of treatment, the cells were harvested by centrifugation, frozen, lyophilized, and subjected to lipid extraction.
Total lipids were extracted twice with chloroform/methanol 2:1 (v/v), after resuspending lyophilized cells in 25 μl of water. Then, gangliosides and neutral glycolipids were separated by a two-phase partitioning in chloroform/methanol 2:1 (v/v) and 20% water [22]. The aqueous phase containing gangliosides and the organic phase containing neutral glycolipids were counted for radioactivity. Total lipids and gangliosides were analyzed on HPTLC plates developed in the solvent system chloroform/methanol/0.2% aqueous CaCl2, 60:40:9 (v/v) while neutral glycolipids were separated using the solvent mixture chloroform/methanol/water, 55:20:4.5 (v/v). [3H]Sphingolipids were visualized and quantified by radiochromatoscanner (Beta-Imager 2000, Biospace, Paris, France).
The structural characterization of ganglioside mixture from erythroid and K562 cells was determined by reversed phase HPLC-ESI mass spectrometry. ESI mass spectrometry of ganglioside species was carried out in negative mode on a Thermo Quest Finnigan LCQdeca mass spectrometer equipped with a electrospray ion source [46].
2.5 Glycoproteins characterization
Proteins, 30 μg from erythroid cells and K562 cells homogenates, were separated on SDS-10% polyacrylamide gel and transferred onto a PVDF membrane (Amersham Bioscience). Sialoglycoproteins characterization was carried out employing the DIG Glycan Differentiation Kit (Roche) in accordance with the protocol enclosed. Glycoproteins with sialic acid linked α(2–6) were identified using SNA (Sambucus nigra agglutinin) lectin while sialic acid linked α(2–3) was visualized using MAA (Maackia amurensis agglutinin) lectin.
2.6 Determination of total sialic acid cell content
Total sialic acid content was determined by chromatographic micro procedure of [47] on samples of erythroid cells and K562 cells digested for 90 min in 0.05 M sulphuric acid at 80°C. Insoluble material was removed by centrifugation and total ganglioside-bound and protein-bound sialic acid was determined according to Tettamanti et al. [40].
2.7 Statistical analyses
Values are presented as means ± SD. Statistical analyses were made using unpaired Student’s t-test. Significance was attributed at the 95% level of confidence (p < 0.05).
3 Results
3.1 Sialidase gene expression (mRNA contents) and enzyme activities in CFU-E erythroid precursor and erythroleukemic K562 cells
Real-time PCR experiments were carried out to estimate the amounts of sialidase transcripts in CFU-E and K562 cells. The relative mRNA levels of Neu1, Neu2, Neu3, and Neu4 were normalized with the amount of mRNA encoding the ribosomal protein S14 as internal control. Transcripts encoding Neu1 and Neu3 were easily detectable in freshly isolated CFU-E and K562 cells whereas no mRNAs of Neu2 and Neu4 were measurable in either the cell types, as shown in Fig. 1a. Interestingly, the mRNA levels of lysosomal and plasma membrane-associated sialidases showed a remarkable increase (4.14 ± 0.21 and 7.07 ± 0.35-fold, respectively) in the erythroleukemic cells compared with the erythroid precursors.

Sialidase levels in CFU-E erythroid precursors and K56 cells. (a) Total RNA was isolated from CFU-E erythroid and K562 erythroleukemic cells. 25 ng of RNA was used as template of Neu1, Neu2, Neu3 and Neu4 in SYBR-green semi-quantitative real-time PCR. The relative expression is shown in relation to the expression of ribosomal protein S14 that was measured as an internal control. (b) Sialidase activity in CFU-E and K562 cells was determined in the pellet and supernatant obtained after centrifugation of crude cellular extract. Neu1 sialidase activity was measured in the presence of MU-NeuAc (as substrate), while sialidase Neu3 was measured with ganglioside GD1a. For details see Materials and methods. The data are the mean values of five experiments ± SD values
These observations were confirmed by measurement of Neu1 and Neu3 sialidase activity. As shown in Fig. 1b, the comparison between Neu1 and Neu3 enzyme levels in CFU-E and K562 cells showed an increase of 3.77 ± 0.14 and 8.45 ± 0.43-fold, respectively. As anticipated, we could not detect any cytosolic sialidase activity.
3.2 Glycolipid pattern in CFU-E erythroid precursor and erythroleukemic K562 cells
The shingolipid mixtures from CFU-E and K562 cells were analyzed by HPLC-ESI mass spectrometry and the percentage of each species was determined by radioimaging and densitometry after HPTLC separation. Briefly, quantification was carried out after administration of [1-3H]sphingosine to cultured cells. The radioactive precursor was incorporated by the cells and led to an extensive labeling of the sphingolipids, namely gangliosides, neutral glycosphingolipids, sphingomyelin, and ceramide [48]. In addition, part of the tritiated sphingosine was catabolized by the cells to radioactive ethanolamine and recycled for the biosynthesis of radioactive phosphatidylethanolamine (PE). As already reported in the case of erythrocyte membrane [49], sialosyl-lacto-N-norhexaosylceramide (SnHc) and sialosylparagloboside (SPg), both containing sialosyl α2→3 linkages, as well as GM3 and GM2 are the main gangliosides of the CFU-E erythroid precursor and covered the 50.3, 20.9, 22.7 and 6.1% of the total ganglioside mixture respectively (Fig. 2a). Each ganglioside species contained C18 sphingosine and by HPTLC analysis split in two spots. The molecular species containing palmitic acid showed a lower mobility, while the molecular species containing C24:0 and C24:1 fatty acids showed a slightly higher mobility in our solvent system.

Glycolipid pattern in CFU-E erythroid precursors and K562 cells. CFU-E (1 × 107) and K562 (2 × 106) cells were pulsed for 2 h with [1-3H]sphingosine, 3 × 10−8 M (corresponding to 0.4 μCi), final concentration. After 24 h of chase, cells were harvested and treated for lipid analysis. (a) Left panel: HPTLC separation of the aqueous phase obtained by lipid extraction and fractionation. Solvent system: chloroform/methanol/0.2% aqueous CaCl2 60:40:9 (v/v). Lane 1 (St), Ganglioside Standard ; lane 2, CFU-E cells; lane 3, K562 cells. In the two right panels: ganglioside relative content (%) in CFU-E and K652 cells, respectively. (b) Left panel: HPTLC separation of the organic phase obtained by lipid extraction and fractionation. Solvent system chloroform/methanol/water 55:20:4.5 (v/v). Lane 1 (St), Neutral lipid Standards; lane 2, CFU-E cells; lane 3, K562 cells. In the two right panels: neutral lipid relative content (%) in CFU-E and K652 cells, respectively. The data are the mean values of five different experiments, carried out in triplicate
The ganglioside pattern of erythroleukemic cells is shown in Fig. 2a. Gangliosides GD1a, SnHc, GM1, SPg, GM2, and GM3 were the main gangliosides detected on HPTLC and covered 34.8, 3.78, 30.2 (corresponding to the sum of GM1 and SPg which co-migrate in our solvent system), 15.9, and 15.5 % on the total main ganglioside mixture, respectively. HPLC-ESI mass spectrometry analysis confirmed the heterogeneity of the acyl moiety already observed in case of CFU-E erythroid precursor as well as the presence of GM1 as one of the major gangliosides, that in our separation system co-migrated with SPg and prevented the quantification of each molecular species. Overall, the ganglioside composition of K562 cells was significantly different from the one detected in CFU-E erythroid precursors. First of all, GD1a was the most abundant ganglioside, followed by GM1, that was not detected in CFU-E cells. Secondly, SnHc and SPg were present only in small amounts, whereas they accounted for over than 50% of the total ganglioside in erythroid precursors.
Finally, an analysis of the neutral lipid pattern was carried out. As shown in (Fig. 2b), sphingomyelin (SM), PE, glucosyl-ceramide (GlcCer) and ceramide (Cer) were the main neutral lipids of CFU-E cells and covered 67.1, 5.1, 14.3, and 13.5%, respectively, of the total lipid mixture. In K562 cells, together with SM, PE, GlcCer and Cer, globoside 3 (Gb3), and lactosyl-ceramide (LacCer) were detectable in HPTLC and covered 32.8, 26.6, 23.2, 7.9, 2.7 and 6.8 respectively, of the total lipid mixture.
Quantification of LacCer was carried out after alkaline methanolysis of the lipid sample to remove PE.
3.3 Variations of sialidase expression levels in CFU-E and K562 cells during differentiation
In order to assess whether sialidase expression levels change during differentiation, CFU-E erythroid precursors and K-562 cells were treated with 1 U/ml erythropoietin and 1 mM of sodium butyrate, respectively. In these culture conditions, CFU-E cells differentiated in 13 days to mature erythroblasts whereas K-562 cells reduced their proliferation rate and started to synthesize hemoglobin and some erythroid markers. Cell samples were harvested at different times and analyzed for their content in sialidase transcripts as well as the corresponding enzyme activities. The results of the sialidase transcript quantification by real-time PCR are reported in (Fig. 3). In CFU-E erythroid precursor (Fig. 3a) the NEU1 transcript decreased during differentiation, and at day 13 of erythropoietin treatment the relative expression became about 20% of the initial value. The relative content of the transcript encoding the plasma membrane-associated sialidase NEU3 showed a roughly 44% increase after 3 days of differentiation, then rapidly decreased and continued this trend to about 1/10 of the initial level on day 13. Surprisingly, we were also able to detect the cytosolic sialidase NEU2 mRNA. The transcript appeared only in the late phase of the differentiation process, from day 10 to 13 with only minor changes in the relative content. Overall, the relative amount of the NEU2 mRNA is very low compared to the one of Neu1 and Neu3. In fact, the cycle threshold (CT) for NEU1 and NEU3 amplification corresponds to 25.6 ± 0.33 and 32.7 ± 0.44, respectively, whereas for NEU2 corresponds to 38.4 ± 0.76.

Sialidase expression levels in CFU-E erythroid precursors and K562 cells during differentiation. SYBR-green semi quantitative real time RT-PCR of CFU-E cells treated with 1 U/ml erythropoietin (a) and K562 cells treated with 1 mM of sodium butyrate (b) at different days of differentiation. The relative expression in Neu1, Neu2, Neu3 and Neu4 is shown in relation to the expression of ribosomal protein S14, that was measured as an internal control. The data are the mean values of five experiments ± SD values
During the differentiation of K-562 erythroleukemic cells only the transcripts of the lysosomal and plasma membrane-associated sialidase were detectable (Fig. 3b). After 6 days of differentiation in the presence of butyrate, NEU1 and NEU3 transcripts relative content was reduced to roughly the 60 and 72% of the initial value, respectively.
The sialidase enzyme activities during differentiation are reported in Table 1. Surprisingly, in the case of CFU-E erythroid cells, the Neu1 and Neu3 sialidase activity showed a marked increase. In fact, the erythropoietin treatment induces an increase to about the double of the initial enzyme activities. Thus, there is a difference between enzyme expression and activity. These data could be explained based on the stability and/or activation of the enzymes in their cellular environment. Moreover, despite the appearance of the corresponding gene transcript, no Neu2 enzyme activity was detected in the late phase of the differentiation process, even by Western-blot with rabbit anti-Neu2 polyclonal antiserum (data not shown).
The measurement of the sialidase enzyme activities in K562 cells confirms the results obtained by real-time PCR. In fact, a 22 and 53% decrease of Neu1 and Neu3 mRNAs expression levels was detected at day 6.
3.4 Glycolipid pattern modification in CFU-E erythroid precursors and K562 cells during differentiation
Sphingosine labeling of differentiating cells allowed the characterization of the glycolipid pattern during the process. As shown in (Fig. 4a), the addition of erythropoietin in the culture media of CFU-E cells lead to a 1.4-, 3- and 1.3-fold increase of SnHc, SPg and GM3, respectively, while GM2 content did not show any significant variation. Neutral glycolipid analysis (Fig. 4b) revealed a 1.7- and 3.1-fold increase of PE and Cer, respectively, whereas no marked variations were detected in SM and GlcCer cell content. In addition, the cell membranes of differentiating erythroid precursors were characterized by the presence of LacCer, a molecular species lacking in freshly isolated CFU-E cells.

Glycolipid pattern modification in CFU-E erythroid precursors cells during differentiation. CFU-E cells (1 × 107) were pulsed for 2 h with [1-3H]Sphingosine, 3 × 10−8 M (corresponding to 0.4 μCi), final concentration. After 24 h of chase, cells were harvested and treated for lipid analysis. (a) Left panel: HPTLC separation of the aqueous phase. Solvent system chloroform/methanol/0.2% aqueous CaCl2 60:40:9 (v/v). Lane 1, Ganglioside Standards (St) ; lanes 2, 3, 4 and 5 CFU-E cells before and after 3, 6, 9 and 13 days of erythropoietin administration. In the right panel: ganglioside content (dpm/mg protein × 102) in CFU-E cells during differentiation. (b) Left panel: HPTLC separation of the organic phase obtained by lipid extraction and fractionation. Solvent system chloroform/methanol/water 55:20:4.5 (v/v). Lane 1, Neutral lipid Standards (St); lanes 2, 3, 4 and 5, CFU-E cells before and after 3, 6, 9 and 13 days of erythropoietin administration. In the right: neutral lipid content (dpm/mg protein × 103) in CFU-E cells during differentiation. For details see Materials and methods. The data are the mean values of five different experiments, carried out in triplicate
The corresponding sialoglycolipid patterns detectable in K562 cells cultured up to 6 days in presence of butyrate showed a 2.4- and 1.9-fold increase of GD1a and GM2, respectively. GM1+SPg and GM3 did not show significant variations while SnHc remains the less abundant ganglioside (Fig. 5a), even if at the end of the process it shows a 2-fold increase. The analysis of neutral glycolipid pattern during differentiation shows a roughly 1.5-fold increase of PE and Cer, while no significant variations are detectable in the case SM, GlcCer, LacCer, and Gb3 (Fig. 5b).

Glycolipid pattern modification in K562 cells during differentiation. K562 cells (2 × 106) were pulsed for 2 h with [1-3H]Sphingosine, 3 × 10−8 M (corresponding to 0.4 μCi), final concentration. After 24 h of chase, cells were harvested and treated for lipid analysis. (See Materials and methods). (a) Left panel: HPTLC separation of the aqueous phase. Solvent system chloroform/methanol/0.2% aqueous CaCl2 60:40:9 (v/v). Lane 1, Ganglioside Standards (St); lanes 2 and 3, K 562 cells before and after 6 days of sodium butyrate administration. In the right panel: ganglioside content (dpm/mg protein × 102) in K562 cells during differentiation. (b) Left panel: HPTLC separation of the organic phase obtained by lipid extraction and fractionation. Solvent system chloroform/methanol/water 55:20:4.5 (v/v). Lane 1, Neutral lipid Standards (St); lanes 2 and 3, K562 cells before and after 6 days of sodium butyrate administration. In the right panel: neutral lipid content (dpm/mg protein × 103) in K562 cells during differentiation. The data are the mean values of five different experiments, carried out in triplicate
3.5 Modification of glycoprotein pattern in CFU-E erythroid precursors and K562 cells during differentiation
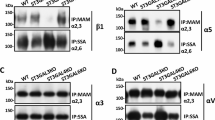
Finally, a characterization of the sialoglycoprotein pattern of CFU-E erythroid precursors and K562 cells during differentiation was carried out. As shown in (Fig. 6a, c), the staining with lectins that specifically recognized α(2–3) and α(2–6) sialosyl-linkages of crude homogenates derived from differentiating erythroid precursors revealed a significant increase of cell sialoglycoproteins. In addition, a densitometry analysis of the membranes clearly showed the appearance of a band with a molecular weight of about 40 kDa.

SDS-PAGE of sialoglycoproteins in CFU-E erythroid precursors and K562 cells during differentiation. Densitometry analysis and SDS-PAGE of membrane sialoglycoproteins in CFU-E (a) and (c) and K562 (b) and (d) during differentiation. Sialoglycoproteins were characterized with DIG Glycan Differentiation Kit and were identified using MAA (Maackia amurensis agglutinin) for sialic acid α(2–3) panels (a) and (b) and SNA (Sambucus nigra agglutinin) lectin for sialic acid α(2–6) panels (c) and (d) For details see Materials and Methods
In K562 erythroleukemic cells, the treatment with butyrate dramatically changed the sialo-glycoprotein pattern (Fig. 6b, d). Starting from a pattern characterized by a quite low cell sialo-glycoprotein content, with a predominance of high molecular weight polypeptides containing α(2–6) linked sialic acid, the differentiation induced the synthesis of a series of sialoglycoproteins with a molecular weight ranging from 45 to 90 kDa that closely resemble those detectable in CFU-E cells (Fig. 6a, c).
4 Discussion
In the last few years increasing evidence suggests the involvement of sialidase enzymes and of sialoconjugates in both cell differentiation [23–25, 27, 28] and malignant transformation [15, 50]. These phenomena are strictly interconnected because one characteristic of cancer cells is their inability to spontaneously undergo a differentiation process. In fact, many chemotherapeutic drugs successfully slow down tumour growth by inducing cell differentiation. [51–54]. We therefore decided to study sialidase expression levels in K562 erythroleukemic cells, in comparison to CFU-E erythroid precursors in normal culture conditions, as well as during cell differentiation. As we anticipated, K562 cells are characterized by an altered sialidase expression both in terms of mRNA levels and enzyme activity. In fact, both lysosomal sialidase Neu1 and plasma membrane sialidase Neu3 are over-expressed in these cells. These aberrations confirm the notion that sialidases are involved in the neoplastic process. In fact, enhanced plasma membrane-associated sialidase mRNA levels have recently been detected in human colon cancer, where the enzyme is involved in the protection against programmed cell death [55]. In this perspective, high Neu3 levels could represent one of the typical features of cancerous cells.
Another aspect closely related to sialidase expression, is the comparative analysis of the sialoglycoconjugates present in K562 and CFU-E cells. In agreement with previous studies [56], K562 cells exhibit a high GD1a content as well as of the mono sialylated gangliosides GM1 and GM2. On the contrary, erythroid precursor membranes are deficient in GD1a, and its place is taken by typical erythrocyte gangliosides such as SnHc and SPg, followed by GM3, and a small amount of GM2. Similarly, we found differences among neutral glycolipids. These variations were less marked but equally important: K562 cells exhibited a considerable content of PE, which is indicative of a metabolism increase typical of cancer cells, and also contain Gb3 and LacCer, both lacking in CFU-E cells. In particular, LacCer was the major diglycosyl ceramide, found in high levels in cells with increased Neu3 sialidase activity as a result of its activity on gangliosides [55]. Moreover, LacCer showed an interesting biological activity leading to apoptosis suppression in human cancer cells [55].
Finally, also the sialoglycoprotein content of K562 was quite different compared to the one of CFU-E. In fact, CFU-E cells were characterized by abundant highly sialilated proteins with a molecular weight around 30 and 60 kDa, almost negligible in K562 cells (Fig. 6a, b).
After this initial analysis, we decided to investigate the expression of sialidases and sialoglycoconjugates during differentiation. The administration of butyrate to K562 cells slowed down their proliferation and induces a partial erythroid differentiation characterized by hemoglobin synthesis. Interestingly, this treatment also influenced sialidase enzyme levels. In fact, we detected a marked decrease of both NEU1 and NEU3 mRNAs and enzymatic activities. This phenomenon was accompanied by a parallel great increase of GD1a and GM2, SnHc, PE, and Cer. The increase of GD1a could be easily associated to NEU3 down-regulation, because of its high specificity toward ganglioside substrates. [19, 25, 57] In addition, the glycoproteins content was greatly increased and modified, giving rise to some sialylated proteins typical of the erythroid precursors. According to this data, the total sialic acid content showed a 2.5-fold increase during differentiation (Table 1). This increase was likely due to a concomitant down-regulation of both Neu1 and Neu3 sialidases, which could also be responsible for the observed differences in the ganglioside and sialoglycoprotein pattern. Moreover, it is also plausible that other processes may occur, including a specific modification of the sialic acid residues to became resistant to sialidase action [58]. Conversely, erythropoietin treatment induced in vitro differentiation of CFU-E cells into mature erythroblasts. The analogous differentiation of K562 cells presented several differences. In fact, in CFU-E cells, this process is initially associated with an increase of Neu1 and Neu3 enzyme levels, which progressively decreased during differentiation, while Neu1 and Neu3 decreased constantly.
On the other hand, Neu2 and Neu4 could not be detected in K562, even during and after differentiation. Interestingly, RT-PCR assays revealed the presence of small amounts of NEU2 transcript at the end of the differentiation process in CFU-E cells, but we were not able to detect the presence of the corresponding protein nor its activity. Based on these results, it would be speculative to implicate an involvement of NEU2 in these processes, even though Neu2 involvement in muscle cell differentiation has been reported elsewhere [27].
The differentiation process also induced a massive enhancement of erythrocyte gangliosides (SnHc and SPg), LacCer, and ceramide, followed by a great increase of specific sialoglycoproteins, similar to the ones observed during K562 differentiation. These biochemical modifications were associated with a minor variation of the total sialic acid content that corresponds to a small fraction (from 4 to 13%) of the values detectable in tumor cells. In spite of the marked increase of LacCer and Cer, which can be related to the sialidase content, the general metabolic picture detectable in these cells during differentiation can be associated with an activation of the metabolic pathways leading to mature erythrocytes, more than to catabolic pathways.
Overall, the aberrant glycosilation profile detectable in K562 cells confirmed previous experimental evidences of a characteristic glycoconjugate pattern in most types of human cancer. Aberrant glycosilation is strictly related to neoplastic transformation and could represent a key event in the induction of invasion and metastasis [50]. Moreover, the abundance of particular sphingolipids located mainly on the plasma membrane lipid rafts at close proximity of receptors and signaling molecules, could modify the net signal transduction in the cell, leading to considerable effects on proliferation and apoptosis [59]. In this context, NEU1 and NEU3 sialidases, which are able to modify both sphingolipids and glycoproteins, are present in high levels in cancer cells and seem to be directly involved in their malignancy. In particular, the plasma membrane associated sialidase Neu3 is able to act through cell-to-cell interaction [22], and is probably one of the most potent regulator of the cell surface gangliosides. The lysosomal sialidase Neu1 plays a pivotal role in the catabolism of sialoglycoconjugates within the organelle, and its involvement in suppression of metastasis in murine melanoma cells has been demonstrated [60]. Interestingly, the results obtained by over-expression of NEU1, are associated with an increase of desialylated glycoproteins on the cell surface.
Studies on the regulation of sialidase expression through molecular biology are currently underway, and they may further clarify sialidase involvement in cancer transformations.
Abbreviations
- GM3:
-
NeuAc(α2→3)Gal(β1→4)Glc-Cer
- GM2:
-
GalNAc(β1→4)NeuAc(α 2→3)Gal(β1→4)Glc-Cer
- GM1:
-
Gal(β1→3)GalNAc(β1→4)NeuAc(α 2→3)Gal(β1→4)Glc-Cer
- GD1a:
-
NeuAc(α 2→3) Gal(β1→3)GalNAc(β1→3)NeuAc(α 2→3)Gal(β1→4)Glc-Cer
- SPg:
-
NeuAc(α 2→3)Gal(β1→4)GlcNAc(β1→3)Gal(β1→4)Glc-Cer
- SnHc:
-
NeuAc(α 2→3)Gal(β1→4)GlcNAc(β1→3)Gal(β1→4)GlcNAc(β1→3)Gal(β1→4)Glc-Cer
References
Schauer, R., Kelm, S., Reuter, G., Roggentin, P. Shaw, L.: Biochemistry and role of sialic acids. In: Rosenberg, A. (ed.) Biology of the Sialic Acids, pp. 7–67. Plenum Press, New York (1995)
Corfield, T.: Bacterial sialidases—roles in pathogenicity and nutrition. Glycobiology 2, 509–521 (1992)
Saito, M.Y., Saito, R.K.: Biochemistry and function of sialidases. In: Rosenberg, A. (ed.) Biology of the Sialic Acids, pp. 261–313. Plenum Press, New York (1995)
Kannagi, R., Izawa, M., Koike, T., Miyazaki, K., Kimura, N.: Carbohydrate-mediated cell adhesion in cancer metastasis and angiogenesis. Cancer Sci. 95, 377–384 (2004)
Fogel, M., Altevogt, P., Schirrmacher, V.: Metastatic potential severely altered by changes in tumor cell adhesiveness and cell-surface sialylation. J Exp Med. 157, 371–376 (1983)
Collard, J.G., Schijven, J.F., Bikker, A., La Riviere, G., Bolscher, J.G., Roos, E.: Cell surface sialic acid and the invasive and metastatic potential of T-cell hybridomas. Cancer Res. 46, 3521–3527 (1986)
Hakomori, S.: Tumor malignancy defined by aberrant glycosylation and sphingo(glyco)lipid metabolism. Cancer Res. 56, 5309–5318 (1996)
Tettamanti, G., Preti, A., Cestaro, B., Venerando, B., Lombardo, A., Ghidoni, R., Sonnino, S.: Gangliosides, neuraminidase and sialyltransferase at the nerve endings. Adv. Exp. Med. Biol. 125, 263–281 (1980)
Sillence, D.J., Allan, D.: Repair of BHK cell surface ganglioside GM3 after its degradation by extracellular sialidase. Mol. Membr. Biol. 15, 229–235 (1998)
Azuma, Y., Taniguchi, A., Matsumoto, K.: Decrease in cell surface sialic acid in etoposide-treated Jurkat cells and the role of cell surface sialidase. Glycoconj. J. 17, 301–306 (2000)
Basu, S.B., Basu, M., Basu, S.S.: Biological specificity of sialyltransferases. In: Rosenberg, A. (ed.) Biology of the Sialic Acids, pp. 69–94. Plenum Press, New York (1995)
Kijima-Suda, I., Miyazawa, T., Itoh, M., Toyoshima, S., Osawa, T.: Possible mechanism of inhibition of experimental pulmonary metastasis of mouse colon adenocarcinoma 26 sublines by a sialic acid: nucleoside conjugate. Cancer Res. 48, 3728–3732 (1988)
Dall’Olio, F., Chiricolo, M.: Sialyltransferases in cancer. Glycoconj J. 18, 841–850 (2001)
Yoshida, S., Fukumoto, S., Kawaguchi, H., Sato, S., Ueda, R., Furukawa, K.: Ganglioside GD2 in small cell lung cancer cell lines: enhancement of cell proliferation and mediation of apoptosis. Cancer Res. 61, 4244–4252 (2001)
Miyagi, T., Wada, T., Yamaguchi, K., Hata, K.: Sialidase and malignancy: a minireview. Glycoconj. J. 20, 189–198 (2004)
Achyuthan, K.E., Achyuthan, A.M.: Comparative enzymology, biochemistry and pathophysiology of human exo- alpha-sialidases (neuraminidases). Comp. Biochem. Physiol., Part B Biochem. Mol. Biol. 129, 29–64 (2001)
Monti, E., Preti, A., Venerando, B., Borsani, G.: Recent development in mammalian sialidase molecular biology. Neurochem. Res. 27, 649–663 (2002)
Pshezhetsky, A.V., Ashmarina, M.: Lysosomal multienzyme complex: biochemistry, genetics, and molecular pathophysiology. Prog. Nucleic Acid Res. Mol. Biol. 69, 81–114 (2001)
Monti, E., Bassi, M.T., Papini, N., Riboni, M., Manzoni, M., Venerando, B., Croci, G., Preti, A., Ballabio, A., Tettamanti, G., Borsani, G.: Identification and expression of NEU3, a novel human sialidase associated to the plasma membrane. Biochem. J. 349, 343–351 (2000)
Kalka, D., von Reitzenstein, C., Kopitz, J., Cantz, M.: The plasma membrane ganglioside sialidase cofractionates with markers of lipid rafts. Biochem. Biophys. Res. Commun. 283, 989–993 (2001)
Wang, Y., Yamaguchi, K., Wada, T., Hata, K., Zhao, X., Fujimoto, T., Miyagi, T.: A close association of the ganglioside-specific sialidase Neu3 with caveolin in membrane microdomains. J. Biol. Chem. 277, 26252–26259 (2002)
Papini, N., Anastasia, L., Tringali, C., Croci, GL., Bresciani, R., Yamaguchi, K., Miyagi, T., Preti, A., Prinetti, A., Prioni, S., Sonnino, S., Tettamanti, G., Venerando, B., Monti, E.: The plasma membrane-associated sialidase MmNEU3 modifies the ganglioside pattern of adjacent cells supporting its involvement in cell-to-cell interactions. J. Biol. Chem. 279, 16989–16995 (2004)
Kopitz, J., von Reitzenstein, C., Sinz, K., Cantz, M.: Selective ganglioside desialylation in the plasma membrane of human neuroblastoma cells. Glycobiology 6, 367–376 (1996)
Kopitz, J., Muhl, C., Ehemann, V., Lehmann, C., Cantz, M.: Effects of cell surface ganglioside sialidase inhibition on growth control and differentiation of human neuroblastoma cells. Eur. J. Cell Biol. 73, 1–9 (1997)
Hasegawa, T., Yamaguchi, K., Wada, T., Takeda, A., Itoyama, Y., Miyagi, T.: Molecular cloning of mouse ganglioside sialidase and its increased expression in neuro2a cell differentiation. J. Biol. Chem. 275, 14778 (2000)
Tringali, C., Papini, N., Fusi, P., Croci, G., Borsani, G., Preti, A., Tortora, P., Tettamanti, G., Venerando, B., Monti, E.: Properties of recombinant human cytosolic sialidase HsNEU2. The enzyme hydrolyzes monomerically dispersed GM1 ganglioside molecules. J. Biol. Chem. 279, 3169–3179 (2004)
Sato, K., Miyagi, T.: Involvement of an endogenous sialidase in skeletal muscle cell differentiation. Biochem. Biophys. Res. Commun. 221, 826–830 (1996)
Fanzani, A., Giuliani, R., Colombo, F., Zizioli, D., Presta, M., Preti, A., Marchesini, S.: Overexpression of cytosolic sialidase Neu2 induces myoblast differentiation in C2C12 cells. FEBS Lett. 547, 183–188 (2003)
Comelli, E.M., Amado, M., Lustig, S.R., Paulson, J.C.: Identification and expression of Neu4, a novel murine sialidase. Gene 321, 155–161 (2003)
Monti, E., Bassi, M.T., Bresciani, R., Civini, S., Croci, G.L., Papini, N., Riboni, M., Zanchetti, G., Ballabio, A., Preti, A., Tettamanti, G., Venerando, B., Borsani, G.: Molecular cloning and characterization of NEU4, the fourth member of the human sialidase gene family. Genomics 83, 445–453 (2004)
Seyrantepe, V., Landry, K., Trudel, S., Hassan, J.A., Morales, C.R., Pshezhetsky, A.V.: Neu4, a novel human lysosomal lumen sialidase, confers normal phenotype to sialidosis and galactosialidosis cells. J. Biol. Chem. 279, 37021–37029 (2004)
Lozzio, C.B., Lozzio, B.B.: Human chronic myelogenous leukemia cell-line with positive Philadelphia chromosome. Blood 45, 321–334 (1975)
Gahmberg, C.G., Jokinen, M., Andersson, L.C.: Expression of the major red cell sialoglycoprotein, glycophorin A, in the human leukemic cell line K562. J. Biol. Chem. 254, 7442–7448 (1979)
Villeval, J.L., Pelicci, P.G., Tabilio, A., Titeux, M., Henri, A., Houesche, F., Thomopoulos, P., Vainchenker, W., Garbaz, M., Rochant, H., Breton-Gorius, J., Edwards, P.A., Testa, U.: Erythroid properties of K562 cells. Effect of hemin, butyrate and TPA induction. Exp. Cell. Res. 146, 428–435 (1983)
Fukuda, M.: K562 human leukaemic cells express fetal type (i) antigen on different glycoproteins from circulating erythrocytes. Nature 285, 405–457 (1980)
Lozzio, C.B., Lozzio, B.B., Machado, E.A., Fuhr, J.E., Lair, S.V., Bamberger, E.G.: Effects of sodium butyrate on human chronic myelogenous leukaemia cell line K562. Nature 281, 709–710 (1979)
Testa, U.: Apoptotic mechanisms in the control of erythropoiesis. Leukemia 18, 1176–1199 (2004)
Carter, H.E., Rothfus, J.A., Gigg, R.H.: Biochemistry of the sphingolipids: XII. Conversion of cerebrosides to ceramides and sphingosine; structure of Gaucher cerebroside. J. Lipid Res. 228–234 (1961)
Toyokuni, T., Nisar, M., Dean, B., Hakomori, S.: A facile and regiospecific titration of sphingosine: synthesis of (2S,3R,4E)-2-amino-4-octadecene-1,3-diol-1-3H. J. Labelled Compd. Radiopharm. 29, 567–574 (1991)
Tettamanti, G., Bonali, F., Marchesini, S., Zambotti, V.: A new procedure for the extraction, purification and fractionation of brain gangliosides. Biochim. Biophys. Acta 296, 160–170 (1973)
Sonnino, S., Ghidoni, R., Gazzotti, G., Kirschner, G., Galli, G., Tettamanti, G.: High performance liquid chromatography preparation of the molecular species of GM1 and GD1a gangliosides with homogeneous long chain base composition. J Lipid Res. 25, 620–629 (1984)
Sonnino, S., Nicolini, M., Chigorno, V.: Preparation of radiolabeled gangliosides. Glycobiology 6, 479–487 (1996)
Sambrook, J., Russell, D.: Molecular Cloning: A Laboratory Manual, 3rd edn, pp. 2344. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY (2001)
Fibach, E., Rachmilewitz, E.A.: The two-step liquid culture: a novel procedure for studying maturation of human normal and pathological erythroid precursors. Stem Cells 11(Suppl 1), 36–41 (1993)
Hasegawa, T., Feijoo Carnero, C., Wada, T., Itoyama, Y., Miyagi, T.: Differential expression of three sialidase genes in rat development. Biochem. Biophys. Res. Commun. 280, 726–32 (2001)
Zhu, J., Li, Y.T., Li, S.C., Cole R.B.: Structural characterization of gangliosides isolated from mullet milt using electrospray ionization-tandem mass spectrometry. Glycobiology 9, 985–993 (1999)
Caimi, L., Lombardo, A., Preti, A., Wiesmann, U., Tettamanti, G.: Optimal conditions for the assay of fibroblast neuraminidase with different natural substrates. Biochim. Biophys. Acta 571, 137–146 (1979)
Chigorno, V., Riva, C., Valsecchi, M., Nicolini, M., Brocca, P., Sonnino, S.: Metabolic processing of gangliosides by human fibroblasts in culture—formation and recycling of separate pools of sphingosine. Eur. J. Biochem. 250, 661–669 (1997)
Watanabe, K., Powell, M.E., Hakomori, S.I.: Isolation and characterization of gangliosides with a new sialosyl linkage and core structures. II. Gangliosides of human erythrocyte membranes. J. Biol. Chem. 254, 8223–8229 (1979)
Hakomori, S.: Glycosylation defining cancer malignancy: new wine in an old bottle. Proc. Natl. Acad. Sci. U. S. A. 99, 10231–10233 (2002)
Spira, A.I., Carducci. M.A.: Differentiation therapy. Curr. Opin. Pharmacol. 3, 338–343 (2003)
Freemantle, S.J., Spinella, M.J., Dmitrovsky, E.: Retinoids in cancer therapy and chemoprevention: promise meets resistance. Oncogene 22, 7305–7315 (2003)
Moqattash, S., Lutton, J.D.: Leukemia cells and the cytokine network: therapeutic prospects. Exp. Biol. Med. (Maywood) 229, 121–137 (2004)
Wong, C.F., Guminski, A., Saunders, N.A., Burgess, A.J.: Exploiting novel cell cycle targets in the development of anticancer agents. Curr. Cancer Drug Targets 5, 85–102 (2005)
Kakugawa, Y., Wada, T., Yamaguchi, K., Yamanami, H., Ouchi, K., Sato, I., Miyagi, T.: Up-regulation of plasma membrane-associated ganglioside sialidase (Neu3) in human colon cancer and its involvement in apoptosis suppression. Proc. Natl. Acad. Sci. U. S. A. 99, 10718–10723 (2002)
Kannagi, R., Papayannopoulou, T., Nakamoto, B., Cochran, N.A., Yokochi, T., Stamatoyannopoulos, G., Hakomori, S.: Carbohydrate antigen profiles of human erythroleukemia cell lines HEL and K562. Blood 62, 1230–1241 (1983)
Hata, K., Wada, T., Hasegawa, A., Kiso, M., Miyagi, T.: Purification and characterization of a membrane-associated ganglioside sialidase from bovine brain. J. Biochem. (Tokyo) 123, 899–905 (1998)
Mandal, C., Chatterjee, M., Sinha, D.: Investigation of 9-O-acetylated sialoglycoconjugates in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 110, 801–812 (2000)
Hakomori, S.: Inaugural article: the glycosynapse. Proc. Natl. Acad. Sci. U. S. A. 99, 225–232 (2002)
Kato, T., Wang, Y., Yamaguchi, K., Milner, C.M., Shineha, R., Satomi, S., Miyagi, T.L: Overexpression of lysosomal-type sialidase leads to suppression of metastasis associated with reversion of malignant phenotype in murine B16 melanoma cells. Int. J. Cancer 92, 797–804 (2001)
Author information
Authors and Affiliations
Corresponding author
Additional information
The ganglioside nomenclature proposed by Svennerholm L. (1980) Adv. Exp. Mod. Biol. 125, 11.
Rights and permissions
About this article
Cite this article
Tringali, C., Anastasia, L., Papini, N. et al. Modification of sialidase levels and sialoglycoconjugate pattern during erythroid and erytroleukemic cell differentiation. Glycoconj J 24, 67–79 (2007). https://doi.org/10.1007/s10719-006-9013-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10719-006-9013-0