
Abstract
Human activities have caused worldwide loss and fragmentation of natural habitats, resulting in the decline and isolation of wild populations, consequently increasing their risks of extinctions. We investigated the genetic consequences of anthropogenic effects on the Sichuan snub-nosed monkeys (Rhinopithecus roxellana) in the Shennongjia Nature Reserve (SNR), which is a small and isolated distribution of R. roxellana in China and would continue to be threatened by habitat degradation and loss, using extensive sampling and 16 microsatellite loci. High level of genetic variation was observed from 202 individuals collected from three R. roxellana populations (SNR population, Sichuan-Gansu population and Shaanxi population). However, R. roxellana in SNR showed the lowest genetic diversity. The likelihood analysis of migration/drift equilibrium indicated that the SNR population suffered much stronger effect of drift than the other two populations, indicating that small populations are prone to be affected by drift. The STRUCTURE analysis identified two clusters, separating the SNR population from the other two populations, suggesting an increasing drift-induced differentiation between SNR and the other two populations. Bottleneck tests revealed that R. roxellana in SNR experienced a severe population decline (37-fold) during the past 500 years as a consequence of human population expansion. The current effective population size (Ne) in SNR is less than 100 and the ratio of Ne to the census population size is approximately 0.08. Based on our findings, we suggest that the SNR population should be monitored systematically and considered as an important conservation and management unit.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Habitat loss and fragmentation as a result of human activities have been regarded as major threats in the long-term survival of species, as they can result in small and isolated populations. Isolation can accelerate loss of genetic diversity within, and differentiation among populations through restricted gene flow, increase the levels of genetic drift and inbreeding in small and isolated habitat remnants (Frankham et al. 2002). Lower genetic diversity and inbreeding will decrease the potential of small populations to respond to environmental changes, leading to an increased risk of extinction by demographic or/and environmental stochasticity (Burkey 1995; Willi et al. 2006). If a small and isolated population that cannot shift geographic range is to persist, it must adapt to a changing environment (Miller and Waits 2003). The extent to which it can adapt is directly proportional to its genetic variability (Lande and Shannon 1996).
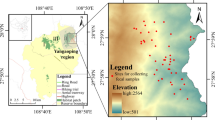
The Sichuan snub-nosed monkey (Rhinopithecus roxellana) is an arboreal primate endemic to China. Despite a wide distribution in China during the Pleistocene (Jablonski 1998), wild R. roxellana populations now only occur in three isolated mountainous regions at elevations ≥1,400–3,000 m, including the provinces of Sichuan, Gansu, Shaanxi, and Hubei (Fig. 1). The current census population size is ca. 22,000, with ca. 16,000 individuals in Sichuan-Gansu (SG) population, ca. 5,500 individuals in Shaanxi (SX) population, and ca. 1,000 individuals in the Shennongjia Nature Reserve (SNR), Hubei province (Zhang et al. 2002). The SNR is the easternmost distribution of R. roxellana in China. Because of large-scale commercial timber exploitation between the 1950s and 2000, the habitats of R. roxellana in SNR became fragmented, and illegal hunting led to the capture of at least 130 monkeys in the 1970s and 1980s (Zhu 2003). In SNR, the population size was estimated to be more than 2,000 individuals prior to 1970 (Quan and Xie 2002). However, the population had dropped to an estimated 500 individuals in 1989 (Zhu 2003).

Location of the Shennongjia Nature Reserve (SNR). Shaded areas indicate approximate distributions of R. roxellana in the populations of Sichuan-Gansu (SG), Shaanxi (SX), and Hubei (SNR)
Effective conservation management of threatened populations needs to incorporate the current pattern of genetic diversity, the knowledge of the demographic history and their present status of genetic structure (Zhang et al. 2007). Several studies investigated the genetic diversity and population history of R. roxellana using the mtDNA control region and indicated a low level of genetic diversity and no evidence of bottleneck for the SNR population (Li et al. 2007a, b; Luo et al. 2012). However, very few samples from the SNR population were used (Li et al. 2007a, b; Luo et al. 2012; Pan et al. 2009). Moreover, the mtDNA genome behaves as a single genetic locus, which thus traces the outcome of a single coalescent process (Rosenberg and Nordborg 2002). More reliable inferences of population history are expected using multiple independent genetic markers (Lucchini et al. 2004). Therefore, a more comprehensive population genetic analysis is required to infer more accurately the population history and to document relative amount of genetic diversity of R. roxellana in SNR.
In this study, we applied a Bayesian coalescent-based method using the information from the full alleletic distributions (Storz and Beaumont 2002) to infer the extent and timing of historical demographic changes of R. roxellana in SNR, using extensive field sampling and 16 microsatellite loci. We specifically attempt to (i) investigate the current level of genetic diversity of the SNR population and compare it with that in other populations; (ii) identify the extent of the SNR population isolation; (iii) to elucidate the patterns of demographic change and possible causes. Our findings will provide a baseline for continued monitoring of genetic diversity of R. roxellana in SNR in general and in the conservation and management unit in particular.
Materials and methods
Sample collection, DNA extraction and amplification
Research was conducted in the Shennongjia Nature Reserve (31°21’20’’–31º36’20’’N, 110º03’05’’–110º33’50’’E) in Hubei province, China (Fig. 1). The sampling strategy was as described in Chang et al. (2012). In total, we collected 337 fecal samples from four groups using the two-step ethanol-silica procedure (Nsubuga et al. 2004) and stored at room temperature. The geographic coordinates of each group sampled were recorded using a GPS.
To compare with the other two populations, we also collected the samples from the populations of SG and SX. Samples of these two populations were mainly obtained from zoos and field, including five dried skins, 12 blood samples and 32 tissue samples. Skin, blood and tissue samples were collected following a protocol approved by Institute of Zoology, Chinese Academy of Sciences Institutional Animal Care Committee and State Forestry Administration of China. Samples obtained from zoos were traced to their source populations according to archival records. The fecal samples and all the other samples were confirmed as different individuals by microsatellite-based individual identification as followed shown. In sum, 202 individuals were analysed in this study, comprising three geographical populations: SG population (n = 23), SX population (n = 26), and SNR population (n = 153).
Genomic DNA was extracted from blood, tissue and skin using the SDS-phenol/chloroform method (Sambrook et al. 1989), from fecal samples using the QIAmp Stool Kit (QIAGEN). Two negative extraction controls were processed along with genomic DNA extractions.
Three independent amplifications from each DNA extract were performed for 16 loci (Chang et al. 2012) together with a minimum of six negative controls (in which 5 μL H2O rather than DNA was added to the well). A two-step multiplex PCR method was used as described by Arandjelovic et al. (2009). Multiplex PCRs were conducted in a 20 μL reaction volume containing 1× SuperTaq buffer (HT Biotechnology), 1.75 mM MgCl2, 0.15 mM of each forward (unlabelled) and reverse (unlabelled) primer, 110 μM of each dNTP, 16 μg bovine serum albumin, 0.5 U SuperTaq (HT Biotechnology) premixed 2:1 with TaqStart Antibody (BD Biosciences), and 5 μL template DNA. The PCR protocol was as follows: initial denaturation for 9 min at 94 °C, 30 cycles of 20 s at 94 °C, 30 s at 55 °C, and 30 s at 72 °C, and a final extension of 5 min at 72 °C. Singleplex PCRs were carried out as above but with the following modifications: 5 μL of 1:100 diluted multiplex PCR product was used as template, half the amount of MgCl2 (0.875 mM) was added, and only 0.35 U of SuperTaq (HT Biotechnology) premixed 2:1 with TaqStart Antibody was used. Moreover, each singleplex PCR only contained a single primer pair: 0.25 mM of a FAM, HEX, or NED fluorescently labeled forward primer and 0.25 mM of a reverse primer. To guard against contamination, all steps of the PCR set-up for DNA samples (except the addition of template) were performed under a hood that was irradiated with ultraviolet before and after use. Up to three different PCR products were combined and electrophoresed on an ABI PRISM 3100 Genetic Analyser and allele size was determined relative to an internal size standard (ROX-labeled HD400) using GeneMapper version 3.7 (Applied Biosystems). Genotypes were confirmed by observation of each allele twice in two or more independent reactions for heterozygote genotypes while homozygous genotypes were ascertained by up to five independent observations depending on the quality of DNA in the extract. For the final result of individual identification, we utilized MICRO-CHECKER to check possible genotyping errors due to stuttering, short allele dominance, and null alleles, running 1,000 Monte Carlo simulations and calculating 95 % confidence intervals (Van Oosterhout et al. 2004).
Genetic diversity
Genetic diversity was assessed as the number of alleles per locus (A), observed (H O) and expected (H E) heterozygosity which were implemented in MS-tools add-in to Excel (Park 2001). To account for differences in sample sizes among populations, allelic richness (AR) for each population and for all populations was calculated using FSTAT (version 2.9.3.2) (Goudet 2001). Differences in AR, H E and H O between populations were tested with Wilcoxon signed-ranked test. Deviations from Hardy-Weinberg equilibrium across all loci for each population were assessed using an exact probability test implemented in GENEPOP 4.0 (Rousset 2008). Linkage disequilibrium between loci was tested for each population performed in FSTAT. Significance values were adjusted by Bonferroni correction for multiple testing, as implemented in the software. We also used the FSTAT to calculate the inbreeding coefficient (FIS) as defined by Weir and Cockerham (1984).
Analysis of population structure
To assess levels of genetic differentiation among populations, we used FSTAT to estimate FST across all loci for each pairwise comparison of populations. To test whether the values were significantly different from zero, we applied the permutation procedure implemented in FSTAT, without assuming Hardy-Weinberg equilibrium. For the sake of comparison, we also calculated overall and pairwise Dest values across all loci with SMOGD 1.2.5 (Crawford 2009). This estimate is increasingly considered more accurate than traditional ones such as FST when using highly polymorphic microsatellite markers.
A Bayesian approach implemented in STRUCTURE 2.3.3 was used to identify the number of distinct genetic cluster (K) among populations. We assumed the model with population admixture and correlated allele frequencies (Pritchard et al. 2000). We conducted 10 independent runs for each assumed number of cluster K between 1 and 7. We used the burn-in of 200,000 iterations and collected data for 1,000,000 iterations. We took two approaches to choose the best K. First, we calculated the most likely number of K using the maximum average log likelihood Ln P(X|K). We also determined the optimal value of K by calculating ∆K (Evanno et al. 2005), a measure of second-order rate of change in the likelihood of K. To visualize the distribution of genetic variation across individuals, we performed a factorial correspondence analysis (FCA) in Genetix 4.05 (Belkhir et al. 1996–2004).
Changes of population size
Two different but complementary approaches were used to detect whether R. roxellana in SNR experienced a recent population bottleneck. First, a heterozygosity excess test (Cornuet and Luikart 1996) was performed under the two-phased mutation model, as microsatellite loci are unlikely to strictly follow either the infinite allele model or stepwise mutation model (Di Rienzo et al. 1994; Piry et al. 1999). We conducted analyses using 10, 20 and 30 % multistep mutations and a variance of 12 for the multistep mutations. Significance of heterozygosity excess based on 10,000 iterations of simulation was tested with a Wilcoxon signed rank test. This approach was applied to each population and implemented in the BOTTLENECK version 1.2 (Cornuet and Luikart 1996).
To estimate the parameters of current population size (N0), ancient population size (N1), and T, the time since the population size changed, we applied a Bayesian method which uses the information from the full allelic distributions (Storz and Beaumont 2002). The Storz and Beaumont method uses a Markov Chain Monte Carlo (MCMC) approach to estimate the posterior probability distributions of N0, N1, and T. This method is implemented in the program MSVAR 1.3, assuming a strict single-step mutation model, with a rate θ = 2N0μ, where μ is the locus mutation rate. Prior distributions for N0, N1, T, and θ, are assumed to be log-normal. The means and standard deviations of these prior log-normal distributions are themselves drawn from prior (or hyperprior) distributions. Change in population size was evaluated using exponential model. The generation time of R. roxellana was set to 5 years (Li et al. 2003; Pan et al. 2009). Different runs were performed by allowing N1 to be either larger than or equal to N0, which did not affect the posterior distribution. Similar to Goossens et al. (2006), runs with the same priors (assuming that N1 and N0 were of the same size) were repeated five times as a test for convergence. The total number of iterations was 2 × 109 and thinning value was 2 × 104. The first 10 % of the output was discarded to alleviate possible bias from the starting value. The Bayesian method is highly computer intensive and thus was applied to one subset of the SNR population (DW group: n = 26), as did by Goossens et al. (2006).
Estimate of current effective population size (Ne)
The program LDNe (Waples and Do 2008) was used to estimate Ne of R. roxellana in SNR from genotypic data based on the linkage disequilibrium method. We used the jackknife method and assumed a random mating model. The program calculates separate estimates after excluding all alleles with frequencies less than user-specified critical values. We tested the following critical values (Pcrit): 0.05; 0.02; 0.01. In addition, Ne was also estimated with the program ONeSAMP 1.2 (Tallmon et al. 2008), using summary statistics and approximate Bayesian computation to estimate Ne. The upper and lower bounds of the prior distribution for Ne were 2 and 200, respectively. Priors for Ne of 2–100 and 2–300 were also tested to verify if the results were robust to changes in these assumed values.
Tests of population history
The program 2mod updated version (Ciofi et al. 1999) was used to test whether the R. roxellana populations had attained migration-drift equilibrium. Deviation from migration/drift equilibrium was tested by comparing the relative likelihoods of ‘gene flow’ and ‘drift only’ models, using a MCMC procedure with 6 × 105 iterations and a burn-in of 6 × 104. The gene flow model assumes that the gene frequencies within islands are determined by a balance between genetic drift and immigration. The drift only model assumes that an ancestral panmictic population separated into several independent units which start diverging purely by genetic drift. Both models assume that the effects of microsatellite mutations are negligible and calculate a parameter F, the probability that two alleles in a given population share a common ancestor. Data were analysed under both models, and Bayes factor was calculated to infer decisive power estimate of the most likely model (Jeffreys 1961). We also reported the mode and 90 % Highest Posterior Density limits for the posterior distribution of F for each population.
Results
Genetic variation
Of the 337 fecal samples collected from SNR, 287 fecal samples produced sufficiant information containing more than 10 loci. Among these 287 fecal samples, 153 unique individuals from four groups were identified and constituted approximately one sixth of the total estimated population (Chang et al. 2012). Also, 49 samples collected from SG and SX were successfully genotyped at more than 11 loci. MICRO-CHECKER analysis provided no evidence of genotyping errors because of stuttering or large allele dropout in any of the loci for each population, with the exception of D13S321 in SNR, which showed scoring errors caused by stuttering. We thus excluded this locus in the subsequent analysis. Test of Hardy-Weinberg equilibrium showed that the SX population deviated significantly from equilibrium. After applying Bonferroni correction, no linkage disequilibrium was seen for any pair of loci in any population. The 16 microsatellite loci used in this study are polymorphic (Table 1 and Supplementary materials Table S1). When all samples from three populations were pooled, R. roxellana showed a mean H O and H E of 0.613 and 0.669, respectively (Table 1). The mean number of alleles per locus (MNA) ranged from 4.50 (SNR) to 6.38 (SX), with an overall average of 7.44 (Table 1). The values of H E, H O, and AR in SNR were lowest among the three populations, ranging from 0.589 (SNR) to 0.727 (SG) for H E, from 0.591 (SNR) to 0.720 (SG) for H O, and from 3.951 (SNR) to 6.240 (SX) for AR, respectively. Moreover, AR, H O and H E in SNR significantly differ from those in either SG or SX (Wilcoxon signed rank test, all P < 0.05), while these values were not significantly different between SG and SX (Wilcoxon signed rank test, all P > 0.05).
Population structure
Genetic differentiation between populations were relatively high, ranging between 0.109 and 0.177. Samples from different populations were strongly differentiated (P < 0.05) (Table 2). Dest estimates were markedly high in R. roxellana populations, ranging from 0.178 to 0.328, with an overall Dest value of 0.298 (Table 2). The STRUCTURE analysis showed a peak in ∆K at K = 2, corresponding to two clusters, which separated the SNR populaton from the populations of SG and SX (Fig. 2a, b). To detect lower level structure, we repeated the analysis within the cluster of SG and SX. In this case, the ∆K also exhibited a peak at K = 2 (Fig. 2c), indicated a substructure within this cluster. This conclusion was also suggested by the FCA analysis where axis 1 explained most of the variance and clearly differentiated SNR from SG and SX (Fig. 2d).

a The optimal value of K simulated by STRUCTURE; b STRUCTURE output of two genetic clusters identified (K = 2); c STRUCTURE output of cluster obtaining SG and SX; d Result of the factorial correspondence analysis (FCA), showing the first two axes. SNR, SG, and SX are depicted in white, yellow and blue, respectively
Changes of population size
The heterozygosity excess test showed a significant signal of population bottleneck of R. roxellana in SNR as well as in SG and SX (Table 3). The Storz and Beaumont method showed that the posterior distributions for log(N0) and log(N1) had limited overlap with medians of approximately 100 and approximately 3,700, for N0 and N1, respectively (Fig. 3), confirming the result of BOTTLENECK. The Storz and Beaumont method also revealed that the population decline of R. roxellana in SNR was very likely commence 500 years before present (BP) (Fig. 4; 10, 50 and 90 % of the distribution mass were approximately 30, 500, 5,000 years, respectively). Multiple independent runs produced similar results regardless of the prior distributions (stable or decreased population size), which had very limited effect on the posterior distribution of population demographic parameters (Supplementary materials Fig. S1 and S2).

Ancestral and present population size of R. roxellana in SNR. The posterior distributions are represented in a logarithmic scale. N0: present population size, N1: ancestral population size

Time since population size decrease (T) of R. roxellana in SNR. The posterior distribution for the time since the population decrease is represented on a logarithmic scale. These distributions have a median around 500 years. 10, 50 and 90 % of the distribution mass are approximately 30, 500, 5,000 years, respectively
Current effective population size
Separate estimates using different criteria in LDNe produced similar results (data not shown). Also, the results obtained from ONeSAMP were robust to changes in the prior (data not shown). Both LDNe and ONeSAMP methods provided similar estimates and showed that the current Ne of R. roxellana in SNR is less than 100 individuals and approximately 80 individuals (Table 4). Considering the census size of SNR population (N) is ca. 1,000, the ratio of Ne/N is approximately 0.08.
Tests of population history
Between the two models in the 2mod program, the likelihood of the gene flow model was 39.7 times higher than that of the drift only model (P(gene flow model) = 0.975; Bayes factor = 39.7). This indicates that R. roxellana populations have been at migration-drift equilibrium for a substantial period of time. Nonetheless, the values of F varied across populations suggesting the effects of drift were more stronger in some populations than others. Among the three populations, SG and SX were the two populations least affected by drift effect (F = 0.10, F = 0.06, respectively), while SNR was the most affected (F = 0.30) (Table 1). These results of 2mod also supported the idea that smaller populations had been more strongly influenced by drift.
Discussion
Habitat fragmentation can have important consequences for population genetic diversity and structure due to the effects of genetic drift and reduced gene flow (Martinez-Cruz et al. 2007). Understanding the genetic consequences of habitat fragmentation has became an important theme for conservation biologists and wildlife managers (Keyghobadi 2007). Our analysis suggests that habitat destruction and population decline due to anthropogenic effects have resulted in significant loss of genetic diversity of R. roxellana in SNR. Meanwhile, the STRUCTURE and 2mod analysis indicated that habitat fragmentation led to an increasing drift-induced differentiation of R. roxellana in SNR.
Genetic diversity of R. roxellana has been investigated using different genetic markers (Protein, mtDNA) (Li et al. 2001, 2003, 2007a, b; Pan et al. 2009). Protein and cytochrome-b gene analysis suggested a very low level of genetic diversity in R. roxellana (Li et al. 2001, 2003). However, analysis of mtDNA control region indicated that R. roxellana should not be regarded as a taxon with low genetic diversity (Li et al. 2007a, b; Pan et al. 2009). Our assessment based on microsatellite data also revealed a relatively high level of genetic diversity in R. roxellana. At the species level, the genetic diversity in R. roxellana is slightly higher than or similar to that of giant panda (Ailuropoda melanoleuca, MNA = 7.1, H E = 0.642; Zhang et al. 2007) and Yunnan snub-nosed monkey (Rhinopithecus bieti, MNA = 7.5, H E = 0.703; Liu et al. 2009). However, genetic variation was highly variable among the three R. roxellana populations. Analysis of mtDNA and microsatellite data consistently showed that the SNR population had the lowest level of genetic diversity among the three populations (Table 1; Li et al. 2007a, b; Luo et al. 2012; Pan et al. 2009). The level of AR and H E in SNR were significantly lower than those in SX and SG. Comparisons of nucleotide diversity at mtDNA [0.0034 (SNR) vs.0.021(SX) vs.0.039 (SG)] were consistent but more extreme (unpublished data). Although R. roxellana populations had been at migration-drift equilibrium, the impact of genetic drift on the SNR population was much more stronger than the other two populations, resulting in a large amount of loss of genetic variability in SNR. Moreover, small population size because of demographic bottleneck further accelerated the loss of genetic variability of R. roxellana in SNR. Although all of the three populations experienced demographic bottleneck, the SNR population sufferred much more serious influence of reduction in Ne than either SX or SG, because in natural populations, genetic variation is related to population size (Frankham 1996).
Previous studies indicated that R. roxellana may have experienced a bottleneck during the Late Pleistocene based on evidences of dental morphology (Jablonski 1992) and allozyme analysis (Li et al. 2003). However, our results indicated that the Pleistocene climate change should not be responsible for the population decline of R. roxellana in SNR, even the prehistoric human activity could not reasonably explain the detected genetic signature of population decrease, because dates older than 10,000 years had a posterior distribution support of only 4.13 %. The most possible explantation for the population decline is the recent anthropogenic exploitation of habitat, because the posterior distribution of population decline of R. roxellana overlapped with human population expansions. Li et al. (2002) pointed that the human population in China began to explode and caused a rapid increase in cultivated lands during the past 400 years. As a consequence, R. roxellana may have been forced to reduce their use of space to a small core area, utilize higher elevations, and inhabit more remote areas. Such anthropogenic pressure on R. roxellana sharply increased during the past 200 years, and many its populations became extinct in the plains and in some mountainous regions after the 1800s (Li et al. 2002). However, our results revealed that the population decline of R. roxellana in SNR very likely commenced 500 years BP. Thus, we speculated that the time of strong anthropogenic effects on R. roxellana in SNR may be more earlier than the estimate by Li et al. (2002). Similar to R. roxellana, many other endangered wildlife (such as giant panda, red panda, et al) in China had also been impacted by strong anthropogenic factors during the past several centuries. The giant panda (Ailuropoda melanoleuca) population had experienced a strong demographic reduction starting approximately 250 years BP because of explosion of the human population and subsequent deforestation (Zhu et al. 2010), which was also detected in the red panda (Ailurus fulgens) sympatric with A. melanoleuca (Hu et al. 2011). Li et al. (2007a, b) found that the population size of François’ langurs (Trachypithecus francoisi) in Guangxi had experienced a 90 % decrease since the early 1980s and suggested that the primary threat to the T. francoisi was hunting, which resulted in a relatively low genetic diversity (Sun et al. 2010). In contrast, proboscis monkeys (Nasalis larvatus) in Malaysia still retained substantial genetic variation (haplotype diversity = 0.90; nucleotide diversity = 0.022) although the population size had experienced dramatically reduction during the past decades (Bennett 1988; Munshi-South and Bernard 2011).
MtDNA analysis suggested that the substantial historical gene flow existed from SX to SNR and between SG and SX (Luo et al. 2012). In this study, significant genetic differentiation among populations was detected in microsatellite data, indicating a recent subdivision within R. roxellana population. The analysis of STRUCTURE showed that the SNR population was separated from the other two populations and even genetic differentiation between SX and SG was also significant, suggesting a more isolated status for the SNR population. Small and isolated populations are vulnerable to extinction through stochastic processes due to loss of genetic diversity and inbreeding. Studies of wild (Madsen et al. 1999; Saccheri et al. 1998) and laboratory (Bijlsma et al. 2000; Reed and Bryant 2000) animals have indicated that a minimum Ne > 50 is needed to avoid the negative short-term effects of inbreeding. To maintain long-term evolutionary potential in the form of additive genetic variance, Ne should be at least 500–5,000 (Franklin and Frankham 1998; Lynch and Lande 1998). However, Ne in SNR is only about 80, suggesting that genetic diversity will decline in such an isolated population with a Ne less than 100.
Implications for conservation
As an object of public interest and national pride, the Sichuan snub-nosed monkey has been the focus of conservation biologists. Although the size of SNR R. roxellana population has doubled during the past 20 years (Chang et al. 2012), the outlook of this population is still worrisome because of its low genetic diversity and small effective population size. In the long-term, genetic variability can play a decisive role in allowing a population to persist and adapt in a changing environment (Lande and Shannon 1996). The very low genetic diversity of R. roxellana in SNR will reduce its ability to respond to extreme environmental changes. Genetic diversity can be maintained over longtime spans only through gene flow. Meanwhile, local extinctions and population declines can also be offset by gene flow and connectivity through dispersal between populations (Lawton et al. 2011). However, considering the current habitat conditions of R. roxellana, it is impossible that gene flow will occur naturally within several decades among populations. Thus, it is possible that genetic factors will have a substantial effect on the viability of R. roxellana in SNR over the next several decades. In addition, demographic and environmental effects may push such small populations to extinction even before genetic factors. Therefore, we suggest that translocations of monkeys into SNR population should be considered in the near future. However, compared to low genetic diversity, the more immediate threats to R. roxellana in SNR (also other R. roxellana populations) are habitat destruction and loss. Sustainable development should be planned to reduce the pressure of human activities on the habitats of monkeys. We also argue that conservation management should focus on keeping the SNR population at or above its current size and enlarging the area of the reserve if possible.
References
Arandjelovic M, Guschanski K, Schubert G et al (2009) Two-step multiplex polymerase chain reaction improves the speed and accuracy of genotyping using DNA from noninvasive and museum samples. Mol Ecol Resour 9:28–36
Belkhir K, Borsa P, Chikhi L, et al (1996–2004) Genetix 4.05, logiciel sous Windows TM pour la génétique des populations. Laboratoire Génome, Populations, Interactions, CNRS UMR 5171, Université de Montpellier II, Montpellier, France
Bennett EL (1988) Proboscis monkeys and their swamp forests in Sarawak. Oryx 22:69–74
Bijlsma R, Bundgaard J, Boerema AC (2000) Does inbreeding affect the extinction risk of small populations?: predictions from Drosophila. J Evol Biol 13:502–514
Burkey TV (1995) Extinction rates in archipelagos: implications for populations in fragmented habitats. Conserv Biol 9:527–541
Chang ZF, Liu ZJ, Yang JY, Li M, Vigilant L (2012) Noninvasive genetic assessment of the population trend and sex ratio of the Shennongjia population of Sichuan snub-nosed monkeys (Rhinopithecus roxellana). Chin Sci Bull 57:1135–1141
Ciofi C, Beaumont MA, Swingland IR et al (1999) Genetic divergence and units for conservation in the Komodo dragon Varanus komodoensis. Proc R Soc B-Biol Sci 266:2269–2274
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Crawford NG (2009) SMOGD: software for the measurement of genetic diversity. Mol Ecol Resour 10:556–557
Di Rienzo A, Peterson AC, Garza JC et al (1994) Mutational processes of simple-sequence repeat loci in human populations. Proc Natl Acad Sci USA 91:3166–3170
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Frankham R (1996) Relationship of genetic variation to population size in wildlife. Conserv Biol 10:1500–1508
Frankham R, Ballou JD, Briscoe DA (2002) Introduction to conservation genetics. Cambridge University Press, Cambridge
Franklin IR, Frankham R (1998) How large must populations be to retain evolutionary potential. Anim Conserv 1:69–70
Goossens B, Chikhi L, Ancrenaz M et al (2006) Genetic signature of anthropogenic population collapse in Orang-utans. PLoS Biol 4:285–291
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). Available from http://www.unil.ch/izea/softwares/fstat.html
Hu YB, Guo Y, Qi DW et al (2011) Genetic structuring and recent demographic history of red pandas (Ailurus fulgens) inferred from microsatellite and mitochondrial DNA. Mol Ecol 20:2662–2675
Jablonski NG (1992) Dental agenesis as evidence of possible genetic isolation in the Colobine monkey, Rhinopithecus roxellana. Primates 33:371–376
Jablonski NG (1998) The response of Catarrhine primates to Pleistocene environmental fluctuations in east Asia. Primates 39:29–37
Jeffreys H (1961) Theory of probability third edition. Oxford classic texts in the physical sciences. Oxford University Press, Oxford
Keyghobadi N (2007) The genetic implications of habitat fragmentation for animals. Can J Zool 85:1049–1064
Lande R, Shannon S (1996) The role of genetic variation in adaptation and population persistence in a changing environment. Evolution 50:434–437
Lawton RJ, Messmer V, Pratchett MS et al (2011) High gene flow across large geographic scales reduces extinction risk for a highly specialised coral feeding butterflyfish. Mol Ecol 20:3584–3598
Li M, Liang B, Feng ZJ et al (2001) Molecular phylogenetic relationships among Sichuan snub-nosed monkeys (Rhinopithecus roxellanae) inferred from mitochondrial cytochrome-b gene sequences. Primates 42:153–160
Li BG, Pan RL, Oxnard CE (2002) Extinction of snub-nosed monkeys in China during the past 400 years. Int J Primatol 23:1127–1244
Li HP, Meng SJ, Men ZM et al (2003) Genetic diversity and population history of golden monkeys (Rhinopithecus roxellana). Genetics 164:269–275
Li M, Liu ZJ, Gou JX et al (2007a) Phylogeography and population structure of the golden monkeys (Rhinopithecus roxellana): inferred from mitochondrial DNA sequences. Am J Primatol 69:1195–1209
Li YB, Huang CM, Ding P, Tang Z, Wood C (2007b) Dramatic decline of François’ langur Trachypithecus francoisi in Guangxi Province, China. Oryx 41:38–43
Liu ZJ, Ren BP, Wu RD et al (2009) The effect of landscape features on population genetic structure in Yunnan snub-nosed monkeys (Rhinopithecus bieti) implies an anthropogenic genetic discontinuity. Mol Ecol 18:3831–3846
Lucchini V, Galov A, Randi E (2004) Evidence of genetic distinction and long-term population decline in wolves (Canis lupus) in the Italian Apennines. Mol Ecol 13:523–536
Luo MF, Liu ZJ, Pan HJ et al (2012) Historical geographic dispersal of the golden snub-nosed monkey (Rhinopithecus roxellana) and the influence of climatic oscillations. Am J Primatol 74:91–101
Lynch M, Lande R (1998) The critical effective size for a genetically secure population. Anim Conserv 1:70–72
Madsen T, Shine R, Olsson M et al (1999) Conservation biology: restoration of an inbred adder population. Nature 402:34–35
Martinez-Cruz B, Godoy JA, Negro JJ (2007) Population fragmentation leads to spatial and temporal genetic structure in the endangered Spanish imperial eagle. Mol Ecol 16:477–486
Miller CR, Waits LP (2003) The history of effective population size and genetic diversity in the Yellowstone grizzly (Ursus arctos): implications for conservation. Proc Natl Acad Sci USA 100:4334–4339
Munshi-South J, Bernard H (2011) Genetic diversity and distinctiveness of the proboscis monkeys (Nasalis larvatus) of the Klias peninsula, Sabah, Malaysia. J Hered 102:342–346
Nsubuga AM, Robbins MM, Roeder AD et al (2004) Factors affecting the amount of genomic DNA extracted from ape feces and the identification of an improved sample storage method. Mol Ecol 13:2089–2094
Pan D, Hu HX, Meng SJ et al (2009) A high polymorphism level in Rhinopithecus roxellana. Int J Primatol 30:337–351
Park SDE (2001) Trypanotolerance in West African Cattle and the population genetic effects of selection [Ph.D. thesis] University of Dublin
Piry S, Luikart G, Cornuet JM (1999) BOTTLENECK: a computer program for detecting recent reductions in effective population size from allele frequency data. J Hered 90:502–503
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Quan GQ, Xie JH (2002) Research on the golden monkeys. Science and Education Publishing House, Beijing
Reed DH, Bryant EH (2000) Experimental tests of minimum viable population size. Anim Conserv 3:7–14
Rosenberg NA, Nordborg M (2002) Genealogical trees, coalescent theory and the analysis of genetic polymorphism. Nat Rev Genet 3:380–390
Rousset F (2008) GENEPOP’007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8:103–106
Saccheri I, Kuussaari M, Kankare M et al (1998) Inbreeding and extinction in a butterfly metapopulation. Nature 392:491–494
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Press, Cold Spring Harbor Laboratory, Cold Spring Harbor
Storz JF, Beaumont MA (2002) Testing for genetic evidence of population expansion and contraction: an empirical analysis of microsatellite DNA variation using a hierarchical Bayesian method. Evolution 56:154–166
Sun T, Wang BS, Liu ZJ et al (2010) Identification and characterization of microsatellite markers via cross-species amplification from François’ langur (Trachypithecus francoisi). Acta Theriologica Sinica 30:351–353
Tallmon DA, Koyuk A, Luikart GH et al (2008) ONeSAMP: a program to estimate effective population size using approximate Bayesian computation. Mol Ecol Resour 8:299–301
Van Oosterhout C, Hutchinson WF, Wills DPM (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Waples RS, Do C (2008) LDNE: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Resour 8:753–756
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Willi Y, Buskirk JV, Hoffmann AA (2006) Limits to the adaptive potential of small populations. Annu Rev Ecol Evol Syst 37:433–458
Zhang RZ, Chen LW, Qu YW et al (2002) The primates of China: biogeography and conservation status—past, present and future. Edit by Department of Wildlife Conservation, State Forestry Administration, the People’s Republic of China. China Forestry Publishing House, Beijing
Zhang BW, Li M, Zhang ZJ et al (2007) Genetic viability and population history of the Giant panda, putting an end to the “evolutionary dead end”? Mol Biol Evol 24:1801–1810
Zhu ZQ (2003) Ecological research of golden monkeys (Rhinopithecus roxellana) in Shennongjia Nature Reserve. Hubei For Sci Tech Z1:46–52 (in Chinese)
Zhu LF, Zhan XJ, Wu H et al (2010) Conservation implications of drastic reductions in the smallest and most isolated populations of giant pandas. Conserv Biol 24:1299–1306
Acknowledgments
This project was supported by the Natural Science Foundation of China (No. 31130061), the Project of Public Benefit (No. 201104073), National Basic Research Program of China (973 Program: 2007CB411600), the Innovation Project of the Chinese Academy of Sciences (KSCX2-YW-R-091), Hubei Province Key Laboratory of Conservation Biology of Shennongjia Golden Monkey, and the joint doctoral program of the Max Planck Society and the Chinese Academy of Sciences (Z-F. C.). We thank Dr. K. Guschanski for experimental guidance. We also thank G. Schubert, M. Arandjelovic, K. Langergraber, J. Bhagavatula, L. F. Zhu, and Y. B. Hu for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Chang, Z.F., Luo, M.F., Liu, Z.J. et al. Human influence on the population decline and loss of genetic diversity in a small and isolated population of Sichuan snub-nosed monkeys (Rhinopithecus roxellana). Genetica 140, 105–114 (2012). https://doi.org/10.1007/s10709-012-9662-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-012-9662-9