
Abstract
A rice mini-core collection consisting of 217 accessions has been developed to represent the USDA core and whole collections that include 1,794 and 18,709 accessions, respectively. To improve the efficiency of mining valuable genes and broadening the genetic diversity in breeding, genetic structure and diversity were analyzed using both genotypic (128 molecular markers) and phenotypic (14 numerical traits) data. This mini-core had 13.5 alleles per locus, which is the most among the reported germplasm collections of rice. Similarly, polymorphic information content (PIC) value was 0.71 in the mini-core which is the highest with one exception. The high genetic diversity in the mini-core suggests there is a good possibility of mining genes of interest and selecting parents which will improve food production and quality. A model-based clustering analysis resulted in lowland rice including three groups, aus (39 accessions), indica (71) and their admixtures (5), upland rice including temperate japonica (32), tropical japonica (40), aromatic (6) and their admixtures (12) and wild rice (12) including glaberrima and four other species of Oryza. Group differentiation was analyzed using both genotypic distance Fst from 128 molecular markers and phenotypic (Mahalanobis) distance D2 from 14 traits. Both dendrograms built by Fst and D2 reached similar-differentiative relationship among these genetic groups, and the correlation coefficient showed high value 0.85 between Fst matrix and D2 matrix. The information of genetic and phenotypic differentiation could be helpful for the association mapping of genes of interest. Analysis of genotypic and phenotypic diversity based on genetic structure would facilitate parent selection for broadening genetic base of modern rice cultivars via breeding effort.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rice (Oryza sativa L.) is the most important food crop in the world and serves as the staple food for about three billion people (Liang et al. 2004). However, the increase in rice yield has decreased from 2.4% (after the green revolution in the late 1980s) to 0.9% per year (Hossain 2007). One of the possible causes in the low genetic gains of the rice crop is the continuous use of genetically related elite germplasm by breeding programs. In this way, the genetic base of modern rice cultivars is narrowing and causing the crop to become more vulnerable to a broad set of biotic and abiotic stresses. According to Tanksley and McCouch (1997), this practice leads to a decline in genetic variability, reducing the chance of selecting new allelic combinations.
Exploitating of the genetic diversity stored in genebanks which is available for introduction into the breeding programs represents the best way to increase the diversity in cultivated crops (Tanksley and McCouch 1997). More than 400,000 rice germplasm accessions are conserved in genebanks around the world (Hamilton and Raymond 2005) and the great number of accessions in these germplasm collections makes comprehensive and accurate descriptions impractical, although the descriptions on both phenotypic and genotypic levels are absolutely fundamental for use. An alternative to the efficient exploitation of genetic resources is the establishment of core collections, which retain most of the genetic diversity of the original collection in a smaller number of accessions, (Brown 1989).
In the US, Yan et al. (2007) assembled a rice core collection consisting of 1,794 entries originating from 114 countries and representing approximately 10% of the 18,709 accessions (verified in April, 2010) in the USDA Rice World Collection maintained by the National Small Grains Collection. Information derived from the core collection could be used to assess the whole collection with an 88% certainty. Recently, the core collection was applied in exploring the association between genetic marker and straighthead resistance (Agrama and Yan 2009). However, such a core collection would still be too large for accurate descriptions of the important traits related to the nutritional value, biotic and abiotic resistance, and bio-energy and bio-chemical production which require more resources and technology than agronomic traits. Furthermore, it is also too large to be genotyped with a high density of molecular markers which fully cover the entire genome, such as single nucleotide polymorphism (SNP) markers. Accurate descriptions of phenotyping and genotyping are essential for broadening the genetic diversity in food production.
Consequently, Upadhyaya and Oritz (2001) suggested the concept of a mini-core collection including 10% of the core and 1% of the whole collection. Development of a mini-core subset from the core collection by further reduction of accessions becomes a more effective to obtain detailed genotypic information at a reasonable cost, This is specifically advantageous for those traits that require high technology and greater resources for characterization. As a result, a mini-core has been established in chickpea (Cicer arietinum L.) (Upadhyaya and Oritz 2001), sorghum (Sorghum bicolor L.) (Upadhyaya et al. 2009), pigeonpea (Cajanus cajan L.) (Upadhyaya et al. 2006) and Peanut (Arachis hypogaea L.) at both ICRISAT (International Crop Research Institute for Semi-Arid Tropics) (Upadhyaya et al. 2002) and in the US (Holbrook and Dong 2005). Using the mini-core collections, resistance to grain mold and downy mildew has been described in sorghum (Sharma et al. 2010), drought resistance and genes for oleic acid metabolism have been characterized in peanut (Chu et al. 2007; Upadhyaya 2005) and germplasm resources resistant to multiple diseases were identified in chickpea (Pande et al. 2006).
Recently, a mini-core collection has been established to represent rice in Brazil (Borba et al. 2009) and the US (Agrama et al. 2009) collections. The Brazilian mini-core has 24 accessions selected from a 550 core collection which came from a whole collection of 10,000 accessions distributed across three strata, Brazilian landraces (308 accessions) breeding materials, and foreign countries (242) (Abadie et al. 2005). The USDA rice mini-core (URMC) subset was developed from 1,794 accessions in its core collection representing over 18,000 accessions in the USDA global genebank of rice (Yan et al. 2007). This URMC contains 217 accessions which originated from 76 countries covering 15 geographic regions, a similar global distribution with the core collection. Full coverage of phenotypic variations measured by 26 traits and genotypic diversity measured by 70 molecular markers was achieved in this mini-core, indicating a rich gene pool harboring valuable genes. The URMC has the second most accessions after sorghum (242), followed by chickpea (211), ICRISAT peanut (184), pigeonpea (146) and US peanut (112).
Genetic diversity in a gene pool indicates the possibility that a specific gene of interest can be extracted from the pool (Garris et al. 2005). Population structure plays an essential role in accurately mapping genes associated with phenotypic traits of interest to avoid type II error (Zhu and Yu 2009). Genetic distance increases along with an increase in heterosis or hybrid vigor, but it also increase genetic incompatibility between certain populations or parents (Yan et al. 2009). A balance between the heterosis and genetic compatibility is a challenge to plant breeders. Mapping genes responsible for phenotypic traits helps breeders to use molecular markers in their selection, which improves breeding efficiency especially for those quantitative traits that are difficult to be characterized and are easily affected by environment.
As a result, our objective in this study was to characterize the 217 accessions in the URMC for genetic diversity, population structure and genetic relationships using both genotypic and phenotypic descriptors.
Materials and methods
Materials
The 217 accessions of URMC are presented and described by Agrama et al. (2009). Of these accessions, 203 accessions belong to Oryza sativa, eight to O. glaberrima, two each to O. nivara and O. rufipogon, and one each to O. glumaepatula and O. latifolia. Two accessions, NSGC 5944 (PI 590413) and WC 10253 (PI 469300), were from unknown countries of origin. Each of these accessions is listed in the Genetic Stock Oryza (GSOR) collection at www.ars.usda.gov/spa/dbnrrc/gsor. Information is listed on cultivar name or designation, accession number, registration year, place of origin, longitude and latitude of origin, pedigree or genetic background (if available), morphological characteristics and references. The GSOR provides seeds for research purposes to national and international users upon to request.
Phenotyping
Evaluations were conducted at the Dale Bumpers National Rice Research Center, Stuttgart, Arkansas. The 217 mini-core accessions were arranged by a randomized complete block design, with three replications and nine plants spaced 0.3 × 0.6 m in each plot. The experiment was drill-seeded on April 23, 2009 followed by a regular management practice for weed control, water and fertilizer application. Data collection followed procedures described by Yan et al. (2005a, b) with modifications. Heading was recorded as number of days when 50% of panicles in a plot had began to emerge from the boot of panicles. Meanwhile, three plants were selected and their main panicles were marked, then each plant was bagged at the top to avoid panicle damage and supported by a bamboo pole to avoid lodging. Each plant was cut at ground level when mature and air-dried for 2 months before waiting to obtain plant weight (g). Then, plant height (cm) was measured from the base to the panicle tip, the main panicle was removed at the panicle node and tillers of the plant were recorded before being threshed. Grain yield (g) was measured as total weight after the threshed grains were cleaned by an Almaco seed cleaner. Harvest index (%) was calculated as the ratio of grain yield to plant weight. Each main panicle was measured for its length (cm), and primary and secondary branches were counted before manually threshing. All kernels from the panicle were placed in a cup half full of water and the cup was stirred with a spoon. Blank kernels floated to the top in the water and filled kernels sank to the bottom. The number of each was recorded after they were dried at 50°C for 12 h. Seed weight (mg) was determined by the filled kernel weight divided by its number, and seed set (%) was expressed by a ratio of the filled kernels to the total kernels in the panicle. Panicle length and branch data were used to generate kernels/cm panicle and kernels/branch panicle using total kernels including both filled and unfilled.
Genotyping
Five-plant bulk tissue was collected from each accession as described by Brondani et al. (2006) and total genomic DNA was extracted using a rapid alkali extraction procedure (Xin et al. 2003). The bulked DNA allowed identification of the origin of heterogeneity, which can result from the presence of heterozygous individuals or from a mix of individuals with different homozygous alleles (Borba et al. 2005). The 128 molecular markers covering the entire rice genome, which averaged one marker per 15 cM, were used to genotype the 217 accessions in the URMC, including 122 SSRs obtained from the Gramene database (www.gramene.org). The other 5 SSR markers were amplified using the following primers: AP5652-1, F = GTA CAG CGC AAA AGT GGT AG, R = CAT GGG ACT TGA TGT AGG AG; AP5652-2, F = TTG ACT TAT AGA AGT TGA ATT TGG, R = TGT GTC AGT CAA GCA GAC AG; AL606682-1, F = TAG AGC TCC CTC AGC TGC TC, R = CGC GCA TGC ATG TAC AGT AG; con673. F = CGT ACT TGC CAC CGT AAG, R = TTG ATA GGC AAT GTT TCT CC and LJSSR1, F = CCT CCG ACC TCC GAG CTA, R = AGC CGC ATC AGT AGT CAT CA. The remaining marker was an indel at the Rc locus, named Rid 12 responsible for rice pericarp color (Sweeney et al. 2006). These markers were arranged in such a way where markers that amplified allele sizes with at least 20 bp of difference, and labeled with different fluorochromes, were selected to be part of the same multiplex set to ensure that their fragments’ sizes would not overlap. Polymerase chain reaction (PCR) marker amplifications were performed as described by Agrama et al. (2009). DNA samples were run on an ABI Prism 3730 DNA analyzer according to the manufacturers instructions (Applied Biosystems, Foster City, CA, USA). Fragments were sized and binned into alleles using GeneMapper version 3.7 software.
Statistical analysis
Genetic distance was calculated using Nei distance (Nei and Takezaki 1983). Phylogenetic reconstruction was based on the unweighted pair-group method using arithmetic average (UPGMA) method implemented in PowerMarker version 2.7 (Liu and Muse 2005; www.powermarker.net). The tree to visualize the phylogenetic distribution of accessions and ancestry groups was constructed using MEGA version 4 (Tamura et al. 2007). The model-based program STRUCTURE (Pritchard et al. 2000) was used to infer population structure using a burn-in of 10,000, run length of 100,000, and a model allowing for admixture and correlated allele frequencies. The number of groups (K) was set from 1 to 10, with ten independent runs each. The most probable structure number of (K) was calculated based on Evanno et al. (2005) using an ad hoc statistic D(K), assisted with L(K), L′(K) and (L″K). The D(K) perceives the rate of change in log probability of the data between successive (K) values rather than just the log probability of the data. Determination of mixed ancestry (an accession unable to be clearly assigned to only one group) was based on 60% (Q) as a threshold to consider an individual with its inferred ancestry from one single group. Principal coordinate analysis (PCoA), that summarizes the major patterns of variation in multi-locus data set, was performed with NTSYSpc software version 2.11 V (Rohlf 2000). Two principal coordinates were used to visualize the dispersion of the mini core accessions in a graphic. PowerMarker was also used to calculate the average number of alleles, gene diversity, and polymorphism information content (PIC) values. Fst indicative of ancestral relationship between genetic groups was calculated using an AMOVA approach in Arlequin V2.000 (Weir 1996; Schneider and Excoffier 1999). The number of private alleles was estimated by Genetic Data Analysis (GDA) program (Lewis and Zaykin 2001).
Fourteen phenotypic characteristics were used to calculate Mahalanobis distance as a measurement of genetic differentiation among the groups (Kouame and Quesenberry 1993). The Mahalanobis distance and Canonical discriminant analysis were performed by the procedures PROC CANDISC of the SAS version 9.1 statistical packages (SAS Institute 2002). Eventually, the correlation of genetic structure differentiation resulting from the genotypic markers with phenotypic traits was assessed using the Mantel test (Mantel 1967) performed by PowerMarker. Analysis of variance (ANOVA) was conducted by PROC ANOVA (SAS Institute 2002) to test differences among the groups generated by the cluster analysis for 14 traits. To determine the differences between the pairs of groups (group means), t tests (LSD) were conducted.
Results
Profile of SSR markers
The whole set of 128 markers with a genome-wide distribution detected a total of 1,729 alleles across 217 accessions in the URMC. The average number of alleles per locus was 13.5 ranging from 2 for RM338 to 57 for con673. PIC varied from 0.30 for AP5625-1 to 0.97 for con673 among these markers, with an average of 0.71. Since every accession was analyzed as a bulk of five plants, 54 (42.19%) loci showed heterozygosity for the set of 128 loci and 38 (17.51%) accessions showed heterogeneity for at least one locus. Nei and Takezaki (1983) genetic distance was estimated for each pair of the 217 rice accessions which ranged from 0.021 to 1.000, with an average 0.752.
Structural analysis
UPGMA tree showed that 202 accessions of Oryza sativa were classified in two main branches (Fig. 1a). The other 12 accessions of related species in Oryza were not grouped into either of the two main branches. Eight accessions of O. glaberrima stayed together, distinguishable from each of O. latifolia, and O. glumaepatula on one side, but each of O. nivara and O. sativa (PI 430909) together on another side of the tree in the wild rice group. Although PI 430909 from Pakistan was classified O. sativa in the Germplasm Resources Information Network (GRIN) at www.ars-grin.gov, it had spreading plant type, black hull with full and long awns and small, red and shattering kernels, typical characteristics of wild rice. Adversely, PI 590422 from Myanmar in 1995 and PI 346371 from Brazil in 1969 were classified as O. rufipogon in the GRIN, but the former was clustered with indica (Q-indica = 0.77) and the latter with an admixture of aus and indica (Q-aus = 0.59, Q-indica = 0.41). The disagreement of cluster analysis in the study with traditional classification in the GRIN is worthy of further attention.

a UPGMA tree and b principal coordinate analysis based on 128 nuclear SSRs and 127 accessions in the USDA rice mini-core collection, both visualizing six main groups (AUS aus, IND indica, ARO aromatic, TRJ tropical japonica, TEJ temperate japonica, WD wild rice) and two admixed groups (ADA accessions having the ancestry of AUS and IND, ADJ accessions having the ancestry of TRJ and TEJ)
In order to verify the subdivision, a model-based clustering method for multi-loci genotype data was employed to infer the population structure and assign individuals to populations using STUCTURE. The most probable structure number of (K) was calculated based on Evanno et al. (2005) using an ad hoc statistic D(K), assisted with L(K), L′(K) and L″(K) (supplemental material). These statistical parameters uniformly demonstrated that there were probably six subdivisions. Consistency of structuring with clustering results proved that six groups sufficiently explain genetic differentiation in the URMC. Then, ancestry index or Q value from the model-based grouping was estimated for each of 217 URMC accessions.
The ancestry of each accession was inferred from the Q value and classified into one of the six groups which corresponded to aromatic (ARO), aus (AUS), indica (IND), temperate japonica (TEJ), tropical japonica (TRJ) and wild rice (WD) based on reference cultivars reported previously by Garris et al. (2005), Agrama and Eizenga (2008), Agrama and Yan (2009). The classification was clear for a single group when the Q value was greater than 60%, otherwise admixed ancestry with another group was signed. Totally, 21 accessions (9.68%) in the URMC had admixed ancestry either between temperate and tropical japonica (ADJ) or between aus and indica (ADA) principally (Figs. 1a, b). Two main branches to differentiate 205 of the accessions were equivalent to lowland and upland cultivars, respectively. Ecogeographically, indica is primarily known as lowland rice grown throughout tropical Asia, while japonica is typically found in temperate East Asia, upland geographic regions of Southeast Asia, and high elevations of South Asia (Garris et al. 2005). The lowland branch was further distinguished into two minor groups corresponding to aus and indica accessions, while the upland branched into three groups, temperate japonica, tropical japonica and aromatic (Fig. 1a).
The first-two axes in PCoA with 83.2% of total variation sufficiently discriminated the six main groups and two admixture groups (Fig. 1b). Each main group was distinguishable from another, but overlaps existed either among temperate and tropical japonica and their admixtures, or among indica, aus and their admixtures. Basically, wild rice and aromatic accessions stayed alone individually. The PCoA visualization and UPGMA tree were in agreement, which demonstrates a correct division of genetic structure in the URMC.
Genotypic diversity and differentiation
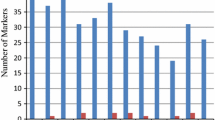
In the URMC, the majority of accessions were IND (33%), followed by TRJ and AUS (18% each), TEJ (15%), WD (6%) and ARO with six accessions only (Fig. 2). All the 128 loci were polymorphic in IND (Table 1). TRJ had 99% polymorphic loci, followed by WD, AUS, TEJ and ARO. IND had the most alleles per locus, TRJ and AUS the second most, TEJ and WD the third most and ARO had the fewest alleles. However, the largest number of private alleles per locus (alleles unique in one group and not found in another group) were found in WD (41.89%), followed by IND (23.78%) and AUS (17.66%). TRJ and TEJ had about equal number of private alleles, and the least was found in ARO. Gene diversity averaged 0.47 among the groups ranging from 0.37 in ARO to 0.52 in both IND and AUS. TRJ and WD had the same diversity (0.50), slightly greater than TEJ (0.43).

a Dendrogram of differentiation based on AMOVA.(Fst) using 128 molecular markers for b six main groups, c estimated group structure with each individual represented by a horizontal bar and d two admixture groups. Abbreviation: AUS aus in green, IND indica in yellow, ARO aromatic in blue, TEJ temperate japonica in light blue, TRJ tropical japonica in red, WD wild rice in purple, ADA admixtures of aus and indica, ADJ admixtures of temperate japonica and tropical japonica
Results from the AMOVA showed that 37.92% of total variation was due to differences among groups, 61.21% within groups and 0.88% within individuals. Pair-wise estimates of Fst using the AMOVA approach indicated a high degree of differentiation among the six main model-based groups (Fig. 2a). The mean Fst of all group pairs was 0.39 ranging from 0.24 between TRJ and TEJ to 0.48 between ARO and WD (Table 2). All pair-wise Fst values for the six groups were significant. The greatest genetic distance (0.990) among the 217 mini-core accessions was observed for PI 590413, an O. glumaepatula accession from the WD with 22 IND accessions and three accessions admixed AUS and IND, followed by the distance of 0.981 for PI 590413 with 9 AUS and 28 IND accessions, and the distance of 0.981 for PI 269727, an O. latifolia accession from the WD with 4 TEJ accessions. Two IND accessions, PI 202864 and PI 214077 had the shortest distance (0.021).
Phenotypic diversity and differentiation
Canonical discriminant analysis of 14 phenotypic traits for 217 mini-core accessions clearly separated the six plus two admixture model-based genetic groups derived from molecular data (Fig. 3a). The first four significant (P < 0.001) canonical discriminant functions (CAN) explained 92.02% of the total variance, 54.87% by the first CAN and 18.08% by the second CAN function, respectively (Fig. 3b). The accessions in group of AUS, ARO, IND, TEJ, TRJ and WD were clustered into their groups with various overlaps. The upland (ARO, TEJ and TRJ) were obviously discriminated from the lowland (AUS, IND), and the admixed groups ADA was scattered across AUS and IND while ADJ across TEJ and TRJ.

a Dendrogram of differentiation based on Mahalanobis distance and b Canonical discriminant analysis (CDA) using 14 phenotypic traits among structural groups for 217 accessions in the USDA rice mini-core collection: AUS aus, IND indica, ARO aromatic, TRJ tropical japonica, TEJ temperate japonica, WD wild rice, ADA admixtures of aus and indica, ADJ admixtures of temperate japonica and tropical japonica
All fourteen traits were significantly different among the eight (six plus two admixtures) model-based genetic groups (Table 3). However, only three traits, plant weight (biomass), tillers and grain yield, had larger variation among groups than within groups. Therefore, they are considered the main discriminatory characters (r 2 ≥ 0.49) in differentiating these genetic groups. The first canonical loading was 0.81 for grain yield and tillers and 0.78 for plant weight. Nevertheless, the second canonical loading was dominated by panicle length (0.59), heading days (0.55) and seed weight (0.51).
The most tillers were observed in AUS accessions PI 385697 and 352687, and in WD PI 450396 with 93, 86 and 82 tillers, respectively. The lowest readings were recorded from TRJ accessions PI 584567, 154464 and 434632 with 9, 10 and 10 tillers, respectively. On average, WD had the most tillers (60), followed by AUS (46), ADA (44), AUS (46), IND (38), ARO (27), TEJ (24), ADJ (21) and TRJ (18).
The greatest plant weight was 731 g for PI 549215 (IND), followed by 634 g for PI 450421(WD) and 564 g for PI 373536 (IND). The lowest weight was 37 g for PI 281630 (TEJ), followed by 41 g for PI 245694 (TEJ) and 58 g for PI 615198 (TEJ). The most group mean of plant weight was observed in WD (442 g), followed by ADA (379 g), AUS (295 g), IND (274 g), ARO (172 g), ADJ (167 g), TRJ (157 g) and TEJ (127 g).
PI 373335 (IND) had the highest grain yield per plant at 175 g, followed by 169 g for PI 373347 (AUS) and 154 g for PI 389267 (IND). The lowest grain yield was 11 g for PI 389933 (IND), followed by 12 g for PI 269630 (WD) and 17 g for PI 281630 (TEJ), respectively. The highest group mean of grain yield was observed in ADA (127 g per plant), followed by AUS (118 g), IND (96 g), WD (90 g), TRJ (62 g), ADJ (61 g), ARO (60 g) and TEJ (55 g). The grain yield of the WD was an average of the five accessions of O. glaberrima and the accession of O. sativa PI 430909 because the other six accessions in the WD failed to produce seeds.
Relationships between genotypic and phenotypic differentiation
The dendrogram based on the Mahalanobis distance (D2) using the 14 phenotypic traits (Fig. 3a) matched up very well with the dendrogram based on the Fst genetic differentiation from AMOVA using 128 markers (Fig. 2a). The two dendrograms differentiated the lowland including IND, AUS and their admixtures from the upland having TEJ, TRJ, ARO and their admixtures. The WD or non-sativa accessions remained independent from the others.
Analysis developed by Mantel (1967) is widely used to describe the genetic relationship between genotypic and phenotypic measurements (Gaudeul et al. 2000, Gizaw et al. 2007). In our study, genetic distance derived from the 128 markers among the six plus two admixture model-based groups was highly and significantly correlated with the distance derived from 14 phenotypic traits (r = 0.85, P < 0.001). This explains the correspondence of the two dendrograms in Fig. 2a and Fig. 3a, and similar pattern of D2 and Fst in Table 2.
Discussion
Genetic diversity of the USDA rice mini-core (URMC) collection
The average number of alleles per locus among the 217 accessions of the URMC genotyped by a set of 128 markers was 13.5, higher than the reported other rice collections or populations. Part of the alleles cane from other species, such as O. latifolia, O. glumaepatula, O. rufipogon, O. nivara and O. glaberrima. In previous studies, the average number of alleles per locus was 5.1 in Cho et al. (2000), 7.8 in Jain et al. (2004), 11.9 in Xu et al. (2004) and 11.8 in Garris et al. (2005). Recently, 13 alleles per locus were reported in the rice population studied by Thomson et al. (2007), 5.5 by Thomson et al. (2009) and 12.4 by Borba et al. (2009). The PIC in the URMC was 0.71, larger than it in the population studied by Cho et al. (2000) (0.56 PIC), Jain et al. (2004) (0.60), Xu et al. (2004) (0.66), Garris et al. (2005) (0.67), Thomson et al. (2007) (0.66), and Thomson et al. (2009) (0.45). The PIC was slightly less in our study than in the population studied by Borba et al. (2009) (0.75). Both the average allele number and PIC values are indicative of genetic diversity or gene richness in a germplasm collection. The higher the genetic diversity is in a collection, which indicates the larger content of genes, the greater opportunity for a gene of interest to be mined from the collection.
Greater genetic diversity in the URMC is probably due to its global originations. All the studied rice collections are either for a country (Thomson et al. 2007), or for certain groups (Jain et al. 2004) and regions in a country (Thomson et al. 2009), or for special interests (Xu et al. 2004; Garris et al. 2005). The International Rice Research Institute (IRRI) is the only another collector of global rice, but a standard material transfer agreement (SMTA) is required for the availability to the public. However, each accession in the USDA rice world collection is available for the public without any attachment, which allowed Brazil to introduce the entire collection in 2008.
Genotypic and phenotypic characterization of genetic differentiation
Assessment of genotypic and phenotypic differetiation and their relationship has long been attractive to the scientific community. In this study, the six plus two admixture model-based groups were similarly differentiated by both genotypic analysis using 128 SSR markers and phenotypic analysis using 14 morphological traits. The phenotypic dendrogram based on the Mahalanobis distance (D2) matched up very well with the genotypic dendrogram based on the Fst genetic differentiation from AMOVA. Genetic differentiation assessed by the genotypic markers among the model-based groups was highly and significantly correlated with the phenotypic differentiation assessed by the phenotypic traits (Mantel test, r = 0.85, P < 0.001). These results suggested that the clear differentiation of genetic structure was significantly reflected in these morphological traits.
In rice, ancestry structure and genetic diversity are widely studied using molecular markers to genotype each accession in a germplasm collection with SSR (Cho et al. 2000; Jain et al. 2004; Xu et al. 2004; Garris et al. 2005; Thomson et al. 2007; 2009; Borba et al. 2009), RAPD (Mackill 1995) and isozyme (Glaszmann 1987) markers. Phenotypic characteristics are hardly ever used to analyze genetic diversity or structure in rice germplasm collections. Zeng et al. (2003) collected samples from each of six genetic groups for a diversity analysis using 31 phenotypic traits, but failed to reveal their genetic differentiations. Elias et al. (2001a) reported a significantly positive association between genotypic and phenotypic distances (r = 0.204, P = 0.054) using eight SSRs and 14 characters for 38 accessions of cultivated cassava (Manihot esculenta Crantz). The correlation was increased (r = 0.283, P < 0.01) in a set of 29 cassava accessions genotyped with AFLP markers and phenotyped for 14 morphological and four agronomic traits (Elias et al. 2001b). The low correlation in other study can be explained by the sensitivity of some traits to environmental influence, which results in low heritability. The low heritability makes it hard to discriminate lines in the population. A non neutral evolutionary history for some traits can also give a low correlation between phenotypic distance and genetic distance (Merilaè and Crnokrak 2001).
Potential benefit of genotypic and phenotypic diversity and population structure
In this study, 14 traits and 128 markers with high genetic diversity among the cultivars made it possible to identify superior trait alleles in the URMC collection. A pair of cultivars with distinct morphological traits could be used as potential parents to facilitate high resolution QTL mapping and validate candidate genes responsible for quantitatively agronomic characters. Genetic structure should be taken into consideration for fertility traits, in order to prevent incompatibility caused widely crossing. Moreover, using this framework of genetic groups revealed in this study, it may be possible to explore the rice gene pools more effectively with population genetics-based approaches, such as association mapping.
In a practical way, this diversity information based on genetic structure is extremely important in selecting parents for inter-group crosses to broaden the genetic base of modern rice cultivars. These crosses may have a certain degree of sterility. As a viable alternative, crosses involving genitors of the same group from different regions should be performed. This can produce novel favorable gene/allele combinations for traits of agronomic interest for breeding purposes.
References
Abadie T, Cordeiro CMT, Fonseca JR, Alves RBN, Burle ML, Brondani C, Rangel PHN, Castro EM, Silva HT, Freire MS, Zimmermann FJP, Magalhaes JRSO (2005) Constructing a rice core collection for Brazil. Pesquisa Agropecu Bras 40:129–136
Agrama HA, Eizenga GC (2008) Molecular diversity and genome-wide linkage disequilibrium patterns in a worldwide collection of Oryza sativa and its wild relatives. Euphytica 160:339–355
Agrama HA, Yan WG (2009) Genetic diversity and relatedness of rice cultivars resistant to straighthead disorder. Plant Breed. doi:10.1111/j.1439-0523.2009.01688.x
Agrama HA, Yan WG, Lee F, Fjellstrom R, Chen MH, Jia M, McClung A (2009) Genetic assessment of a mini–core subset developed from the USDA rice Genebank. Crop Sci 49:1336–1346
Borba TCO, Brondani RPV, Rangel PHN, Brondani C (2005) Evaluation of the number and information content of fluorescent–labeled SSR for rice germplasm characterization. Crop Breed Appl Biotechnol 2:157–165
Borba TCO, Brondani RPV, Rangel PHN, Brondani C (2009) Microsatellite marker–mediated analysis of the EMBRAPA rice core collection genetic diversity. Genetica 137:293–304
Brondani C, Borba TCO, Rangel PHN, Brondani RPV (2006) Determination of traditional varieties of Brazilian rice using microsatellite markers. Genet Mol Biol 29:676–684
Brown AHD (1989) Core collections: a practical approach to genetic resources management. Genome 31:818–824
Cheng CY, Motohashi R, Tsuchimoto S, Fukuta Y, Ohstubo H, Ohtsubo E (2003) Polyphyletic origin of cultivated rice: based on the interspersion pattern of SINEs. Mol Biol Evol 20:67–75
Cho YG, Ishii T, Temnykh S, Chen X, Lipovich L, McCouch SR, Park WD, Ayres N, Cartinhour S (2000) Diversity of microsatellites derived from genomic libraries and genbank sequences in rice (Oryza sativa L.). Theor Appl Genet 100:713–722
Chu Y, Ramos L, Holbrook CC, Ozias-Akins P (2007) Frequency of a loss-of-function mutation in oleoyl-PC desaturase (ahFAD2A) in the mini-core of the US peanut germplasm collection. Crop Sci 47:2372–2378
Elias M, Penet L, Vindry P, McKey D, Panaud O, Robert T (2001a) Unmanaged sexual reporoduction and the dynamics of genetic diversity of a vegetatively propagated crop plant, cassava (Manihot esculenta Crantz), in a traditional farming system. Mol Eco 10:1895–1907
Elias M, McKey D, Panaud O, Anstett MC, Robert T (2001b) Traditional management of cassava morphological and genetic diversity by the Makushi Amerindians (Guyana, South America): perspectives for on-farm conservation of crop genetic resources. Euphytica 120:143–157
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Garris AJ, Tai TH, Coburn J, Kresovich S, McCouch SR (2005) Genetic structure and diversity in Oryza sativa L. Genetics 169:1631–1638
Gaudeul M, Taberlet P, Till-Bottraud I (2000) Genetic diversity in an endangered alpine plant, Eryngium alpinum L. (Apiaceae), inferred from amplified fragment length polymorphism markers. Molecular Ecol 9:1625–1637
Gizaw S, Van Arendonk JAM, Komen H, Windig JJ, Hanotte O (2007) Population structure, genetic variation and morphological diversity in indigenous sheep of Ethiopia. Anim Genet 38:621–628
Glaszmann JC (1987) Isozymes and classification of Asian rice varieties. Theor Appl Genet 74:21–30
Hamilton RS, Raymond R (2005) Toward a global strategy for the conservation of rice genetic resources. In: Toriyama K, Heong KL, Hardy B (eds) Rice is life: scientific perspectives for the 21st century. Proceedings of the world rice research conference held in Tsukuba, Japan, CD–ROM, pp 47–49
Holbrook CC, Dong W (2005) Development and evaluation of a mini core collection for the US peanut germplasm collection. Crop Sci 45:1540–1544
Hossain M (2007) Rice facts: a balancing act. Rice Today 6:37
Jain S, Jain RK, McCouch SR (2004) Genetic analysis of Indian aromatic and quality rice (Oryza sativa L.) germplasm using panels of fluorescently–labeled microsatellite markers. Theor Appl Genet 109:965–977
Kouame CN, Quesenberry KH (1993) Cluster analysis of a world collection of red clover germplasm. Genet Res Crop Evo 40:39–47
Lewis PO, Zaykin D (2001) Genetic data analysis: computer program for the analysis of allelic data version 1.0. http://lewis.eeb.uconn.edu/lewishome/software.html. Cited 21 Nov 2001
Liang F, Deng Q, Wang Y, Xiong Y, Jin D, Li J, Wang B (2004) Molecular marker–assisted selection for yield–enhancing genes in the progeny of “9311 × O. rufipogon” using SSR. Euphytica 139:159–165
Liu K, Muse SV (2005) Powermarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
Mackill DJ (1995) Plant genetic resources: classifying japonica rice cultivars with RAPD markers. Crop Sci 35:889–894
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220
Merilaè J, Crnokrak P (2001) Comparison of genetic differentiation at marker loci and quantitative traits. J Evol Biol 14:892–903
Nei M, Takezaki N (1983) Estimation of genetic distances and phylogenetic trees from DNA anlysis. Proc. 5th World Cong. Genet Appl Livstock Prod 21:405–412
Pande S, Kishore GK, Upadhyaya HD, Rao JN (2006) Identification of sources of multiple disease resistance in mini-core collection of chickpea. Plant Dis 90:1214–1218
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rohlf F (2000) NTSYS–PC numerical taxonomy and multivariate analysis system ver 2.11L. Applied Biostatistics, NY
Schneider S, Excoffier L (1999) Estimation of demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: application to human mitochondrial DNA. Genetics 152:1079–1089
Sharma R, Rao VP, Upadhyaya HD, Reddy VG, Thakur RP (2010) Resistance to grain mold and downy mildew in a mini-core collection of sorghum germplasm. Plant Dis 94:439–444
Sweeney MT, Thomson MJ, Pfeil BE, McCouch S (2006) Caught red-handed: Rc encodes a basic helix–loop–helix protein conditioning red pericarp in rice. Plant Cell 18:283–294
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Tanksley S, McCouch SR (1997) Seed banks and molecular maps: unlocking genetic potential from the wild. Science 277:1063–1066
Thomson MJ, Septiningsih EM, Suwardjo F, Santoso TJ, Silitonga TS, McCouch SR (2007) Genetic diversity analysis of traditional and improved Indonesian rice (Oryza sativa L.) germplasm using microsatellite markers. Theor Appl Genet 114:559–568
Thomson MJ, Polato NR, Prasetiyono J, Trijatmiko KR, Silitonga TS, McCouch SR (2009) Genetic diversity of isolated populations of Indonesian landraces of rice (Oryza sativa L.) collected in east Kalimantan on the island of Borneo. Rice 2:80–92
Upadhyaya HD (2005) Variability for drought resistance related traits in the mini core collection of peanut. Crop Sci 45:1432–1440
Upadhyaya HD, Oritz R (2001) A mini-core collection for capturing diversity and promoting utilization of chickpea genetic resources in crop improvement. Theor Appl Genet 102:1292–1298
Upadhyaya HD, Reddy LJ, Gowda CLL, Reddy KN, Singh S (2006) Development of a mini core for enhanced and diversified utilization of pigeonpea germplasm resources. Crop Sci 46:2127–2132
Upadhyaya HD, Pundir RPS, Dwivedi SL, Gowda CLL, Reddy VG, Singh S (2009) Developing a mini core collection of sorghum for diversified utilization of germplasm. Crop Sci 49:1769–1780
Weir BS (1996) Genetic data analysis II: methods for discrete population genetic data. Sinauer assoc., Inc. Sunderland, MA
Xin Z, Velten JP, Oliver MJ, Burke JJ (2003) Highthroughput DNA extraction method suitable for PCR. Biotechniques 34:820–826
Xu YB, Beachell H, McCouch SR (2004) A marker–based approach to broadening the genetic base of rice in the USA. Crop Sci 44:1947–1959
Yan W, Rutger JN, Bockelman HE, Tai TH. (2005a) Agronomic evaluation and seed stock establishment of the USDA rice core collection. In: RJ Norman et al. (ed.) BR wells rice research studies 2004. University of Arkansas, Agri Exp Sta Res Ser 529:63–68
Yan W, Rutger JN, Bockelman HE, Tai TH. (2005b). Evaluation of kernel characteristics of the USDA rice core collection. In: RJ Norman et al. (ed.) BR wells rice research studies 2004. University of Arkansas, Agri Exp Sta, Res Ser 529:69–74
Yan WG, Rutger JN, Bryant RJ, Bockelman HE, Fjellstrom RG, Chen MH, Tai TH, McClung AM (2007) Development and evaluation of a core subset of the USDA rice (Oryza sativa L.) germplasm collection. Crop Sci 47:869–878
Yan WG, Li Y, Agrama HA, Luo D, Gao F, Lu X, Ren G (2009) Association mapping of stigma and spikelet characteristics. Mol Breeding 24:277–292
Zeng Y, Shen S, Li Z, Yang Z, Wang X, Zhang H, Wen G (2003) Ecogeographic and genetic diversity based on morphological characters on indigenous rice (Oryza sativa L.) in Yunnan, China. Genet Res Crop Evo 50:567–577
Zhu C, Yu J (2009) Nonmetric multidimensional scaling corrects for population structure in whole genome association studies. Genetics 182:875–888
Acknowledgments
The authors thank Ellen McWhirter for critical review, Tiffany Sookaserm, Tony Beaty, Yao Zhou, LaDuska Simpson, Curtis Kerns and Sarah Hendrix for technical assistance.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Li, X., Yan, W., Agrama, H. et al. Genotypic and phenotypic characterization of genetic differentiation and diversity in the USDA rice mini-core collection. Genetica 138, 1221–1230 (2010). https://doi.org/10.1007/s10709-010-9521-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-010-9521-5