
Abstract
Bloom syndrome (BS) is a rare genetic disorder caused by biallelic inactivation of the BLM gene, which usually manifests in childhood by significant growth retardation, immune deficiency, characteristic skin lesions, cancer predisposition and other distinguishable disease features. To our knowledge, all prior instances of BS have been identified via intentional analysis of patients with clinical suspicion for this disease or DNA testing of members of affected pedigrees. We describe an incidental finding of BS, which occurred upon routine germline DNA analysis of consecutive breast cancer patients. The person with the biallelic pathogenic BLM c.1642C>T (p.Gln548Ter) variant remained clinically healthy for 38 years until she developed breast cancer. Detailed examination of this woman, which was carried out after the genetic diagnosis, revealed mild features of BS. A sister chromatid exchange (SCE) test confirmed the presence of this syndrome. The tumor exhibited triple-negative receptor status, a high proliferation rate, a low tumor mutation burden (TMB), and a moderate level of chromosomal instability (homologous recombination deficiency (HRD) score = 29). The patient showed normal tolerability to radiotherapy and several regimens of cytotoxic therapy. Thus, some BS patients may remain undiagnosed due to the mild phenotype of their disease. BLM should be incorporated in gene panels utilized for germline DNA testing of cancer patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breast cancer (BC) patients, especially women with clinical signs of BC predisposition (disease onset before age 50 and/or family history of cancer and/or bilateral disease and/or triple-negative receptor phenotype), are advised to undergo comprehensive genetic testing. This DNA analysis must include full-length sequencing of BRCA1 and BRCA2 genes and is often supplemented by testing of other hereditary cancer loci. Slavic populations are characterized by high incidence of recurrent pathogenic variants in the CHEK2, NBN and BLM genes, which confer a moderate (approximately 2-fold) increase in BC risk in heterozygotes. Therefore, these protein-truncating variants (CHEK2: c.1100delC, del exons 9–10, c.444 + 1G > A; NBN: c.657_661delACAAA; BLM: c.1642C>T) are often included in routine DNA testing procedures in countries with predominantly Slavic populations [1, 2]. Within years 2016–2023, we have performed genetic analysis of 10,690 consecutive Slavic BC patients and identified 56 (0.5%) subjects with the Slavic BLM c.1642C>T (p.Gln548Ter) truncating allele. Surprisingly, one of these women was biallelic for this pathogenic variant; however, the accompanying medical information did not indicate that she was affected by Bloom syndrome (BS). We contacted her primary physician, and she reported that this BC patient remained healthy until the onset of cancer. BS is a severe disease manifesting by significant growth retardation, immune deficiency, characteristic skin lesions, etc.; therefore, we made a special effort to investigate this case in more detail.
Case presentation
The patient was diagnosed with breast cancer at the age of 38. She underwent sectoral resection of her left breast along with left axillary lymphadenectomy. The excised tumor was classified as invasive carcinoma of no special type, pT1cN2M0. Immunohistochemical analysis revealed a lack of expression of estrogen, progesterone and HER2 receptors coupled with a high proliferative rate of tumor cells (Ki-67 index: 65–70%). Surgery was followed by radiation therapy (48 Gy to the mammary gland and 44 Gy to the lymph nodes) and conventional adjuvant chemotherapy (4 cycles of doxorubicin plus cyclophosphamide followed by 12 cycles of weekly paclitaxel at a standard dose). The last cycle of chemotherapy was administered in January 2021; however, regular examination carried out in July 2022 revealed the emergence of infraclavicular lymph node metastasis. The patient received chemotherapy with capecitabine at a standard dose 2000 mg/m2/day, which was accompanied by skin toxicity (hand-foot syndrome, grade 2). A metastatic lesion in the liver was detected one year later. The patient was treated with bevacizumab and docetaxel but eventually developed multiple lung metastases. In 2023, targeted sequencing of hereditary cancer genes was performed. Full-length BRCA1/2 analysis did not reveal pathogenic variants; however, surprisingly, a homozygous BLM c.1642C>T genotype was identified in the blood-derived DNA. The analysis of tumor cells and adjacent normal tissues confirmed the presence of biallelic BLM c.1642C>T substitution.
DNA analysis of the patient’s parents confirmed that they were heterozygous for this allele. This woman was born as the only child of a nonconsanguineous Slavic couple. There was no history of intrauterine growth restriction; she was delivered at 36 weeks of pregnancy with a weight of 2800 g and a body length of 48 cm. Early development occurred without significant abnormalities, however, the poor growth rate was consistently reported by pediatricians. Her intellectual development was normal. She had suffered from chronic gastritis since the age of 8 and was diagnosed with a peptic ulcer at the age of 23. She had no other complaints. Family history was negative for growth deficiency or breast cancer. Her mother reported late-onset diabetes mellitus, and her paternal grandfather had skin cancer at the age of 60. Her menarche started at her 15, and her menstrual cycle was regular. Despite the use of cytotoxic therapy for the treatment of breast cancer, no menopause has occurred. She had no history of pregnancy. At the time of examination, the patient was 158 cm tall, her mother’s height was 174 cm, and her father’s height was 180 cm. Her head circumference was 51 cm, i.e., below the 3rd centile for adults. Visual examination of the patient revealed no skin erythema. Importantly, this woman had never experienced any sun-induced facial erythema in her life, even after intense sun exposure during vacations. In agreement with the genetic diagnosis of BS, she had an elongated face with a prominent nose (Fig. 1. a, b). Furthermore, a generalized deficiency of subcutaneous fat was evident, and skin examination revealed two large café-au-lait lesions and some confetti-like spots on the inner surface of the right thigh (Fig. 1. c).

BS patient homozygous for the BLM c.1642C>T pathogenic variant: a, b. Facial features of the patient; c. Skin café-au-lait spots
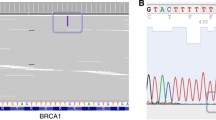
To prove the presence of functional defects characteristic of BS, we performed a sister chromatid exchange (SCE) test [3]. In brief, PHA-stimulated peripheral blood lymphocytes from a healthy donor and a patient were cultured in the presence of 5-bromo-2’-deoxyuridine (BrdU) for two cycles of DNA replication. Differential staining of metaphase chromosome sister chromatids was carried out using acridine orange. At least 10 metaphase plates were analyzed for each individual. A parametric Student’s t-test with Welch’s correction was used to compare the data sets. The number of SCEs in the healthy donor cells varied from 4 to 11 (mean number 7.36 ± 2.06 per metaphase plate) compared with 91 to 178 (126 ± 30.15 per metaphase plate) in the patient (p < 0.0001) (Fig. 2. a, b). The obtained data were in agreement with the results of DNA testing performed by various methods (Fig. 2. c, d).

Laboratory confirmation of BS diagnosis. SCE assay in the healthy donor (a.) and the BS patient (b.); arrows indicate examples of SCE. c. Sanger sequencing for the blood DNA sample from the BS patient. Homozygous BLM c.1642C>T substitution (NM_000057.3) is marked by arrow. d. NGS reads of the BLM gene fragment in a breast carcinoma sample obtained from the patient. e. Copy number alteration profile of the breast tumor
No additional BLM mutations was detected in breast carcinoma sample (Fig. 2. d). Tumor-derived DNA was further analyzed with the TruSight Tumor 170 panel (Illumina) for somatic mutations and the SeqCap EZ CNV/LOH Backbone Design panel (Roche) for somatic copy number alterations, as described previously [4]. The tumor mutation burden (TMB) was low. The homologous recombination deficiency (HRD) score approached 29, indicating a moderate level of chromosomal instability. The tumor harbored RB1 p.Glu137fs and TP53 p.Arg248Gln somatic mutations, amplifications of the EGFR and AR genes, and gains at chromosomal loci 6q16.3-6q21 (ATG5, AIM1) and 19q13.12-19q13.2 (PAK4, AKT2, CIC) (Fig. 2. e).
Discussion
Predisposition to tumor development is well described for BS patients. The cumulative incidence of any malignancy by the age of 40 years reaches 83%, with leukemia and lymphoma being the most common tumor types [5]. Our patient was diagnosed with breast cancer at the age of 38, which is slightly older than the median age of 33 reported in women with BS [6]. Notably, radiation therapy was well tolerated; this observation is in accordance with published data [7]. It is generally advised that cancer patients with BS should be considered for a reduction of chemotherapy dose, although the appropriate experience supporting this recommendation is limited [8]. The patient described in this report showed normal tolerability to cytotoxic drugs. For example, capecitabine-induced skin toxicity, which was observed in this woman, is characteristic for more than 60% of patients receiving this drug [9].
Although sun-induced facial erythema is still considered a salient feature of BS, there is a growing body of evidence indicating that some patients may lack this symptom [10,11,12,13]. Intrafamilial variability of dermatological manifestations was reported in two Turkish siblings with BS, where only the girl but not the boy had sun-sensitive skin lesions [14]. Three reports [10, 11, 13] described patients who were homozygous for the BLM c.1642C>T Slavic allele. Trizuliak et al. described a 35-year-old woman with short stature, facial dysmorphism, premature ovarian failure, endocrine abnormalities, bronchiectasis, and recurrent malignant lymphoma diagnosed at 15 and 34 years of age. She was homozygous for the BLM c.1642C>T allele. In contrast, her brother, who also had short stature and facial dysmorphism and was found to be homozygous for the same allele, had telangiectasias in sun-exposed areas and no history of cancer. Vojtková et al. reported BS in three Slovak siblings (a 13-year-old boy, an 11-year-old girl, and a 4-year-old boy). None of the patients had a history of cancer at the time of diagnosis. Two of the patients had no facial photosensitivity or telangiectasia, while one boy had mild erythema on the cheek and above the upper lip. Moreover, some patients with other BLM genotypes, e.g., c.2207_2212delinsTAGATTC (p.Tyr736Leufs*5) / c.3681del (p.Lys1227Asnfs*52) [12], also did not show characteristic facial erythema. Despite the absence of the above symptom, our patient had a few other mild manifestations suggestive for BS, for example, short stature, long thin face and café-au-lait spots in the skin (Fig. 1). However, the diagnostic significance of this features has become evident only upon the receipt of the results of germline DNA testing, as this woman was never considered by her doctors as a candidate for genetic counselling.
Previous studies involving homozygous carriers of the Slavic BLM c.1642C>T (p.Gln548Ter) allele did not consider the SCE count. Historically, SCE analysis has been used as a main laboratory assay for the confirmation of BS; however, it has been replaced by DNA testing in recent years. We decided to perform SCE quantitation for our patient because, unlike the majority of other patients with BS, she remained in good health before cancer onset. This test revealed an elevated number of SCEs, thus confirming the pathogenic role of the Slavic BLM c.1642C>T (p.Gln548Ter) variant. In theory, this elevation may be attributed to prior chemotherapy. Tekcan et al. performed an analysis of SCEs in 25 breast cancer patients receiving chemotherapy. The SCE values were 8.25 ± 3.67, 10.19 ± 2.95 and 11.52 ± 3.33 in the pretreatment period, during treatment and remission, respectively, while this estimate approached 7.01 ± 1.24 in healthy controls [15]. However, the sample of our patient obtained in the remission phase demonstrated an SCE value of 126 ± 30.15, which is an order of magnitude greater than the normal variation, and therefore is very unlikely to be attributed solely to prior treatment.
Protein truncating pathogenic variants usually have significant functional consequences, leading to a more severe phenotype. Typically, manifestations are more subtle when the affected codon is near the end of translation. This is definitely not the case for BLM p.Gln548Ter, as the canonical transcript consists of 1417 amino acids.
Heterozygous carriers of BLM pathogenic variants have normal phenotype; however, they may be at increased risk of developing breast cancer [2, 16]. Importantly, the penetrance of monoallelic BLM germline mutations is at best moderate [2], with some studies failing to confirm an association between BLM heterozygosity and elevated tumor susceptibility [1]. Consequently, while all gene panels utilized for DNA germline testing always consider highly penetrant genes, e.g., BRCA1, BRCA2, PALB2 and TP53, there is no consistency with regard to the genes associated with an approximately 2-fold increase in cancer risk, such as BLM or NBN [17]. Importantly, all these diagnostic efforts are usually targeted at the identification of heterozygous carriers of tumor-predisposing pathogenic variants. This case report highlights a previously unrecognized concern: indeed, systematic genetic analysis of cancer patients has the potential to reveal instances of biallelic inactivation of genes that are associated with increased cancer risk but otherwise healthy status in a heterozygous state, however, usually cause very severe disorders while being fully inactivated at the germline. The relevant examples may include ataxia-telangiectasia (ATM), Nijmegen breakage syndrome (NBN), Bloom syndrome (BLM), and Fanconi anemia (FANCA, FANCC, FANCG, etc.). Recent studies have revealed instances of an unusually mild manifestation of serious autosomal recessive conditions such as cystic fibrosis [18], congenital myotonia [19], and myopathy [20], and our observation of well-compensated patient with Bloom syndrome adds to this list.
This study indicates that some patients with BS may remain relatively healthy at least until adulthood. Factors affecting the severity of BS should be investigated. BLM gene analysis may be considered a part of standard DNA germline testing for patients with cancer.
Data availability
All the data generated or analyzed during this study are included in this article. Further enquiries can be directed to the corresponding author.
References
Cybulski C, Kluźniak W, Huzarski T, Wokołorczyk D, Kashyap A, Rusak B et al (2019) The spectrum of mutations predisposing to familial breast cancer in Poland. Int J Cancer 145:3311–3320. https://doi.org/10.1002/ijc.32492
Sokolenko AP, Iyevleva AG, Preobrazhenskaya EV, Mitiushkina NV, Abysheva SN, Suspitsin EN et al (2012) High prevalence and breast cancer predisposing role of the BLM c.1642 C > T (Q548X) mutation in Russia. Int J Cancer 130:2867–2873. https://doi.org/10.1002/ijc.26342
Dracopoli NC, Haines JL, Korf BR, Morton CC, Seidman CE, Rosenzweig A, Seidman JG, Smith DR (2004) Short protocols in Human Genetics: a compendium of methods from current protocols in Human Genetics. Wiley, Hoboken
Sokolenko AP, Gorodnova TV, Bizin IV, Kuligina ES, Kotiv KB, Romanko AA et al (2021) Molecular predictors of the outcome of paclitaxel plus carboplatin neoadjuvant therapy in high-grade serous ovarian cancer patients. Cancer Chemother Pharmacol 88:439–450. https://doi.org/10.1007/s00280-021-04301-6
Sugrañes TA, Flanagan M, Thomas C, Chang VY, Walsh M, Cunniff C (2022) Age of first cancer diagnosis and survival in Bloom syndrome. Genet Med 24:1476–1484. https://doi.org/10.1016/j.gim.2022.03.008
Cunniff C, Djavid AR, Carrubba S, Cohen B, Ellis NA, Levy CF et al (2018) Health supervision for people with Bloom syndrome. Am J Med Genet A 176:1872–1881. https://doi.org/10.1002/ajmg.a.40374
Schoenaker MHD, Takada S, van Deuren M, Dommering CJ, Henriët SSV, Pico I et al (2021) Considerations for radiotherapy in Bloom Syndrome: a case series. Eur J Med Genet 64:104293. https://doi.org/10.1016/j.ejmg.2021.104293
Langer K, Cunniff CM, Kucine N (2006) Bloom Syndrome. https://www.ncbi.nlm.nih.gov/books/NBK1398/
Blum JL, Barrios CH, Feldman N, Verma S, McKenna EF, Lee LF et al (2012) Pooled analysis of individual patient data from capecitabine monotherapy clinical trials in locally advanced or metastatic breast cancer. Breast Cancer Res Treat 136:777–788. https://doi.org/10.1007/s10549-012-2288-x
Vojtková J, Čiljaková M, Jeseňák M, Mišovicová N, Bánovčin P (2016) Bloom syndrome without typical sun-sensitive skin lesions in three Slovak siblings. Int J Dermatol 55:687–690. https://doi.org/10.1111/ijd.13009
Suspitsin EN, Sibgatullina FI, Lyazina LV, Imyanitov EN (2017) First two cases of Bloom Syndrome in Russia: lack of skin manifestations in a BLM c.1642C > T (p.Q548X) homozygote as a likely cause of Underdiagnosis. Mol Syndromol 8:103–106. https://doi.org/10.1159/000454820
Bouman A, van Koningsbruggen S, Karakullukcu MB, Schreuder WH, Lakeman P (2018) Bloom syndrome does not always present with sun-sensitive facial erythema. Eur J Med Genet 61:94–97. https://doi.org/10.1016/j.ejmg.2017.10.010
Trizuljak J, Petruchová T, Blaháková I, Vrzalová Z, Hořínová V, Doubková M et al (2020) Diagnosis of Bloom Syndrome in a patient with short stature, recurrence of malignant lymphoma, and Consanguineous Origin. Mol Syndromol 11:73–82. https://doi.org/10.1159/000507006
Boduroğlu K, Tunçbilek E (1999) Two siblings with Bloom’s syndrome exhibit different clinical features: possible effect of sex. Turk J Pediatr 41:107–111
Tekcan A, Elbistan M, Ulusoy AN (2012) Sister chromatid exchanges in breast cancer patients who underwent chemotherapy. J Toxicol Sci 37:235–243. https://doi.org/10.2131/jts.37.235
Thompson ER, Doyle MA, Ryland GL, Rowley SM, Choong DY, Tothill RW et al (2012) Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles. PLoS Genet 8:e1002894. https://doi.org/10.1371/journal.pgen.1002894
Easton DF, Pharoah PD, Antoniou AC, Tischkowitz M, Tavtigian SV, Nathanson KL et al (2015) Gene panel sequencing and the prediction of breast cancer risk. N Engl J Med 372:2243–2257. https://doi.org/10.1056/NEJMsr1501341
Tümmler B (2019) Mild cystic fibrosis in carriers of two nonsense mutations - a case of genetic compensation response? J Cyst Fibros 18:e51–e52. https://doi.org/10.1016/j.jcf.2019.06.012
Gurgel-Giannetti J, Senkevics AS, Zilbersztajn-Gotlieb D, Yamamoto LU, Muniz VP, Pavanello RC et al (2012) Thomsen or Becker Myotonia? A novel autosomal recessive nonsense mutation in the CLCN1 gene associated with a mild phenotype. Muscle Nerve 45:279–283. https://doi.org/10.1002/mus.22252
Tajsharghi H, Hilton-Jones D, Raheem O, Saukkonen AM, Oldfors A, Udd B (2010) Human disease caused by loss of fast IIa myosin heavy chain due to recessive MYH2 mutations. Brain 133:1451–1459. https://doi.org/10.1093/brain/awq083
Funding
The study was supported by the Russian Science Foundation [grant no. 22-15-00266].
Author information
Authors and Affiliations
Contributions
Conceptualization: E.S. and E.I. Medical management of the patient: D.E. and E.S. Formal analysis and investigation: O.C., E.B., S.A., and A.S. Writing – original draft preparation: E.S., A.S., and E.I. Writing – review and editing: all authors. Funding acquisition: A.S.
Corresponding author
Ethics declarations
Ethical approval
This study protocol was reviewed and approved by the local Ethics Committee of the N.N. Petrov Institute of Oncology, approval number [No. 20/25 23.01.2020].
Consent to participate
The patient provided written informed consent to participate in the study.
Consent to publish
Written informed consent was obtained from the patient for publication of this case report and any accompanying images.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Suspitsin, E., Eliseyeva, D., Chiryaeva, O. et al. Asymptomatic Bloom syndrome diagnosed by chance in a patient with breast cancer. Familial Cancer (2024). https://doi.org/10.1007/s10689-024-00420-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10689-024-00420-0