
Abstract
This study investigated the genetics of seedlessness in grape (Vitis vinifera L.) by the application of specific locus amplified fragment sequencing (SLAF-seq) and genome-wide association mapping. SLAF-seq was used to provide dense genome-wide marker coverage for the 199 grape accessions to screen for genetic controls over seedlessness. After comparison of genome sequences with the reference genome, single nucleotide polymorphisms (SNPs) were developed. A phylogenetic tree of the 199 accessions was then constructed and investigated using principal component analysis (PCA). Linkage disequilibrium between the SNPs was estimated and a genome wide association study (GWAS) was performed to detect trait loci and candidate genes associated with seedlessness. In total, 414,223 SNPs were identified, and were used for the fine mapping of seed development genes. The r2 decay to half of its initial value was 11.57 kb. The phylogenetic tree showed that the 199 accessions were clustered into 12 branches; PCA indicated that most grape accessions were clustered. Analysis using a general linear model identified 294 SNPs significant correlation with seedlessness, while the compressed mixed linear model identified 82 SNPs. Candidate genes involved in seed development were identified at loci near the significant SNPs. These candidates included ubiquitin protein, abscisic aldehyde oxidase, ethylene responsive transcription factor, zinc finger protein, somatic embryogenesis receptor, and MADs-box genes. This study demonstrates that integrating SLAF-seq and GWAS are powerful complementary approaches for dissecting complex traits in the grape.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Grape (Vitis vinifera L.) is one of the most economically important fruit crops worldwide. For table grapes, large berries and seedlessness are the most appreciated traits (Doligez et al. 2013). Two types of genetically mediated seedlessness exist in grape, parthenocarpy and stenospermocarpy (Bouquet and Danglot 1996; Cabezas et al. 2009; Ingrosso et al. 2011; Sarikhani et al. 2009). In parthenocarpy, the ovule develops without fertilization, yielding small berries that completely lack seeds. In stenospermocarpy, although pollination occur, the seeds fail to develop due to the early degeneration of the endosperm and abnormal development of integuments (Pratt 1970; Ledbetter and Ramming 2011). Unlike parthenocarpy, stenospermocarpy can produce berries of a size compatible with commercial requirements (Ledbetter and Burgos 1994). However, the development of new stenospermocarpy varieties involves the generation and selection of large numbers of hybrids each year, which is both expensive and time consuming (Karaagac et al. 2012). Therefore, breeding stenospermocarpy varieties tends to focus on identifying and exploiting molecular markers associated with genes that control seedlessness (Striem et al. 1994, 1996).
Many models for the genetic basis of seedlessness in grape have been proposed based on the traits associated with the molecular markers. Currently, the most widely accepted model involves three independent and complementary recessive genes, which are controlled by a dominant regulator locus called seed development inhibitor (SDI) (Bouquet and Danglot 1996). The seedless phenotype is determined by the presence of a dominant allele at the SDI locus, which has been located on chromosome 18 by quantitative trait locus (QTL) mapping (Cabezas et al. 2009; Doligez et al. 2002; Mejia et al. 2007). The VvAGL11 gene is an orthologue of a MADS-box gene involved in ovule differentiation, which has been suggested as the most probable candidate gene at the SDI locus for seedlessness (Mejía et al. 2011; Bergamini et al. 2013).
Various studies have sought to identify genes for seedlessness through the comparison of gene expression profiles in seeded and seedless grape varieties. Based on differential gene expression patterns in seeded and seedless cultivars, Wang et al. (2011) found that VvCBP1 was essential for embryo development in seedless grape. In addition, Nwafor et al. (2014) produced a list of genes that were differentially expressed in a seedless mutant compared to the wild-type. Although numerous studies have been carried out, our understanding of the genetic basis of seedlessness remains limited.
Genome-wide association study (GWAS) has been widely used to identify QTL and candidate genes due to its ability to analyse genetic controls of complex traits (Yan et al. 2011; Verslues et al. 2014; Mamidi et al. 2013). In this study, we performed specific-locus amplified fragment sequencing (SLAF-seq) and identified single nucleotide polymorphisms (SNPs) in 199 grape accessions. Subsequently, we applied GWAS for detection of trait loci and candidate genes associated with seedlessness.
Materials and methods
Mapping population
In the present study, 199 grape accessions were evaluated (Supplement Table 1). These accessions were obtained from the National Grape Germplasm Repository at Zhengzhou Fruit Research Institute of Chinese Academy of Agricultural Sciences (113°42′E and 34°42′N). The accessions for each treatment contained three plants, and a completely randomized selection with twenty replicates was used. For each accession, the row spacing was 2.5 m, and the planting distance was 1.0 m. The plants were cultivated under the same environmental conditions, without any growth regulator, and all plants were 12 years old. Seeds were scored repeatedly during four growing seasons (2012–2015 years) according to criteria of the OIV (International Organisation of Vine and Wine; Anonymous 1983). Among 199 accessions, 124 were seeded and 75 were seedless (OIV descriptor 241).
SLAF-seq and SNP identification
Genomic DNAs from each grape accession were isolated from fresh leaves using a previously described method (Murray et al. 1980). SNP genotyping was performed using SLAF-seq. Different restriction enzyme combinations were selected using in silico digestion-site prediction to obtain more than 400,000 sequencing tags of 300–500 bp per genome that were distributed evenly in unique genomic regions. Sequencing tags of 314–464 bp lengths were selected as SLAF labels. Double digestion with the enzymes RaseI and HaeIII (New England Biolabs, NEB, USA) were found to be the most suitable. The SLAF tags were evenly distributed through the genome. The sequence reads at both ends of each fragment in each library were generated using Illumina HiSeq™2500 (Illumina, Inc.; San Diego, CA, USA), with a barcode approach to identify each sample.
All reads were processed for quality control and then filtered with Seqtk (https://github.com/lh3/seqtk). High-quality paired-end reads were mapped onto the reference grape genome (PN40024) (Jaillon et al. 2007) using the Short Oligonucleotide Alignment Program 2 (SOAP2, Version_2.20) (Li et al. 2009b; Li and Durbin 2010). The sequences were downloaded from the grape database, which was available at ftp://ftp.ensemblgenomes.org/pub/release-23/plants/fasta/vitis_vinifera/dna/. After comparing the reference genome with the obtained genomic sequences using the Burrows Wheeler Aligner (BWA, Version_0.7.10-r789) software package (Li and Durbin 2010), SNPs were developed with the Genome Analysis Toolkit (GATK, Version_3.2) (Mckenna et al. 2010) and SAMtools (Version_1.1) (Li et al. 2009a) software packages. The SNPs were filtered according to minor allele frequency (MAF) >0.05 and integrity of each SNP >0.8.
Population structure evaluation
The SNPs were used for population structure evaluation filtered after the integrity of each SNP more than 0.8 and the minor allele frequencies (MAF) more than 0.05. A phylogenetic tree of the grape accessions based on SNPs was constructed using the neighbour-joining algorithm (Saitou and Nei 1987) and the MEGA5 software package (Tamura et al. 2011). Population structure of the grape accessions was analysed by the admixture software package (Alexander et al. 2009). Principal component analysis (PCA) (Steinherz et al. 1999) was performed using the cluster software package (De Hoon et al. 2004).
Linkage disequilibrium (LD) analysis and genome-wide association mapping
LD between pairs of SNPs was estimated as r2 using TASSEL version 3.0 software package (Bradbury et al. 2007). 414,223 SNPs from the grape accessions after filtered by the integrity of each SNP more than 0.8 and the MAF more than 0.05, were used for the association analysis using a general linear model (GLM) and compressed mixed linear model (MLM) in TASSEL. The p value was adjusted with the Bonferroni method at α ≤ 0.1 (corresponding to P ≤ 2.4 × 10−7) and α ≤ 0.01 (corresponding to P ≤ 2.4 × 10−8) to determine whether the association was significant (Holm 1979).
Results
SLAF-seq and SNP identification of grape accessions
An association map was constructed for the 199 grape accessions to enable fine mapping of seedlessness genes. Sequencing of the SLAF libraries yielded approximately 404 million paired-end reads, and 88.65% of these were successfully mapped to the grape reference genome. The average Q30-value of these reads was 90.93%, and GC content was 39.05%. A total of 421,204 high quality SLAF tags were obtained from the 199 genotypes, of which 327,872 were polymorphic. In total, 4,180,905 SNPs were identified from these SLAF-seq tags; 414,223 SNPs were selected by the criteria MAF >0.05 and integrity of each SNP >0.8 (Table 1). These 414,223 SNPs markers covered all 19 chromosomes. The maximum number of SNPs was located on chromosomes 14 (26,175 SNPs) and 18 (25,948 SNPs); the minimum number of SNPs was observed on chromosomes 2 (16,078 SNPs) and 17 (16,228 SNPs).
LD and population evolution
The extent of LD depended on mapping resolution and the required marker density for GWAS. r2 was quantified between different physical distances. The 414,223 SNPs detected in the 199 accessions were used for pair-wise analyses. The r2 decay to half of its initial value was 11.57 kb (Fig. 1a). On the basis that the reference genome size was 486,265,422 bp, we estimated our SNP density is approximately one SNP per 1.17 kb.


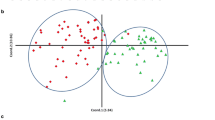
Genetic diversity and population structure of 199 accessions. a Linkage disequilibrium (LD) pattern of the grape genome; b phylogenetic tree; c population structure. Each colour represents a group, and each row represents a stakeholder value. d Diagram shows the value of 199 samples based on clustering from 1 to 20; e principal components analysis (PCA). (Color figure online)
The relationships among the grape accessions were assessed by analysis of the population structure, construction of a phylogenetic tree, and by PCA. The population structure analysis identified 414,223 SNPs in the 199 grape accessions. Little divergence was indicated between the grape groups. The phylogenetic tree showed that the 199 accessions could be clustered into 12 branches (Fig. 1b). Different numbers of K populations were explored to reveal the hierarchical population structure. As shown in Fig. 1c and d, this analysis estimated the most likely number of populations at K = 12. A three-dimensional projection of each sample from the PCA analysis was plotted on a scatter plot. Most accessions were found to cluster despite the fact that several accessions were discriminative (Fig. 1e).
Genome-wide association analyses of loci for seedlessness traits
The genetic basis for seedlessness was investigated using an association panel from the 199 genotypes and 414,223 SNP markers. This analysis suggested that a major genetic locus for seedlessness might be located on Chr 18. The GLM and MLM analyses found 294 and 82 SNPs, respectively, which were significantly related to seed formation, with gene variances of 18% and 24%, respectively (Table 2; Fig. 2). From the MLM analysis, 74 SNPs were found to be located at 25.8–29.1 Mb on Chr 18; rs1826891824 was significantly associated with seed formation and may be an important locus for seed formation. Comparison of the positions of the SNPs detected by GWAS with previous QTL studies showed that the SNP rs1826891824 practically overlapped the simple sequence repeat (SSR) marker VMC7f2, which is associated with seedlessness (Fig. 3).

Genome-wide association scan for seedlessness. a Manhattan plots for the MLM; the x-axis shows SNPs along each chromosome; the y-axis is the −log10 (P-value) for the association. The different colors indicate the 19 different and unmapped chromosomes of grape. Red and blue horizontal lines indicate the genome-wide significance and extreme significance threshold, respectively. b Quantile–quantile plot for MLM. The horizontal axis shows −log10-transformed expected P-values, while the vertical axis shows −log10-transformed observed P-values. c Manhattan plots for the GMLM; d Quantile–quantile plot for GLM. (Color figure online)

Regions of the genome showing strong association signals near previously identified SSR markers. The vertical red line indicates the SNPs of lowest P-value. The blue horizontal dashed lines indicate the genome-wide significance and extreme significance threshold, respectively. (Color figure online)
To confirm the beneficial allele at each peak SNP associated with the seedless phenotype, gene models that located in LD decay distance genomic region upstream and downstream of each peak SNP in the reference grape genome were considered to be seedless gene candidates in the present study. Hundreds of grape genes were identified in the flanking regions of each peak SNP based on MLM, however, most of these had no functional annotation or belonged to unknown function families. The candidate genes that had functional annotations are summarized in Supplementary Table 2.
Discussion
GWAS with LD analysis provides an effective tool for the identification of genetic loci controlling quantitative traits, and has been widely applied to investigate correlation genes. To date, a number of reduced representation sequencing methods have been developed, such as genotyping by sequencing (GBS) (Elshire et al. 2011), type IIB restriction site associated DNA (2b-RAD) (Wang et al. 2012), and SLAF-seq (Sun et al. 2013). We chose the latter for this study because of its advantages, such as lower sequencing costs, higher genotyping accuracy, and efficient detection system. In this study, SNP markers with an average density of one SNP per 11.57 kb were obtained. This marker density was sufficient to achieve association mapping of the seedlessness trait.
Data of this study found that the major genetic locus responsible for seedlessness may be at Chr18, suggesting that Chr 18 may be a very important chromosome for seedlessness in grape. The SSR marker VMC7f2 is located on Chr 18 and has been confirmed to have an inhibitory effect on seed development. Adam-Blondon et al. (2001) reported that SSC8 was a useful marker for seedlessness in grape. Additionally, Korpás et al. (2009) have shown that both SCC8 and SCF27 are linked to SDI, and are necessary but not sufficient loci for the seedless phenotype in grape. Interestingly, association analyses found that the SSR marker VMC7f2 was closely related to this QTL, and was therefore useful for selection of seedlessness. Cabezas et al. (2009) reported a major effect QTL located on Chr 18 which explained 50% of the phenotypic variation for fresh seed weight. In the present study, the SNP rs1826891824 (P-value = 1.43E-9, phenotypic variation 16.3%), was found to be associated with the seedlessness trait and was shown to be located within VMC7f2 and to practically overlap with the seedlessness SSR marker VMC7f2. The SNP rs1826891824 was 3.2 kb distant from VIT_18s0041g01880 (VvAGL11: MADS-box protein SEEDSTICK) (Nwafor et al. 2014).
MADS-box genes encode transcription factors with a highly conserved DNA-binding domain termed MADS domain, which are involved in the regulation of many aspects of plant growth and development, such as embryonic development, floral organ determination, ovule development and fruit ripening (Wang et al. 2015; Immink et al. 2009). MADS-box genes generally comprise large families, among which, VvAGL11 has been proposed to be the major functional candidate gene for seedlessness through variations in its promoter region (Mejía et al. 2011). Additionally, VvAGL11 shows homology to the Seedstick/Agamous-like 11 (STK/AGL11) gene, which is expressed during Arabidopsis seed development. VvAGL11 is the closest to the seedlessness SSR marker VMC7f2, and also contains the SDI locus, suggesting that VvAGL11 may be a functional candidate gene for the seedlessness trait (Costantini et al. 2007; Mejía et al. 2011; Bergamini et al. 2013).
Some of the genes that were detected in the seedlessness association analysis were also associated with berry development and berry size traits fell within the previously reported genes (Boss et al. 2002; Houel et al. 2010; Guillaumie et al. 2011; Doligez et al. 2013; Wang et al. 2016; Muñoz-Espinoza et al. 2016). Two SNP loci (rs1826914496 and rs176825328) associated with the genes VvAG3 (MADS-box ovule identity-MIKC gene that was expressed in flowers and berries) and EXPA (Alpha-expansin with expression linked to berry development) were detected from the seedlessness trait analysis. As berry growth and seed formation are known to be related, the correlation between the subtraits of seedlessness and berry weight/development observed at the phenotypic and genetic levels may be due to pleiotropy and may be directly or indirectly affected by the growth regulator produced by seeds (Mejía et al. 2011). Similarly, the correlation between flowering time and seedlessness traits observed at the SNP locus rs188117546 (VVC2892A: expressed in very early stages of floral development) (Doligez et al. 2013) may be due to that genes control the two trait functions independently of each other.
Minor loci were detected on other chromosomes; these may be due to environment-genotype interactions or to the limited detection power because of a combination of moderate population size and at least one major locus responsible for most of the phenotypic variance. In the present study, some loci were found to be consistent with several previously reported loci or related genes for seed traits. Nwafor et al. (2014) identified the gene VIT_12s0059g00560 associated with seed development and the gene located on the SNP rs125367605 downstream 43 kb. Additionally, Royo et al. (2016) reported that the gene VIT_05s0020g02350 plays an important role in seed development and the gene located on the SNP rs54076883 downstream 40 kb.
Hundreds of positional candidate genes were found using SNPs, some of which might be valuable in grape because functional annotation indicated that their potential orthologues were involved in determining seed contents. The expression patterns of genes encoding ubiquitin protein, abscisic aldehyde oxidase, ethylene responsive transcription factor, zinc finger protein, somatic embryogenesis receptor, and MADS-box gene were also obtained by GWAS. The studies of transgenic tobacco and potato that express ubiquitin extension protein promoter-GUS fusion, showed that the uidA gene is expressed in meristematic tissues, pollen and ovules (Garbarino and Belknap 1994; Callis et al. 1990). Hanania et al. (2009) reported that overexpression of the ubiquitin extension protein S27a in carpels and integuments may lead to embryo abortion and seedlessness in grape. The abscisic aldehyde oxidase gene is found to catalyze the final step in the biosynthesis of abscisic acid, and the abscisic acid plays a major role in seed development (González-Guzmán et al. 2004). Zinc finger protein genes constitute a large and complex gene families in the plant genomes, which have been described to have functions on seed development (Zhou et al. 2012). Furthermore, some genes are expected to control processes associated with seed development (such as ethylene, gibberellin and auxin), sugar biosynthesis and transport, cell division or elongation, and signal transduction (Ledbetter and Ramming 2011; Peng et al. 2007; Lijavetzky et al. 2012). Furthermore, MADS-box transcription factors play an important role in floral and ovule development, which may be associated with seedlessness traits (Wang et al. 2015).
Conclusions
A high-density map of SNPs markers from 199 grape accessions was developed using SLAF-seq technology and used for association mapping to investigate the genetic control of seedlessness traits. A major locus with the largest effect on Chr 18 was found, along with some minor loci on different chromosomes. This study demonstrates that SLAF-seq and GWAS are powerful approaches for the dissection of complex traits in grape.
Abbreviations
- SDI:
-
Seed development inhibitor
- QTL:
-
Quantitative trait locus
- GWAS:
-
Genome-wide association study
- SLAF:
-
Specific-locus amplified fragment sequencing
- SNPs:
-
Single nucleotide polymorphisms
- OIV:
-
International Organisation of Vine and Wine
- MAF:
-
Minor allele frequencies
- LD:
-
Linkage disequilibrium
- GLM:
-
General linear model
- MLM:
-
Compressed mixed linear model
- SSR:
-
Simple sequence repeat
- GBS:
-
Genotyping by sequencing
- 2b-RAD:
-
IIB restriction site associated DNA
References
Adam-Blondon AF, Lahogue-Esnault F, Bouquet A, Boursiquot JM, This P (2001) Usefulness of two SCAR markers for marker-assisted selection of seedless grapevine cultivars. Vitis 40(3):147–155
Alexander DH, Novembre J, Lange K (2009) Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19(9):1655–1664
Anonymous (1983) Descriptor list for grapevine varieties and Vitis species. Office International de la Vigne et du Vin (OIV), Paris
Bergamini C, Cardone MF, Anaclerio A, Perniola R, Pichierri A, Genghi R, Alba V, Forleo LR, Caputo AR, Montemurro C (2013) Validation assay of p3_VvAGL11 marker in a wide range of genetic background for early selection of stenospermocarpy in Vitis vinifera L. Mol Biotechnol 54(3):1021–1030
Boss PK, Sensi E, Hua C, Davies C, Thomas MR (2002) Cloning and characterisation of grapevine (Vitis vinifera L.) MADS-box genes expressed during inflorescence and berry development. Plant Sci 162(6):887–895
Bouquet A, Danglot Y (1996) Inheritance of seedlessness in grapevine (Vitis vinifera L.). Vitis 35(1):35–42
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23(19):2633–2635
Cabezas JA, Cervera MT, Ruiz-García L, Carreño J, Martínez-Zapater JM (2009) A genetic analysis of seed and berry weight in grapevine. Genome 52(12):215
Callis J, Raasch JA, Vierstra RD (1990) Ubiquitin extension proteins of Arabidopsis thaliana. Structure, localization, and expression of their promoters in transgenic tobacco. J Biol Chem 265(21):12486–12493
Costantini L, Battilana J, Emanuelli F, Fanizza G, Grando MS (2007) Berry and phenology related traits in grapevine (Vitis vinifera L.): from QTLs to underlying genes. BMC Plant Biol 8:38
De Hoon MJL, Imoto S, Nolan J, Miyano S (2004) Open source clustering software. Bioinformatics 20(9):1453–1454
Doligez A, Bertrand Y, Farnos M, Grolier M, Romieu C, Esnault F, Dias S, Berger G, François P, Pons T (2013) New stable QTLs for berry weight do not colocalize with QTLs for seed traits in cultivated grapevine (Vitis vinifera L.). BMC Plant Biol 13(1):1–16
Doligez A, Bouquet A, Danglot Y, Lahogue F, Riaz S, Meredith P, Edwards J, This P (2002) Genetic mapping of grapevine (Vitis vinifera L.) applied to the detection of QTLs for seedlessness and berry weight. Theor Appl Genet 105(5):780–795
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6(5):e19379
Garbarino JE, Belknap WR (1994) Isolation of a ubiquitin-ribosomal protein gene (ubi3) from potato and expression of its promoter in transgenic plants. Plant Mol Biol 24(1):119–127
Guillaumie S, Fouquet R, Kappel C, Camps C, Terrier N, Moncomble D, Dunlevy JD, Davies C, Boss PK, Delrot S (2011) Transcriptional analysis of late ripening stages of grapevine berry. BMC Plant Biol 11(1):1–27
Hanania U, Velcheva M, Sahar N, Flaishman M, Or E, Degani O, Perl A (2009) The ubiquitin extension protein S27a is differentially expressed in developing flower organs of Thompson seedless versus Thompson seeded grape isogenic clones. Plant Cell Rep 28(7):1033–1042
Holm S (1979) A simple sequentially rejective multiple test procedure. Scand J Stat 6(2):65–70
Houel C, Bounon R, Chaïb J, Guichard C, Péros JP, Bacilieri R, Dereeper A, Canaguier A, Lacombe T, N’Diaye A (2010) Patterns of sequence polymorphism in the fleshless berry locus in cultivated and wild Vitis vinifera accessions. BMC Plant Biol 10(1):284
Ingrosso I, Bonsegna S, De DS, Laddomada B, Blando F, Santino A, Giovinazzo G (2011) Over-expression of a grape stilbene synthase gene in tomato induces parthenocarpy and causes abnormal pollen development. Plant Physiol Biochem 49(10):1092–1099
Immink RG, Tonaco IA, Folter SD, Shchennikove A, Dijk AD, Busscherlange J et al (2009) SEPALLATA3: the ‘glue’ for MADS box transcription factor complex formation. Genome Biol 10(2):1–16
Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C (2007) The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449(7161):463–467
Karaagac E, Vargas AM, Andrés MTD (2012) Marker assisted selection for seedlessness in table grape breeding. Tree Genet Genomes 8(5):1003–1015
Korpás A, Baránek M, Pidra M, Hradilík J (2009) Behaviour of two SCAR markers for seedlessness within Central European varieties of grapevine. Vitis 48(1):33–42
Ledbetter CA, Burgos L (1994) Inheritance of Stenospermocarpic Seedlessness in Vitis vinifera L. J Hered 27(2):1957–1958
Ledbetter CA, Ramming DW (2011) Seedlessness in grapes. Hortic Rev 11:159–184
Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 25(5):1754–1760
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009a) The sequence alignment-map format and SAMtools. Bioinformatics 25(16):2078–2079
Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J (2009b) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25(15):1966–1967
Lijavetzky D, Carbonellbejerano P, Grimplet J, Bravo G, Flores P, Fenoll J, Hellín P, Oliveros JC, Martínezzapater JM (2012) Berry flesh and skin ripening features in Vitis vinifera as assessed by transcriptional profiling. PLoS ONE 7(6):1634
Mamidi S, Lee RK, Goos JR, Mcclean PE (2013) Genome-wide association studies identifies seven major regions responsible for iron deficiency chlorosis in soybean (Glycine max). PLoS ONE 9(9):e107469–e107469
Mckenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M (2010) The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res 20(9):1297–1303
Mejía N, Soto B, Guerrero M, Casanueva X, Houel C, Miccono ML, Ramos R, Le CL, Boursiquot JM, Hinrichsen P (2011) Molecular, genetic and transcriptional evidence for a role of VvAGL11 in stenospermocarpic seedlessness in grapevine. BMC Plant Biol 11(1):29–33
Mejia N, Gebauer M, Munoz L, Hewstone N, Munoz C, Hinrichsen P (2007) Identification of QTLs for seedlessness, berry size, and ripening date in a seedless × seedless table grape progeny. Am J Enol Vitic 58(4):499–507
Miguel González-Guzmán DA, Salinas Julio, Serrano Ramón, Rodríguez Pedro L (2004) Two new alleles of the abscisic aldehyde oxidase 3 gene reveal its role in abscisic acid biosynthesis in seeds. Plant Physiol 135(1):325–333
Muñoz-Espinoza C, Genova AD, Correa J, Silva R, Maass A, González-Agüero M, Orellana A, Hinrichsen P (2016) Transcriptome profiling of grapevine seedless segregants during berry development reveals candidate genes associated with berry weight. BMC Plant Biol 16(1):1–17
Murray MG, Wf T (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8(19):4321–4325
Nwafor CC, Gribaudo I, Schneider A, Wehrens R, Grando MS, Costantini L (2014) Transcriptome analysis during berry development provides insights into co-regulated and altered gene expression between a seeded wine grape variety and its seedless somatic variant. BMC Genom 15(1):1–22
Peng FY, Reid KE, Liao N, Schlosser J, Lijavetzky D, Holt R, Martínez Zapater JM, Jones S, Marra M, Bohlmann J (2007) Generation of ESTs in Vitis vinifera wine grape (Cabernet Sauvignon) and table grape (Muscat Hamburg) and discovery of new candidate genes with potential roles in berry development. Gene 402(1–2):40–50
Pratt C (1970) Reproductive anatomy in cultivated grapes—a review. Am J Enol Vitic 22(2):92–109
Royo C, Carbonell-Bejerano P, Torres-Pérez R, Nebish A, Martínez Ó, Rey M, Aroutiounian R, Ibáñez J, Martínez-Zapater JM (2016) Developmental, transcriptome, and genetic alterations associated with parthenocarpy in the grapevine seedless somatic variant Corinto bianco. J Exp Bot 67(1):259–273
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4(4):406–425
Sarikhani H, Wakana A, Peterlunger E, Gaspero GD, Cipriani G (2009) Effect of ploidy on parthenocarpy in grape cultivars. Acta Hortic 827(827):433–438
Steinherz T, Intrator N, Rivlin E (2009) Principal components analysis. In: International conference on document analysis and recognition, p 153
Striem MJ, Ben-Hayyim G, Spiegel-Roy P (1994) Developing molecular genetic markers for grape breeding, using polymerase chain reaction procedures. Vitis 33(1):53–54
Striem MJ, Ben-Hayyim G, Spiegel-Roy P (1996) Identifying molecular genetic markers associated with seedlessness in grape. Am Soc Hortic Sci 121(5):758–763
Sun X, Liu D, Zhang X, Li W, Liu H, Hong W, Jiang C, Guan N, Ma C, Zeng H (2013) SLAF-seq: an efficient method of large-scale De Novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 8(3):e58700
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28(10):2731–2739
Verslues PE, Lasky JR, Juenger TE, Liu TW, Kumar MN (2014) Genome-wide association mapping combined with reverse genetics identifies new effectors of low water potential-induced proline accumulation in arabidopsis. Plant Physiol 164(1):144–159
Wang L, Yin X, Cheng C, Wang H, Guo R, Xu X, Zhao J, Zheng Y, Wang X (2015) Evolutionary and expression analysis of a MADS-box gene superfamily involved in ovule development of seeded and seedless grapevines. Mol Genet Genom 290(3):825–846
Wang S, Meyer E, Mckay JK, Matz MV (2012) 2b-RAD: a simple and flexible method for genome-wide genotyping. Nat Methods 9(8):808–810
Wang S, Yu Y, Zhang C, Xu W, Wang Y (2011) Molecular cloning and characterization of a novel gene encoding an EF-hand calcium-binding protein related to fruit seedlessness of grapevine. Sci Hortic 130(4):708–714
Wang Y, Li S, Zhang X, Wang Y, Zhang C (2016) Isolation and analysis of differentially expressed genes during ovule abortion in the seedless grape. Sci Hortic 211:376–383
Yan J, Warburton M, Crouch J (2011) Association Mapping for Enhancing Maize (Zea mays L.) Genetic Improvement. Crop Sci 51(2):433–449
Zhou SF, Liu HQ, Gui-Lei XU, Yan JW, Wang F (2012) Screening and analysis of zinc finger protein genes expressed specially during rice seed development. Chin J Biochem Mol Biol 28(11):1033–1039
Acknowledgements
This research was supported by a grant from Agricultural Science and Technology Innovation Program of CAAS (ASTIP, Project#CAAS-ASTIP-2015-ZFRI); China Agriculture Research System (Project#CARS-30-yz-1); Crop Resources Protection Program of China Ministry of Agriculture (Project#2130135-34); The National Key Technology R&D Program of the Ministry of Science and Technology of China (Grant No. 2013BAD01B04-20). We thank Bioinformatics Division, Biomarker Technologies Corporation by help in bioinformatics and technical support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest to declare.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, H., Fan, X., Zhang, Y. et al. Identification of favorable SNP alleles and candidate genes for seedlessness in Vitis vinifera L. using genome-wide association mapping. Euphytica 213, 136 (2017). https://doi.org/10.1007/s10681-017-1919-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-017-1919-z