
Abstract
Soybean is a primary source of plant oil and protein and has a high nutritional value. Plant height (PH) and flowering time (FT) are two important agronomic traits in breeding programs for soybean. In this study, we mapped QTLs associated with PH and FT in three environments using a population with determinate growth including 236 recombinant inbred lines (NJZY-RIL) derived from a cross between two summer planting varieties, ZXD and NN1138-2. A high-density genetic map with 3255 SLAF-markers was constructed that spanned 2144.85 cM of the soybean genome with an average marker distance of 0.66 cM. Altogether, six QTLs controlling PH and eleven QTLs controlling FT were mapped using mixed-model-based composite interval mapping and composite interval mapping methods. qPH-1-1 and qFT-15-2 were two novel main effect QTLs identified in this study; qFT-6-2, qFT-15-2, qFT-16-1, qPH-1-1, qPH-15-1 and qPH-16-1 were consistently detected across environments and by the two mapping methods. Two pairs of QTLs, qFT-15-2 and qPH-15-1 as well as qFT-16-1 and qPH-16-1, which were located in the same marker interval on chromosomes 15 and 16, respectively, were found to have close linkage or pleiotropy. These results may increase our understanding of the genetic control of PH and FT in soybean and provide support for implementing marker-assisted selection in developing soybean cultivars with high yield and early maturity in summer planting regions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Soybean is a primary source of plant oil and protein for humans due to its high nutritional value (Wilcox 2004). However, the soybean yield per unit is relatively lower than that of cereal crops, such as maize. Molecular marker-assisted selection (MAS) for the breeding of high-yield varieties is an alternative to keep pace with the increasing global demand for soybean products. To date, new molecular marker systems based on next-generation sequencing (NGS) have quickly emerged. Among these, single nucleotide polymorphisms (SNPs) possess the most abundant DNA variation compared with amplified fragment length polymorphism (AFLP), restriction fragment length polymorphism (RFLP) and simple sequence repeat (SSR) markers. Due to the completion of the whole genome sequencing of soybean cv. Williams 82 (Schmutz et al. 2010) and the rapid development in sequencing technology, highly polymorphic SNP markers are beginning to be used in soybeans for large-scale genotyping and high-density genetic map construction (Hyten et al. 2008; Song et al. 2016). Specific length amplified fragment sequencing (SLAF-seq) is a developed, high-resolution strategy for the large-scale de novo discovery and genotyping of SNPs based on NGS technology. This technology has greater genotyping accuracy and relatively lower sequencing cost. SLAF-seq technology has been used in several studies; for example, Zhang et al. (2013) constructed the first high-density genetic map for sesame, Huang et al. (2013) constructed a draft genome of the kiwifruit Actinidia chinensis, and Qi et al. (2014) reported a high-density genetic map for soybean. These new maps can facilitate efficiency in the identification of quantitative trait loci (QTLs) associated with important agronomic traits.
The development of cultivars with suitable maturity and plant type is the basic objective of soybean breeders. As an important plant architecture trait, plant height (PH) is considered one of the main yield-related traits in crops, and there are many studies regarding its genetics and QTL composition in soybean (Wilcox and Sediyama 1981; Cooper 1981, 1985; Ablett et al. 1989; Lee et al. 1996a, b, 2015; Mansur et al. 1996; Orf et al. 1999; Chapman et al. 2003; Zhang et al. 2004; Panthee et al. 2007; Liu et al. 2013). To date, at least 180 QTLs controlling PH have been reported (http://www.soybase.org/). Soybean stem growth habit is a major factor affecting PH and is controlled by two major genes, Dt1 and Dt2 (Bernard 1972). In Dt1Dt1 genetic backgrounds, Dt2Dt2 and dt2dt2 genotypes produce semi-determinate and indeterminate stem termination phenotypes. However, in dt1dt1 genetic backgrounds, the phenotype is determinate. Of these loci, dt1 has a much greater effect on stem growth habit. There is a positive correlation between PH and maturity, and there is a similar correlation between maturity and flowering time (FT) (Lee et al. 1996b, 2015; Zhang et al. 2015). In general, the stem in plants with indeterminate growth continues to grow for a long period, even after flowering, while stem growth in plants with determinate growth is terminated when flowering begins or soon after. Thus, FT is considered important in determining the final PH of determinate cultivars.
FT is great interest to agriculture, as the regulation of FT is crucial for enabling crops to adapt to a particular growing region. FT is also positively correlated with PH in the determinate background (Lin et al. 1988; Curtis et al. 2000). It is difficult to breed a determinate variety with a tall PH and early flowering. Since the first QTL experiment on growth stage traits was reported 25 years ago (Keim et al. 1990), more than 70 loci distributed on 16 soybean chromosomes have been detected using different populations (Mansur et al. 1993, 1996; Orf et al. 1999; Tasma et al. 2001; Zhang et al. 2004; Yamanaka et al. 2005; Kong et al. 2014; Zhou et al. 2015). Ten major genes, E1 through E9 and J [E1 and E2 (Bernard 1971), E3 (Buzzell 1971), E4 (Buzzell and Voldeng 1980), E5 (Mcblain and Bernard 1987), E6 (Bonato and Vello 1999), E7 (Cober and Voldeng 2001), E8 (Cober et al. 2010), E9 (Kong et al. 2014) and J (Ray et al. 1995)], have been reported to control the time of flowering and maturity. Among them, five cloned genes, E1 to E4 and E9, were located on Chr.06 (LG C2), Chr.10 (LG O), Chr.19 (LG L), Chr.20 (LG I), and Chr.16 (LG J), respectively (Cregan et al. 1999; Cober and Voldeng 2001; Abe et al. 2003; Cober et al. 2010; Xia et al. 2012; Kong et al. 2014). E7, E8, and J mapped to Chr.06 (LG C2), Chr.04 (LG C1) and Chr.04 (LG C1), respectively (Cober and Voldeng 2001; Cairo et al. 2002; Lu et al. 2015). E6 has not been mapped, and E5 might not be a unique locus (Dissanayaka et al. 2016).
The genetic maps used in most studies mentioned above for mapping QTLs associated with PH and FT in soybean were constructed with only hundreds of RFLP, AFLP and/or SSR markers and therefore were of relatively low resolution (Mansur et al. 1993, 1996; Orf et al. 1999; Tasma et al. 2001; Zhang et al. 2004; Yamanaka et al. 2005). In the Chinese Huang-Huai River Valley, soybean is always planted in early June after wheat harvest and matures by the end of September or early October. The cultivar in this region has a nearly determinate stem and early flowering and maturity. However, little is known about the genetics of PH and FT in these genotypes. The objective of our study was to map QTLs for PH and FT in a recombinant inbred line (RIL) population with determinate growth using a high-density genetic map based on SLAF markers and then dissect the genetic basis of these two traits in summer soybean.
Materials and methods
Plant materials
An RIL population composed of 236 lines developed from a cross between ZXD (Maturity group III) and NN1138-2 (Maturity group IV) was used in this study. NN1138-2 is an elite cultivar characterized by high yield. ZXD is a landrace characterized by high protein content and good tolerance to flooding, among other traits. The RIL population used here (NJZY-RIL) was developed by 7 cycles of single seed descent (SSD) from an F2 population at Jiangpu Experimental Station of Nanjing Agricultural University in Nanjing, Jiangsu Province.
Experimental design and collection of phenotypic data
NJZY-RIL and its two parents were grown in a randomized complete block design with three replications and one row per plot (10-cm plant spacing, 50-cm row spacing and 1.0-m row length; one plant per hill) from approximately June-October in three environments: Jiangpu Experiment Station, Nanjing, Jiangsu Province, in 2012 and 2014 (JP12 and JP14) and Fengyang Experiment Station, Chuzhou, Anhui Province, in 2012 (FY12). Field management was performed under normal conditions. The data for PH and FT in 2013 were inaccurate due to water logging at the seedling stage and the need to replant some lines. Accordingly, we did not use the data in 2013.
FT was calculated as the number of days from germination to the first bloom (R1, 50% of the plants in a plot had an open flower at one of the top nodes with a fully expanded leaf) (Fehr et al. 1971; Fehr and Caviness 1977; Orf et al. 1999). For the PH measurement, three individuals were randomly selected from the middle of each row, and the length between the cotyledon node and the peak of the main stem was measured. The measurement was averaged over the three individuals across three replications.
Population phenotypic data analysis
Statistical analyses, including the frequency distribution of the PH and FT phenotypic data, the mean of the RIL population, the coefficient of variation (CV), the broad-sense heritability (h 2), and the analysis of variance (ANOVA), were conducted using the SAS PROC UNIVARIATE, PROC GLM and PROC CORR programs (SAS Institute Inc. 2011a, b). The h 2 for PH and FT was estimated using the following equation:
where \(\sigma_{\text{g}}^{2}\) is the genotypic variance, \(\sigma_{\text{ge}}^{2}\) is the variance due to the genotype-by-environment interaction, \(\sigma_{\text{e}}^{2}\) is the error variance, n is the number of environments, and r is the number of replications within an environment (Nyquist and Baker 1991).
Genotyping and construction of genetic linkage map
SLAF-seq was used to genotype a total of 236 individuals and the two parents. Approximately 1 g of fresh leaves obtained from each plant were used to extract the genomic DNA using the cetyltrimethylammonium bromide method (Doyle 1990). SLAF library construction and high-throughput sequencing were performed as described by Sun et al. (2013). Then, all polymorphic SLAF markers were filtered four times and further quality assessed as described by Sun et al. (2013). A SLAF with less than three SNPs and an average depth of each sample above 3 was considered a high-quality SLAF marker. Parental homozygous markers were used to construct a high-density genetic map.
After genotyping the 236 RILs, a high-density genetic map including 20 linkage groups (LGs) was constructed using High Map software (Liu et al. 2014); the Kosambi mapping function (Kosambi 1943) was used to calculate the map distances in cM from the recombination frequencies. MapChart v2.2 was used to draw the linkage map (Voorrips 2002). Chromosomes were named using Gm and the chromosome number; for example, Gm01 represents the first chromosome. Genotypes at the Dt1 loci in ZXD and NN1138-2 were identified by DNA sequencing according to Liu et al. (2010).
QTL mapping for PH and FT in multiple environments
Both mixed-model based composite interval mapping (MCIM) and composite interval mapping (CIM) methods were used to reveal the effects of the QTLs of PH and FT. Generally, more genetic effects were analyzed in MCIM than in CIM, whereas only additive effects were analyzed in CIM. QTL Network software v2.2 and MCIM were used to identify main additive effect QTLs, epistatic QTLs (AA), and genotype-by-environment interaction effects (additive by environment [AE] and AA by environment [AAE]) using MCIM (Wang et al. 1999; Yang et al. 2008; Xu et al. 2012). The CIM method of WinQTLCart version 2.5 (Wang et al. 2007) was also used to detect the main additive QTLs to identify stable QTLs expressed in different environments.
While QTL mapping was performed using the MCIM method, one- and two-dimensional genome scanning for QTLs was performed using a 10-cM testing window, a 0.1-cM walk speed and a 10-cM filtration window. The F thresholds for significant QTLs of each trait were determined by a 1000-permutation test at a 95% confidence level. When QTL analysis was performed by CIM, the window size, the working speed and the control marker number were set at 10, 1 and 5 cM, respectively. Model 6 (standard Model) in CIM was used to identify QTLs for each trait in each environment. Permutation tests of 1000 runs at a significance level of P = 0.05 were used to determine the LOD threshold for declaring whether the presence of a QTL in a certain chromosomal region was significantly associated with a target trait (Churchill and Doerge 1994).
If the confidence intervals of QTLs detected for the same trait in different environments overlapped and had the same sign of additive effects, then they were accepted as the same QTL. We followed the nomenclature suggested by McCouch et al. (1997) to name the QTLs detected in our study.
Results
Phenotypic evaluations of PH and FT
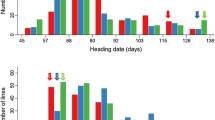
The phenotypic performance of the parents and the RILs is presented in Table 1 and Fig. S1. ZXD and NN1138-2 showed a significant difference in PH but did not greatly differ from each other in FT in all environments. However, a continuous distribution and transgressive segregation were observed for the two traits in the RIL population in all environments. In addition, the kurtosis and the skewness (absolute value) were less than 1 for both traits, except in the JP12 environment. This result indicates that PH and FT are quantitative traits and implies the existence of respective loci in the two parents controlling FT and PH.
The Pearson correlation coefficients (r) of PH and FT were moderately positively correlated in all environments: 0.32 in JP12, 0.52 in FY12, and 0.45 in JP14 (P < 0.01).
The h 2 of the traits was high for PH (0.84) and FT (0.87), which was consistent with the results of previous studies (Lee et al. 1996a). The ANOVA results indicated significant differences in genotype, environment and genotype-by-environment (Table S1), and the mean square (MS) value for the genotype-by-environment interaction was less than that of the genotype.
Marker genotyping and genetic map construction
The RIL population was genotyped using SLAF-seq technology. The average coverage for each SLAF marker was 101.85× for the parents and 3.05× for the RIL lines. In total, we obtained 103,845,237 point reads from the two parents and all of the lines. The reads were then mapped to the reference soybean genome (cv. Williams 82). The reads that could be mapped to a single locus were considered effective SLAFs. In this study, we obtained 71,888 SLAFs, of which 5333 were polymorphic. The polymorphic rate of these SLAFs was 7.4%. Ultimately, a total of 3279 SLAFs were used for high-density linkage map construction after filtration and quality assessment.
Using High Map software, a high-density genetic map was constructed. The 3255 SLAF markers were grouped into 20 LGs. The total genetic distance of this map was 2144.85 cM. The average distance between adjacent markers was 0.66 cM. The mean LG length was 107.24 cM. The LG containing the maximum number of markers was Gm18, with 338 SLAF markers and a length of 140.37 cM. The LG with a minimum number of markers was Gm12, with 48 SLAF markers and a length of 74.90 cM. The number of markers on each chromosome was consistent with its physical length; the longest chromosome was Gm18, and the shortest chromosome was Gm12 (Schmutz et al. 2010). In addition, we found that approximately 96.27% of the intervals between adjacent markers were shorter than 5 cM. Detailed map information is presented in Fig. 1, Table S2 and Figs. S2–S5.

Distribution of markers in 20 linkage groups in the NJZY-RIL population. The black bars in each linkage group represent mapped SLAF-seq markers. The linkage group number is shown on the x-axis, and genetic distance is shown on the y-axis (cM as unit). A detailed map is presented in the supplementary materials (Table S2; Figs. S2–S5)
According to the DNA sequencing at the Dt1 locus, both ZXD and NN1138-2 were of the dt1 type at the Dt1 locus. This finding suggests that the difference in PH was not caused by the Dt1 locus; the genetic background of NJZY-RIL was the determinate growth habit.
The main additive effect QTLs identified by MCIM for PH and FT
In the NJZY-RIL population, a total of 6 main additive effect QTLs for PH distributed on six LGs were identified by MCIM and explained 2.28 to 16.12% of the phenotypic variance (PV) (Table 2). qPH-1-1 (named by a combination of trait and chromosome) was the largest additive effect QTL associated with PH (a = −5.12) and explained 16.12% of the PV. qPH-16-1 and qPH-15-1 were the second and third major QTLs for PH and explained 13.95 and 9.48% of the PV, respectively. The other three QTLs were qPH-7-1, qPH-9-1 and qPH-19-1, which explained 2.10, 2.28 and 3.10% of the PV, respectively. In addition, the positive alleles of qPH-1-1, qPH-7-1 and qPH-19-1 came from ZXD, while those of qPH-9-1, qPH-15-1 and qPH-16-1 came from NN1138-2.
Ten QTLs were detected by MCIM on eight chromosomes associated with FT. The phenotypic variation explained by the detected QTLs ranged from 1.38 to 12.92% (Table 2). qFT-16-1 could be the major QTL because of its high additive effect (a = 0.82), and it explained 12.92% of the PV. qFT-15-2 was another major QTL for FT that explained 11.39% of the PV. qFT-6-2 explained 6.33% of the PV. Other QTLs explained less than 5% of the PV. The additive effects of qFT-5-1, qFT-6-1, qFT-6-2, and qFT-8-1 were negative, suggesting that the positive alleles came from the male parent, ZXD. In contrast, qFT-2-2, qFT-14-1, qFT-15-1, qFT-15-2, qFT-16-1, and qFT-18-1 had positive additive effects, and the positive alleles came from the female parent, NN1138-2.
Epistasis and QTL-by-environment interaction
A total of 3 pairs of epistatic QTLs were identified for PH and FT (Table S3). Two pairs of epistatic QTLs involving four loci on three chromosomes were identified for PH. Pair 1 was composed of two additive QTLs, qPH-1-1 on Chr.01 and qPH-16-1 on Chr.16, and explained 1.3% of the PV for PH. Pair 2, qPH-6-1 and qPH-16-2, had no additive effect and explained 2.7% of the PV. One pair of epistatic QTLs identified for FT was composed of two non-additive QTLs, qFT-2-1 and qFT-16-2, and explained 1.4% of the PV.
In this study, additive QTL-by-environment interactions were relatively weak and explained 0.6% and 0.4–1.0% of the phenotypic variation in PH and FT, respectively (Table 2).
The main additive effect QTLs identified by CIM and a comparative analysis of the main additive effect QTLs detected by CIM and MCIM
A total of ive QTLs associated with PH were detected in at least one of the three environments using CIM based on WinQTLCart (Table 3). qPH-7-1 and qPH-19-1 were detected in a single environment; qPH-1-1, qPH-15-1, and qPH-16-1 were detected in all environments (Fig. 2). Comparing the main additive effect QTLs identified by the two programs, qPH-1-1, qPH-7-1, qPH-15-1, qPH-16-1, and qPH-19-1 were identified both by WinQTLCart and QTLNetwork (Table 2).

Linkage maps of Chr.15 and Chr.16 for two locations associated with flowering time and plant height in the NJZY-RIL population. a Graphical representation of the loci of Chr.15 associated with plant height and flowering time, with LOD plots for three environments (JP12, FY12, and JP14). The hatched lines in the LOD plots indicate the LOD thresholds. b Graphical representation of the loci of Chr.16 associated with plant height and flowering time, with LOD plots for three environments (JP12, FY12, and JP14). The hatched lines in the LOD plots indicate the LOD thresholds
For FT, five QTLs on Chr.03, Chr.06, Chr.15, and Chr.16 were identified in at least one environment using CIM analysis (Table 3). qFT-6-2, qFT-15-2, and qFT-16-1 were detected in all environments (Fig. 2); qFT-6-1 was detected in two environments; and qFT-3-1 was detected in a single environment. Compared with the results of MCIM, all main effect QTLs were identified both by WinQTLCart and QTLNetwork, with the exception of qFT-3-1 (Table 2).
Discussion
Construction of a high-density genetic map based on SLAF-seq markers
QTL mapping has been used as an efficient approach to analyze quantitative traits in plants. The quality of genetic maps has a significant effect on the accuracy of QTL mapping, and increasing marker density can improve the resolution of genetic maps for a given mapping population (Gutierrez-Gonzalez et al. 2011; Zou et al. 2012). Therefore, it is feasible to construct high-density genetic maps and thereby improve the efficiency and accuracy of QTL mapping and MAS. In this study, we used 3279 high-quality SLAF markers to construct a high-density map. Ultimately, a total of 3255 SLAF markers were integrated into 20 LGs, and the average distance between adjacent markers was only 0.66 cM. This high-density genetic map could ensure that a molecular marker and QTL were tightly linked and provided a good foundation for analyzing quantitative traits.
QTLs for plant height in soybean
Because PH is an important yield-related trait in soybean, a series of associated QTLs/loci has already been reported. Liu et al. (2013) used an RIL population and detected 11 QTLs for PH; Lee et al. (2015) mapped six QTLs for PH with an SNP map; and Zhang et al. (2015) detected 27 loci for PH via a genome-wide association study (GWAS). Brummer et al. (1997) reported that in molecular-assisted breeding programs, breeders should use QTLs that are stable in multiple environments. In the present study, a total of six QTLs for PH were identified. qPH-1-1, qPH-15-1, and qPH-16-1 could be considered the major and stable QTLs for PH because they could be detected by the two programs and in all environments with larger LOD values (14.0–17.2, 7.6–11.1 and 8.4–14.8, respectively) and explained more of the PV (18.9–24.6%, 8.6–13.8%, and 10.2–18.9%, respectively). Based on the high-density genetic map, the confidence interval for most of the QTLs was less than 5 cM, and each QTL had two or more closely linked markers (within 0–5 cM). These loci are favorable for the MAS of QTLs by soybean breeding programs.
qPH-15-1 and qPH-16-1 have adjacent or physically overlapping QTLs, as reported by Sun et al. (2006) and Lee et al. (1996b), and might be the same QTL. Because many QTLs associated with PH have been previously reported, it is difficult to identify novel QTLs associated with PH. Based on the QTLs listed in SoyBase (www.soybase.org), two novel QTLs, qPH-1-1 and qPH-9-1, were identified in this study. Compared with the loci/genes that were already known, such as Dt1 and Dt2, novel QTLs, particularly qPH-1-1, which was detected by two programs and in all environments and could be considered the major and stable QTL, in these summer soybean lines would add to the growing knowledge on the genetic control of PH.
Knowledge of epistasis and QTL-by-environment interaction is essential to understand the genetic architecture and the gene networks that underlie complex traits (Wurschum et al. 2011). When epistatic interactions are considered in a QTL mapping model, the precision of QTL mapping is greatly enhanced (Wang et al. 1999), which could help to accurately predict the phenotypic performance in MAS programs. In this study, two pairs of epistatic QTLs (qPH-1-1 × qPH-16-1, qPH-6-1 × qPH-16-2) and some additive QTL-by-environment interactions were detected by QTLNetwork (Table 2, Table S3). qPH-6-1 and qPH-16-2 did not display additive effects; these epistatic QTLs might be considered modifying genes that have no significant effects alone but might affect the expression of PH through epistatic interactions with other loci. In fact, we identified significant additive QTL-by-environment interactions using MCIM (Table 3), although the phenotypic variation for QTL-by-environment interactions was less than that for additive QTLs. This information might be useful for more accurately predicting the breeding value in MAS.
New QTLs for flowering time in soybean
FT is a topic of great interest in soybean breeding programs; it is crucial for the adaptation of crops to a particular growing region and may also affect other traits. In this study, using RILs derived from ZXD and NN1138-2, a total of 11 QTLs for FT were identified. Among these QTLs, qFT-6-1 and qFT-6-2 were located on the same chromosome as E1 and E7. However, qFT-6-1 and qFT-6-2 might not be E1 loci due to their physical position being far from known E1 loci (Xia et al. 2012); qFT-6-2 was located in the same physical range as reported in previous studies and might be the same QTL as qFT-6-2 (Zhang et al. 2004). Corresponding to the physical location of the confidence interval of qFT-16-1, we found the Arabidopsis homolog GmFT5a. Previous studies have shown that GmFT5a controls the soybean photoperiod (Kong et al. 2010); GmFT5a might be a candidate gene for this QTL. Compared with ten major genes, the QTLs listed in SoyBase (www.soybase.org) have been reported to control the time of flowering and maturity. A few novel QTLs were identified for FT in this study, such as qFT-5-1, qFT-6-1, qFT-14-1, qFT-15-1 and qFT-15-2. Compared with E1 through E9 and J, the novel QTLs, particularly qFT-15-2, which explained 11.39% of the PV, would add to the growing knowledge of the genetic control of FT. Among these QTLs, qFT-6-2, qFT-15-2, and qFT-16-1 were detected by two programs and in all environments in this study and could thus be considered major and stable QTLs for further fine mapping and map-based cloning to elucidate the mechanisms of FT. Those loci might be useful in soybean breeding programs.
However, there are many possible FT genes in soybean (Kim et al. 2012). In many previous studies, most soybean materials used for QTL/gene mapping and cloning were from the spring planting type; some major loci, including E1, E2, and E3, play important roles in these lines (Xia et al. 2012; Kong et al. 2014; Lu et al. 2015; Zhang et al. 2015). However, in the NJZY-RIL population, most detected QTLs were novel, indicating the distinct genetic architecture of FT in these two summer soybeans. More diverse germplasms need to be used to reveal the genetic basis of FT in soybean.
Co-location of QTLs for soybean PH and FT
One locus can be related to more than one trait (Zhang et al. 2004); several QTLs of various traits can map to the same locus. In this study, two pairs of QTLs, qPH-15-1 and qFT-15-2 as well as qPH-16-1 and qFT-16-1, were located in the same marker interval (Fig. 2). These loci were found to be related to the two agronomic traits PH and FT. This implies that qFT-15-2 and qFT-16-1 not only control FT but also may affect PH. This phenomenon is consistent with previous reports that found that the QTLs/genes of FT often impact other agronomic traits (Chapman et al. 2003; Kantolic and Slafer 2007; Li et al. 2008; Cober et al. 2010). Furthermore, QTLs clustered in the same LG could suggest moderate correlations among corresponding traits based on field data. Therefore, it is difficult to breed a determinate variety with a tall PH and an early FT. Due to the lines pyramiding different alleles and the additive effects of the QTLs controlling PH and FT from both parents (Tables 2, 3), we identified some lines with a tall PH and an early FT in the field experiments. Thus, we can produce offspring with desirable PH and FT by pyramiding different favorable alleles if we know the QTLs associated with PH and FT in summer soybean. These results would not only add to the growing knowledge of the genetic control of PH and FT but also provide useful information for understanding the molecular mechanisms of PH and FT.
In conclusion, the objective of this study was to detect QTLs for PH and FT, which are two important agronomic traits that should be considered in breeding programs. A high-density genetic map was constructed in this study. The main additive effect QTLs for PH and FT were detected, and the markers closely linked to each QTL were explored. Furthermore, a few novel QTLs, information about epistasis and the QTL-by-environment interaction, and the co-location of QTLs were obtained in this study. These results might be very useful for the fine mapping of soybean genes and provide support for implementing MAS for breeding high-yielding soybean.
References
Abe J, Xu DH, Miyano A, Komatsu K, Kanazawa A, Shimamoto Y (2003) Photoperiod-insensitive japanese soybean landraces differ at two maturity loci. Crop Sci 43(4):1300–1304
Ablett GR, Beversdorf WD, Dirks VA (1989) Performance and stability of indeterminate and determinate soybean in short-season environments. Crop Sci 29(6):1428–1433
Bernard RL (1971) Two major genes for time of flowering and maturity in soybeans. Crop Sci 11(2):242–244
Bernard RL (1972) Two genes affecting stem termination in soybeans. Crop Sci 12(2):235–239
Bonato ER, Vello NA (1999) E6, a dominant gene conditioning early flowering and maturity in soybeans. Genet Mol Biol 22(2):229–232
Brummer EC, Graef GL, Orf J, Wilcox JR, Shoemaker RC (1997) Mapping QTL for seed protein and oil content in eight soybean populations. Crop Sci 37(2):370–378
Buzzell R (1971) Inheritance of a soybean flowering response to fluorescent-day length conditions. Can J Genet Cytol 13(4):703–707
Buzzell R, Voldeng H (1980) Inheritance of insensitivity to long daylength. Soybean Genet Newsl 7:26–29
Cairo CA, Stein J, Delgado L, Bortolotti S, Guelman SA, Ortiz JPA, Morandi EN (2002) Tagging the juvenile locus in soybean [Glycine max (L.) Merr.] with molecular markers. Euphytica 124:387–395
Chapman A, Pantalone V, Ustun A, Allen F, Landau-Ellis D, Trigiano R, Gresshoff P (2003) Quantitative trait loci for agronomic and seed quality traits in an F2 and F4:6 soybean population. Euphytica 129(3):387–393
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138(3):963–971
Cober ER, Voldeng HD (2001) A new soybean maturity and photoperiod-sensitivity locus linked to E1 and T. Crop Sci 41(3):698–701
Cober ER, Molnar SJ, Charette M, Voldeng HD (2010) A new locus for early maturity in soybean. Crop Sci 50(2):524–527. doi:10.2135/cropsci2009.04.0174
Cooper RL (1981) Development of short-statured soybean cultivars. Crop Sci 21(1):127–131
Cooper RL (1985) Breeding semidwarf soybeans. Plant Breed Rev 3:289–311
Cregan PB, Jarvik T, Bush AL, Shoemaker RC, Lark KG, Kahler AL, Kaya N, VanToai TT, Lohnes DG, Chung L, Specht JE (1999) An integrated genetic linkage map of the soybean genome. Crop Sci 39(5):1464–1490
Curtis DF, Tanner JW, Luzzi BM, Hume DJ (2000) Agronomic and phenological differences of soybean isolines differing in maturity and growth habit. Crop Sci 40(6):1624–1629
Dissanayaka A, Rodriguez TO, Di S, Yan F, Githiri SM, Rodas FR, Abe J, Takahashi R (2016) Quantitative trait locus mapping of soybean maturity gene E5. Breed Sci 66(3):407–415. doi:10.1270/jsbbs.15160
Doyle JJ (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Fehr WR, Caviness CE (1977) Stages of soybean development. Iowa State University of Science and Technology, Cooperative Extension Service; Agriculture and Home Economics Experiment Station
Fehr WR, Caviness CE, Burmood D, Pennington J (1971) Stage of development descriptions for soybeans, Glycine max (L.) Merrill. Crop Sci 11(6):929–931
Gutierrez-Gonzalez JJ, Vuong TD, Zhong R, Yu O, Lee JD, Shannon G, Ellersieck M, Nguyen HT, Sleper DA (2011) Major locus and other novel additive and epistatic loci involved in modulation of isoflavone concentration in soybean seeds. Theor Appl Genet 123(8):1375–1385. doi:10.1007/s00122-011-1673-x
Huang SX, Ding J, Deng DJ, Tang W, Sun HH, Liu DY, Zhang L, Niu XL, Zhang X, Meng M, Yu JD, Liu J, Han Y, Shi W, Zhang DF, Cao SQ, Wei ZJ, Cui YL, Xia YH, Zeng HP, Bao K, Lin L, Min Y, Zhang H, Miao M, Tang XF, Zhu YY, Sui Y, Li GW, Sun HJ, Yue JY, Sun JQ, Liu FF, Zhou LQ, Lei L, Zheng XQ, Liu M, Huang L, Song J, Xu CH, Li JW, Ye KY, Zhong SL, Lu BR, He GH, Xiao FM, Wang HL, Zheng HK, Fei ZJ, Liu YS (2013) Draft genome of the kiwifruit actinidia chinensis. Nat Commun. doi:10.1038/ncomms3640
Hyten DL, Song Q, Choi IY, Yoon MS, Specht JE, Matukumalli LK, Nelson RL, Shoemaker RC, Young ND, Cregan PB (2008) High-throughput genotyping with the goldengate assay in the complex genome of soybean. Theor Appl Genet 116(7):945–952. doi:10.1007/s00122-008-0726-2
Kantolic AG, Slafer GA (2007) Development and seed number in indeterminate soybean as affected by timing and duration of exposure to long photoperiods after flowering. Ann Bot-London 99(5):925–933. doi:10.1093/aob/mcm033
Keim P, Diers BW, Olson TC, Shoemaker RC (1990) RFLP mapping in soybean—association between marker loci and variation in quantitative traits. Genetics 126(3):735–742
Kim MY, Shin JH, Kang YJ, Shim SR, Lee SH (2012) Divergence of flowering genes in soybean. J Biosci 37(5):857–870. doi:10.1007/s12038-012-9252-0
Kong FJ, Liu BH, Xia ZJ, Sato S, Kim BM, Watanabe S, Yamada T, Tabata S, Kanazawa A, Harada K, Abe J (2010) Two coordinately regulated homologs of flowering locus t are involved in the control of photoperiodic flowering in soybean. Plant Physiol 154(3):1220–1231. doi:10.1104/pp.110.160796
Kong FJ, Nan HY, Cao D, Li Y, Wu FF, Wang JL, Lu SJ, Yuan XH, Cober ER, Abe J, Liu BH (2014) A new dominant gene e9 conditions early flowering and maturity in soybean. Crop Sci 54(6):2529–2535. doi:10.2135/cropsci2014.03.0228
Kosambi DD (1943) The estimation of map distances from recombination values. Ann Eugen 12(1):172–175
Lee SH, Bailey MA, Mian MAR, Shipe ER, Ashley DA, Parrott WA, Hussey RS, Boerma HR (1996a) Identification of quantitative trait loci for plant height, lodging, and maturity in a soybean population segregating for growth habit. Theor Appl Genet 92(5):516–523. doi:10.1007/Bf00224553
Lee SH, Bailey MA, Mian MAR, Carter TE, Ashley DA, Hussey RS, Parrott WA, Boerma HR (1996b) Molecular markers associated with soybean plant height, lodging, and maturity across locations. Crop Sci 36(3):728–735
Lee S, Jun TH, Michel AP, Mian MAR (2015) SNP markers linked to QTL conditioning plant height, lodging, and maturity in soybean. Euphytica 203(3):521–532. doi:10.1007/s10681-014-1252-8
Li W, Zheng D-H, Van K, Lee S-H (2008) QTL mapping for major agronomic traits across two years in soybean (Glycine max L. Merr.). J Crop Sci Biotechnol 11:171–190
Lin MS, Nelson RL (1988) Relationship between plant height and flowering date in determinate soybean. Crop Sci 28(1):27–30
Liu BH, Watanabe S, Uchiyama T, Kong FJ, Kanazawa A, Xia ZJ, Nagamatsu A, Arai M, Yamada T, Kitamura K, Masuta C, Harada K, Abe J (2010) The soybean stem growth habit gene Dt1 is an ortholog of Arabidopsis TERMINAL FLOWER1. Plant Physiol 153(1):198–210. doi:10.1104/pp.109.150607
Liu YL, Li YH, Reif JC, Mette MF, Liu ZX, Liu B, Zhang SS, Yan L, Chang RZ, Qiu LJ (2013) Identification of quantitative trait loci underlying plant height and seed weight in soybean. The Plant Genome. doi:10.3835/plantgenome2013.03.0006
Liu DY, Ma CX, Hong WG, Huang L, Liu M, Liu H, Zeng HP, Deng DJ, Xin HG, Song J, Xu CH, Sun XW, Hou XL, Wang XW, Zheng HK (2014) Construction and analysis of high-density linkage map using high-throughput sequencing data. PLoS ONE. doi:10.1371/journal.pone.0098855
Lu SJ, Li Y, Wang JL, Srinives P, Nan HY, Cao D, Wang YP, Li JL, Li XM, Fang C, Shi XY, Yuan XH, Watanabe S, Feng XZ, Liu BH, Abe J, Kong FJ (2015) QTL mapping for flowering time in different latitude in soybean. Euphytica 206(3):725–736. doi:10.1007/s10681-015-1501-5
Mansur LM, Lark KG, Kross H, Oliveira A (1993) Interval mapping of quantitative trait loci for reproductive, morphological, and seed traits of soybean [Glycine max (L.) Merr.]. Theor Appl Genet 86(8):907–913
Mansur LM, Orf JH, Chase K, Jarvik T, Cregan PB, Lark KG (1996) Genetic mapping of agronomic traits using recombinant inbred lines of soybean. Crop Sci 36(5):1327–1336
Mcblain BA, Bernard RL (1987) A new gene affecting the time of flowering and maturity in soybeans. J Hered 78(3):160–162
McCouch S, Cho Y, Yano M, Paul E, Blinstrub M, Morishima H, Kinoshita T (1997) Report on QTL nomenclature. Rice Genet Newsl 14(11):11–131
Nyquist WE, Baker RJ (1991) Estimation of heritability and prediction of selection response in plant-populations. Crit Rev Plant Sci 10(3):235–322. doi:10.1080/07352689109382313
Orf JH, Chase K, Jarvik T, Mansur LM, Cregan PB, Adler FR, Lark KG (1999) Genetics of soybean agronomic traits: I. Comparison of three related recombinant inbred populations. Crop Sci 39(6):1642–1651
Panthee DR, Pantalone VR, Saxton AM, West DR, Sams CE (2007) Quantitative trait loci for agronomic traits in soybean. Plant Breed 126(1):51–57. doi:10.1111/j.1439-0523.2006.01305.x
Qi Z, Huang L, Zhu R, Xin D, Liu C (2014) A high-density genetic map for soybean based on specific length amplified fragment sequencing. PLoS ONE 9(11):e104871. doi:10.1371/journal.pone.0114349
Ray JD, Hinson K, Mankono JEB, Malo MF (1995) Genetic-control of a long-juvenile trait in soybean. Crop Sci 35(4):1001–1006
SAS Institute Inc. (2011a) Base SAS® 9.3 Procedures Guide , SAS Institute Inc, Cary
SAS Institute Inc. (2011b) SAS/STAT® 9.3 Procedures Guide, SAS Institute Inc, Cary
Schmutz J, Cannon SB, Schlueter J, Ma JX, Mitros T, Nelson W, Hyten DL, Song QJ, Thelen JJ, Cheng JL, Xu D, Hellsten U, May GD, Yu YS, Sakurai T, Umezawa T, Bhattacharyya MK, Sandhu D, Valliyodan B, Lindquist E, Peto M, Grant D, Shu SQ, Goodstein D, Barry K, Futrell-Griggs M, Abernathy B, Du JC, Tian ZX, Zhu LC, Gill N, Joshi T, Libault M, Sethuraman A, Zhang XC, Shinozaki K, Nguyen HT, Wing RA, Cregan P, Specht J, Grimwood J, Rokhsar D, Stacey G, Shoemaker RC, Jackson SA (2010) Genome sequence of the palaeopolyploid soybean. Nature 465(7294):120. doi:10.1038/nature08957
Song Q, Jenkins J, Jia G, Hyten DL, Pantalone V, Jackson SA, Schmutz J, Cregan PB (2016) Construction of high resolution genetic linkage maps to improve the soybean genome sequence assembly glyma1. 01. BMC Genom 17(1):1
Sun DH, Li WB, Zhang ZC, Chen QS, Ning HL, Qiu LJ, Sun GL (2006) Quantitative trait loci analysis for the developmental behavior of soybean (Glycine max L. Merr.). Theor Appl Genet 112(4):665–673. doi:10.1007/s00122-005-0169-y
Sun XW, Liu DY, Zhang XF, Li WB, Liu H, Hong WG, Jiang CB, Guan N, Ma CX, Zeng HP, Xu CH, Song J, Huang L, Wang CM, Shi JJ, Wang R, Zheng XH, Lu CY, Wang XW, Zheng HK (2013) SLAF-seq: an efficient method of large-scale de novo snp discovery and genotyping using high-throughput sequencing. PLoS ONE. doi:10.1371/journal.pone.0058700
Tasma I, Lorenzen L, Green D, Shoemaker R (2001) Mapping genetic loci for flowering time, maturity, and photoperiod insensitivity in soybean. Mol Breed 8(1):25–35
Voorrips RE (2002) Mapchart: software for the graphical presentation of linkage maps and QTLs. J Hered 93(1):77–78. doi:10.1093/jhered/93.1.77
Wang D, Zhu J, Li Z, Paterson A (1999) Mapping QTLs with epistatic effects and QTL × environment interactions by mixed linear model approaches. Theor Appl Genet 99(7–8):1255–1264
Wang S, Basten C, Zeng Z (2007) Windows QTL cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh
Wilcox JR (2004) World distribution and trade of soybean. Soybeans 3:1–14
Wilcox JR, Sediyama T (1981) Interrelationships among height, lodging and yield in determinate and indeterminate soybeans. Euphytica 30(2):323–326
Wurschum T, Maurer HP, Schulz B, Mohring J, Reif JC (2011) Genome-wide association mapping reveals epistasis and genetic interaction networks in sugar beet. Theor Appl Genet 123(1):109–118. doi:10.1007/s00122-011-1570-3
Xia Z, Watanabe S, Yamada T, Tsubokura Y, Nakashima H, Zhai H, Anai T, Sato S, Yamazaki T, Lü S (2012) Positional cloning and characterization reveal the molecular basis for soybean maturity locus E1 that regulates photoperiodic flowering. Proc Natl Acad Sci 109:E2155–E2164
Xu H, Zhu J (2012) Statistical approaches in QTL mapping and molecular breeding for complex traits. Chin Sci Bull 57(21):2637–2644
Yamanaka N, Watanabe S, Toda K, Hayashi M, Fuchigami H, Takahashi R, Harada K (2005) Fine mapping of the QTL locus for soybean flowering time using a residual heterozygous line derived from a recombinant inbred line. Theor Appl Genet 110(4):634–639
Yang J, Hu CC, Hu H, Yu RD, Xia Z, Ye XZ, Zhu J (2008) QTLNetwork: mapping and visualizing genetic architecture of complex traits in experimental populations. Bioinformatics 24(5):721–723. doi:10.1093/bioinformatics/btm494
Zhang WK, Wang YJ, Luo GZ, Zhang JS, He CY, Wu XL, Gai JY, Chen SY (2004) QTL mapping of ten agronomic traits on the soybean (Glycine max L. Merr.) genetic map and their association with est markers. Theor Appl Genet 108(6):1131–1139. doi:10.1007/s00122-003-1527-2
Zhang YX, Wang LH, Xin HG, Li DH, Ma CX, Ding X, Hong WG, Zhang XR (2013) Construction of a high-density genetic map for sesame based on large scale marker development by specific length amplified fragment (SLAF) sequencing. BMC Plant Biol. doi:10.1186/1471-2229-13-141
Zhang JP, Song QJ, Cregan PB, Nelson RL, Wang XZ, Wu JX, Jiang GL (2015) Genome-wide association study for flowering time, maturity dates and plant height in early maturing soybean (Glycine max) germplasm. BMC Genom 16(1):1. doi:10.1186/S12864-015-1441-4
Zhou L, Wang SB, Jian JB, Geng QC, Wen J, Song QJ, Wu ZZ, Li GJ, Liu YQ, Dunwell JM, Zhang J, Feng JY, Niu Y, Zhang L, Ren WL, Zhang YM (2015) Identification of domestication-related loci associated with flowering time and seed size in soybean with the rad-seq genotyping method. Sci Rep. doi:10.1038/Srep09350
Zou G, Zhai G, Feng Q, Yan S, Wang A, Zhao Q, Shao J, Zhang Z, Zou J, Han B (2012) Identification of QTLs for eight agronomically important traits using an ultra-high-density map based on SNPs generated from high-throughput sequencing in sorghum under contrasting photoperiods. Journal of experimental botany 15:5451–5462
Acknowledgements
This work was supported by Grants from the Natural Science Foundation of China (31271750, 31571691), the Special Project for Crop Breeding (2016ZX08004001), the MOE Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT13073), and the Jiangsu JCIC-MCP program.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Yongce Cao and Shuguang Li have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cao, Y., Li, S., He, X. et al. Mapping QTLs for plant height and flowering time in a Chinese summer planting soybean RIL population. Euphytica 213, 39 (2017). https://doi.org/10.1007/s10681-016-1834-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-016-1834-8