
Abstract
In this work, spectrophotometer was used as a detector for the determination of uranium from water, biological, and ore samples with a flow injection system coupled with solid phase extraction. In order to promote the online preconcentration of uranium, a minicolumn packed with XAD-4 resin impregnated with nalidixic acid was utilized. The system operation was based on U(VI) ion retention at pH 6 in the minicolumn at flow rate of 15.2 mL min−1. The uranium complex was removed from the resin by 0.1 mol dm−3 HCl at flow rate of 3.2 mL min−1 and was mixed with arsenazo III solution (0.05 % solution in 0.1 mol dm−3 HCl, 3.2 mL min−1) and driven to flow through cell of spectrophotometer where its absorbance was measured at 651 nm. The influence of chemical (pH and HCl (as eluent and reagent medium) concentration) and flow (sample and eluent flow rate and preconcentration time) parameters that could affect the performance of the system as well as the possible interferents was investigated. At the optimum conditions for 60 s preconcentration time (15.2 mL of sample volume), the method presented a detection limit of 1.1 μg L−1, a relative standard deviation (RSD) of 0.8 % at 100 μg L−1, enrichment factor of 30, and a sample throughput of 42 h−1, whereas for 300 s of the preconcentration time (76 mL of sample volume), a detection limit of 0.22 μg L−1, a RSD of 1.32 % at 10 μg L−1, enrichment factor of 150, and a sampling frequency of 11 h−1 were reported.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Determination of metal ions at low concentration requires initial preconcentration in order to meet the detection limit of the prescribed method and to eliminate the interference of the matrix elements. There are several efficient methods for carrying out the preconcentration. These includes, liquid–liquid extraction, carrier facilitated transport of the species, and solid phase extraction (SPE) (Ayata et al. 2009; Lemos et al. 2009; Venkatesh and Maiti 2004). SPE is advantageous to other preconcentration methods because of its minimal waste generation, reduced matrix effect, and the collection of metal ions on the solid support in a well-defined chemical form making the further processing of the collected species simpler (Camel 2003). SPE combined with flow injection analysis have the advantage of automation, high sensitivity, and improvement of selectivity (Soylak et al. 2005). SPE can be incorporated with online systems by using minicolumns packed with hydrophobic sorbents that collect the components of the aqueous phase on their surface. Hence, this technique is useful for the recovery and preconcentration of strategically important metals from their sources containing the metal ions at low concentration level.
The developments of efficient methods for the recovery of uranium from its low-grade source, like sea water, have attracted the attention primarily due to the application of uranium in nuclear industry. Determination of traces of uranium in environmental sites is also important from environmental safety considerations. Various flow injection (FI) methods have so far been used for the online determination of uranium in diverse samples. Most of the reported FI methods for determining uranium have employed spectrophotometry due to its simplicity, accuracy, precision, inexpensive instrumentation, and wide availability in laboratories (Tzanavaras and Themelis 2007).
The determination of uranium in samples containing both uranium and thorium was done with arsenazo III and a reduction column in order to reduce U(VI) →U(IV). In these systems, two absorption signals were obtained by a nested loop injection system (Pavon et al. 1989) or two sample injections (Pavon et al. 1990). One of the signal was due to the total amount of thorium and uranium in the sample with a reduction step, and the other signal was mainly due to the amount of thorium without the reduction U(VI)→U(IV). The detection limits of these systems were 23.9 and 6.6 μg L−1, respectively. Grudpan et al. determined uranium in tin tailing with 4-(2-pyridylazo)-resorcinol (PAR) using (CyDTA), sodium fluoride, or sulfosalicylic acid to mask the interfering ions. The detection limit of the system was 280 μg L−1 (Grudpan et al. 1995b). Hirano et al. determined uranium with CAS in the presence of thorium by the use of ethylenediaminetetracetic acid as masking agent (Hirano et al. 2003). The detection limit of the system was 10 μg L−1. Sun et al. determined uranium in standard ore samples using chlorophosphonazo-mN as a chromogenic reagent. The detection limit of the system was 500 μg L−1 (Sun et al. 1994). But these systems are not very sensitive and selective.
Preconcentrations and separations are needed in order to enhance the sensitivity and selectivity. The literature revealed that a lot of work has been done for the determination of uranium after online preconcentration on resins using flow injection coupled with spectrophotometer (FI-SPM). Wu and Qi used levextrel CL-5209 resin for the online separation and preconcentration of trace uranium in geological samples and spectrophotometrically determination with arsenazo III using diethylenetriaminepentaacetic acid and sodium fluoride as masking reagent. The detection limit of the system was 3 μg L−1 (Wu and Qi 1988). Pavon et al. used Dowex 50-X8 resin for the preconcentration of uranium in water samples and determined spectrophotometrically with arsenazo III. The detection limit of the system was 0.6 μg L−1 (Pavon et al. 1992). FI manifolds packed with Duolite C-225(H) and U/TEVA Spec. TM resins were used by Grudpan et al. for the online preconcentration of uranium and spectrophotometric determination using PAR (Grudpan et al. 1995a, 1998). These procedures offer detection limits of 0.27 μg and 200 μg L−1, respectively. The resins used in these systems are not available in the market and their preparation is also difficult. In addition, these resins are often unselective towards uranium ions.
Lemos and Gama used XAD-4 resin functionalized with β-nitroso-α-naphthol for the preconcentration of trace uranium in water and effluent samples and determined spectrophotometrically using arsenazo III. The detection limit of the system was 1.8 μg L−1 (Lemos and Gama 2010). But the uses of covalently bound chelating agents have its limitations because they are not easy to synthesize and this in turn limits their availability. So the need of simple, efficient, and low-cost determination methods for uranium still exists.
The present study was undertaken to develop a sensitive and selective online determination method for uranium after preconcentration on XAD-4 resin (Styrene divinylbenzene copolymer) impregnated with nalidixic acid (HNA).
Styrene divinylbenzene copolymer represents an outstanding solid phase which was used to preconcentrate metal ions (Soylak and Akkayya 2003; Aydin and Soylak 2007). It was also used to develop chelating resins via functionalization with selective organic reagents (Metilda et al. 2004, 2005; Pathak and Rao 1996; Jain et al. 2001; Demirel et al. 2003) or by simple impregnation (Venkatesh and Maiti 2004; Tokalioglu et al. 2009; Ayata et al. 2009) for the preconcentration of metal ions. A good review on the use of styrene divinylbenzene copolymer for the preconcentration of metal ions was described by Soylak et al. (2001). The use of impregnated resins is particularly convenient because it is easy to prepare.
Active ingredients of drugs commonly available in the market hold little attention for their application as metal extracting agents. HNA (1-ethyl-1, 4-dihydro-7-methyl-4-oxo-1, 8-naphthyridine-3-carboxylic acid) is an antibacterial drug commonly used for treating several bacterial diseases (Martindale 2002). In our earlier communication, the potentiality of HNA in dichloromethane for the extraction of various metal ions is reported (Ali et al. 2010). However, the use of HNA for the online preconcentration of uranium has not been studied so far in the literature.
In our previous work, XAD-4 resin had been impregnated with N-benzoyl-N-phenylhydroxylamine and dibenzoylmethane had been used for the preconcentration of thorium and uranium, respectively (Ali et al. 2011; Shahida et al. 2011). In the present study, the determination of uranium by spectrophotometry after online preconcentration on XAD-4 resin impregnated with HNA was done. The chemical and hydrodynamic variables that affect the performance of the online system are performed. Moreover, the method is applied for the determination of uranium from a variety of samples having complex matrix.
Experimental
Instrumentation
The flow injection analyses (FIA) system made by Beijing Vital Instruments Co., Ltd., P. R. China, consisted of a multichannel peristaltic pump furnished with Tygon tubes (2.56 and 1.56 mm i.d.) to propel the liquid and a six-port multifunctional rotary valve to select the stages of preconcentration and elution. All the connecting tubes were 0.5 mm (i.d.) Teflon tubes. Polyethylene minicolumn (5-cm long, 5-mm i.d.) packed with XAD-4 resin impregnated with HNA was inserted in the system. Absorbance measurements were performed at 651 nm, (uranium–arsenazo (III) complex) by UV-visible spectrophotometer (Biotech Engineering Management CO. Ltd. UK), which consisted of a flow through cell of 10 μL capacity, having 5 mm path length. A pH meter model 605 from METROHM Ltd, Switzerland was used to measure the pH of solutions.
Reagents
Deionized distilled water (DDW) was used to prepare all solutions. All glassware was kept overnight in 5 % (v/v) nitric acid solution. Reagents of analytical grade were used. The stock solution of U(VI) ions (1,000 μg mL−1) was prepared by dissolving the required amount of uranyl nitrate hexahydrate (BDH, Poole, England) in DDW in 50 mL volumetric flask. Working solutions of uranium(VI) at micrograms per liter levels were prepared by diluting a 1,000-μg mL−1 stock solution. Hydrochloric acid (HCl) solutions were prepared by direct dilution of the concentrated solution (E. Merck, Darmstadt, Germany) with DDW. The pH adjustments were made with 0.01 mol dm−3 NaOH or HCl solution. A 0.1-mol dm−3 HCl solution was used as an eluent (E). Chromogenic reagent (CR) arsenazo III (Fluka, Vienna, Austria) solution was prepared by dissolving the required amount in 0.1 mol dm−3 HCl. Amberlite XAD-4 resin (surface area 750 m2 g−1, bead size 0.3–1.1 mm) was purchased from E. Merck (Darmstadt, Germany).
The mock sea water was prepared by dissolving the appropriate amount of various salts to get the sea water composition (Lyman and Felming 1940) and was stored in clean Polythene bottle. The tap and ground water samples were collected from Nilore, Islamabad, Pakistan.
The uranium ore mock solution was prepared based on the assumption that 0.16 g of a uranium ore reference material (CANMET-DH-1a Canada Center for Mineral and Energy Technology) could be decomposed with acids and diluted to 50 mL. The solution contained 3.0 mg of Al, 32 μg of Ca, 5.0 mg of Fe, 1.2 μg of K, 76 μg of Mg, 38 μg of Na, 80 μg of Th, and 200 μg of U in 50 mL (Hirano et al. 2003).
Purification of HNA
HNA was purified using Negram tablets (500 mg) which were obtained from a local market. The tablets were powdered in a mortar with a pestle, and a known weight of powder was dissolved in 500 mL of an aqueous solution of 0.1 mol dm−3 NaOH. Sodium salt of HNA was soluble in 0.1 mol dm−3 NaOH solution. The insoluble impurities were separated by filtration. The filtrate containing the sodium salt of HNA was mixed with 0.1 mol dm−3 HCl to get undissociated HNA which was insoluble in water and was separated by filtration. It was washed three times with DDW, dried in an oven at 60°C and purified (99 %) by recrystallization from dichloromethane. The detail of purification is given elsewhere (Ali et al. 2011). The steps involved during the purification are given below.

Impregnation of XAD-4 resin with HNA
A known weight (5.0 g) of XAD-4 resin was mixed with 50 mL of ethanol in a 100-mL Pyrex glass beaker and stirred for 2 h at room temperature using an electrical stirrer (China). The resin was filtered and dried in an oven at 40°C then treated with 50 mL of 2.0 mol dm−3 HCl for 30 min, and repeatedly washed with DDW till the washing was free of any traces of acid. The washed resin was dried in an oven at 100°C. Then 1.0 g of washed resin was equilibrated with 50 mL solution of HNA dissolved in ethanol (0.1 mol dm−3) by stirring it for 2 h. The resin was filtered and washed with DDW and dried. The amount of HNA impregnated was determined by the difference of weight of XAD-4 after and before impregnation. The detail of the procedure is described elsewhere (Venkatesh and Maiti 2004). The weight of impregnated HNA was found to be 0.33 g per g of XAD-4 resin. This amount was reconfirmed by desorbing the loaded amount of HNA from the impregnated resin by repeatedly shaking it with fresh dichloromethane. The absorbance of this HNA solution was measured at 332 nm (λ max) (Salim and Shupe 1966) using dichloromethane as a reference, and the amount was determined from a conventional working curve. The HNA loaded resin (170 mg) was packed in the minicolumn (5-cm long and 5-mm i.d.) having small pieces of polyurethane foam (washed with ethanol) at both ends as support.
Preconcentration method
The complete cycle of determination of U(VI) after online preconcentration in the minicolumn containing XAD-4 resin loaded with HNA by FI-SPM using arsenazo III as chromogenic reagent is given in Table 1. It consists of three steps, i.e., preconcentration, elution, and preconditioning steps. The manifold of the flow system is shown in Fig. 1.

Flow injection manifold for the online preconcentration of U(VI) ions in a minicolumn containing XAD-4 loaded with HNA, elution and determination with spectrophotometer using arsenazo III as chromogenic reagent. P1 and P2 peristaltic pumps, V multifunctional valve, column minicolumn, washing aqueous solution of pH 6.0. Reagent arsenazo III, 0.05 % in 0.1 mol dm−3 HCl, eluent HCl solution (0.1 mol dm−3)
In the first stage (Fig. 1a), the sample solution at pH 6.0 was pumped at 15.2 mL min−1 through the minicolumn containing XAD-4 impregnated with HNA for 60 s by running P1, keeping the multifunctional valve at load position. In this step, U(VI) ions were retained in the minicolumn by forming U(VI)–HNA complex.
After the preconcentration stage (Fig. 1b), P1 was stopped and the multifunctional valve was changed to inject position. At this stage, E (0.1 mol dm−3 HCl) and CR (0.05 % arsenazo III in 0.1 mol dm−3 HCl) solutions were passed through minicolumn by running P2, at a flow rate of 3.2 mL min−1 for 15 s. The analyte was thus desorbed from the solid phase owing to a high decrease in pH, which reduced HNA complexing capacity. Afterwards, the sample plug was mixed with CR and driven to the spectrophotometer, where absorbance measurements were continuously made at 651 nm. The peak height was used for quantitative parameter.
After the elution stage (Fig. 1c), the position of an eight-channel valve was changed to load position and the column was preconditioned to optimum pH suitable for complexation of U(VI) with HNA by passing an aqueous solution of pH 6.0 for 10 s at a flow rate of 4.9 mL min−1 by running P2. The E and CR solutions were recycled during this step to minimize the wastage of solutions. After the preconditioning step, the minicolumn was ready for a new preconcentration cycle.
Pretreatment of biological, water, and ore samples
Pretreatment of biological CRM (IAEA-V4)
Biological CRM (0.5 g) was treated with 10 mL concentrated perchloric acid in a Pyrex glass beaker and digested on hot plate to destroy the organic matter till the clear solution was obtained. The contents of the beaker were heated to near dryness and then made the solution to 500 mL, and pH was adjusted to 6.0 by NaOH solution. A known amount of U(VI) was added in it and the added amount of U(VI) was determined by the proposed method.
Water samples
Water and mock sea water samples were stored in plastic bottles with the addition of concentrated HCl till the pH of solution reached 3 (decomposition of HCO −3 /CO 2−3 ). Samples were filtered; pH was adjusted to 6.0 by the addition of NaOH solution. A known quantity of U(VI) was spiked in it and the added amount was measured by the proposed procedure.
Mock ore solution
The pH of the synthetic ore (CANMET-DH-1a Canada Center for Mineral and Energy Technology) solution was adjusted to 6.0 and U(VI) was determined by the proposed method.
HNA as an online complexing agent
HNA is a weak acid and its pka value is 5.9 to 6.35 (Lozano et al. 2002; Lin et al. 2004; Barbosa et al. 1997). Analytical application of this reagent is limited to spectrophotometric determination of iron(III) (Issopoulos 1989), fluorometric determination of Eu(III), Tb(III), and Dy(III) (Xiangqi et al. 1996), and luminescence determination of Eu(lll) using zirconium phosphate as solid support (Meshkova et al. 2004). In our earlier communication, HNA in dichloromethane is used for the solvent extraction of Eu(III) and Nd(III) at pH 6.5 (Ali et al. 2010). The use of HNA for the modification of solid sorbent and its use as online preconcentration of metal ions prior to their determination is not cited so far in the literature. In the present study, HNA is used for the modification of Amberlite XAD-4 resin by impregnation method. The modified resin (170 mg) is used for the selective preconcentration of metal ions in a minicolumn prior to determination with spectrophotometer. The preliminary experiments showed the good tendency of the sorbent to uranyl ions, UO +22 . The impregnated XAD-4 resin is then studied for enrichment of uranyl ions from water, biological, and geological samples. The arsenazo III in 0.1 mol dm−3 HCl is used as the chromogenic reagent due to its fast complex formation kinetics with U(VI). The results clearly demonstrated the high potential of the new sorbent for U(VI) separation from co-existing alkaline, alkaline earth, and transition and rare earth elements.
Results and discussion
A 100-μg L−1 U(VI) solution was used in the optimization of the online preconcentration system at flow rate of 15.2 mL min−1 for a period of 60 s and 0.1 mol dm−3 HCl as E and 0.05 % arsenazo III as CR at flow rate of 3.2 mL min−1 for a period of 15 s. The method was used in order to evaluate the variables that affected the preconcentration of uranium.
The pH of the aqueous phase is a vital condition in the preconcentration procedures, especially in regard to reactions involving complexing agents for metals due to acid–base properties of these compounds. The formation of the complexes is closely related to pH and can vary depending on the interaction between the organic reagent and metal. Thus, the influence of the sample pH on the analytical signal was examined by varying the pH of sample solution in the range of 1.0–7.0. As can be seen in Fig. 2, the peak absorbance increased with the increase of pH and reached to a maximum at pH 6.0 and then decreased with further increase in pH. The decrease in the absorbance signal at pH > 6.0 might be due to the formation of hydroxides of uranium. On the other hand, at pH < 6.0, the chelating capacity of the solid phase decreased due to the protonation of HNA present in the resin. Therefore, the sample pH was always adjusted to 6.0 in all the further experimental work.

Effect of pH of U(VI) sample solution (100 μg L−1) on the peak absorbance. Sample flow rate = 15.2 mL min−1, preconcentration time = 60 s, eluent (E) = HCl solution (0.1 mol dm−3), CR = 0.05 % arsenazo III in 0.1 mol dm−3 HCl, E and CR flow rate = 3.2 mL min−1
Preliminary studies suggested that there was no uptake of U(VI) by virgin XAD-4, and the uptake of U(VI) by the modified resin was only due to the presence of HNA in it.
In order to check the effect of residual U(VI) (not bound with HNA) in the column, a washing step was added between the preconcentration and elution step. This was done by running the preconditioning step in between the step 1 and step 2 of manifold by passing the aqueous solution of pH 6.0 as shown in Fig. 1c. The peak absorbance obtained by adding washing step was compared to the peak absorbance obtained without washing the column, and a very negligible change in the peak absorbance was observed. The reason may be the low concentration of U(VI) used for preconcentration and/or the negligible void volume of minicolumn due to good packing of the resin. Therefore, due to negligible effect of washing step on the peak absorbance, it was eliminated from the preconcentration method. However, a washing step can be added to remove those metal ions from the void volume of the column that might interfere in the determination of U(VI) ions with CR solution.
As column condition became different from that required for the U–HNA complex formation after passing acidic eluent, therefore to resume the optimum pH, the effect of preconditioning step was checked by passing the aqueous solution of pH 6.0 by running the pump 2 at the flow rate of 4.9 mL min−1 after elution step. The preconditioning step was found to increase the peak absorbance considerably. The reason could be the adjustment of the conditions of column by aqueous solution of pH 6.0, suitable for the complexation of U(VI) ions with HNA. Therefore, a preconditioning step was added after the elution step in the whole process.
In the FIA, the flow rate of sample could affect the sensitivity of the proposed method. Therefore, the effect of the sample flow rate on the preconcentration system was studied in the range of 1.8–15.2 mL min−1 and the results are depicted in Fig. 3. An increase in the signal was observed to 15.2 mL min−1 sample flow rate. The sample flow rate higher than 15.2 mL min−1 could not be studied due to increased backpressure in the manifold which caused leakage. Thus a sample flow rate of 15.2 mL min−1 was selected for further studies.

Effect of sample flow rate on the peak absorbance. pH of sample solution (U(VI)), 100 μg L−1) = 6.0, preconcentration time = 60 s, eluent (E) = HCl solution (0.1 mol dm−3), CR = 0.05 % arsenazo III in 0.1 mol dm−3 HCl, E and CR flow rate = 3.2 mL min−1
Hydrochloric acid solution was chosen to displace U(VI) from the column due to its non-oxidizing and less interfering nature. The analyte is desorbed due to the lowering of the pH caused by the acid. In this way, the formation of the uranium–HNA complex was not favored and the metal returned to the aqueous phase. Since the absorbance of the complex as well as CR changes with the change of the pH, the same concentration of HCl was used for E and CR solution preparations so that after mixing E and CR solution, any change in the pH of the final mixture and hence Schlieren effect should be avoided. The molarity of HCl was studied from 0.05 to 0.5 mol L−1. The absorbance increased with the increase of acid molarity up to 0.1 mol L−1 and after that a decrease in the absorbance was observed. This behavior is shown in Fig. 4. For acid concentration of less than 0.1 mol L−1, a decrease in absorbance signal was observed because the acid concentration was not sufficient to completely decrease the pH of the medium and a smaller amount of analyte was desorbed. The decrease in absorbance at higher molarity 0.5 mol L−1 is due to the residual acidity present in the minicolumn, which causes a loss of analyte in the early steps of preconcentration. In subsequent experiments, 0.1 mol L−1 HCl solution was used as the eluent.

Plot of peak absorbance versus concentration of HCl used as eluent and CR medium. Sample flow rate (100 μg L−1 U(VI), pH 6.0) = 15.2 mL min−1, preconcentration time = 60 s, eluent (E) = HCl solution (0.1 mol dm−3), CR = 0.05 % arsenazo III in 0.1 mol dm−3 HCl, E and CR flow rate = 3.2 mL min−1
As the P2 was used for the flow of both E and CR solutions by using the Tygon tubes of the same internal diameter, the effect of flow rate of both the solutions was studied simultaneously on the peak absorbance. Experiments were performed employing a 0.1-mol L−1 eluent solution at flow rates between 1.8 to 6.4 mL min−1 as shown in Fig. 5. The peak absorbance increased with the increase of flow rate and the best results were found at 3.2 mL min−1 and after that the signal was decreased. Slower flow rates (<3.2 mL min−1) cause higher dispersion and thus decrease the peak absorbance (Grudpan et al. 1995a, b). Higher flow rate (>3.2 mL min−1) causes the decrease of peak absorbance due to the dilution of metal ions in the eluent zone (Lozano et al. 2002). Therefore, 3.2 mL min−1 flow rate for E and CR solutions was selected for further studies. The elution profile was found to be better at this flow rate, and only 15 s was found to be sufficient for quantitative elution of metal ions as shown in Fig. 6.

Peak absorbance as a function of E and CR flow rate. Sample flow rate (100 μg L−1 U(VI), pH 6.0) = 15.2 mL min−1, eluent (E) = HCl solution (0.1 mol dm−3), CR = 0.05 % arsenazo III in 0.1 mol dm−3 HCl

Elution profile at the optimized conditions. Sample flow rate (100 μg L−1 U(VI), pH 6.0) = 15.2 mL min−1, preconcentration time = 60 s, eluent (E) = HCl solution (0.1 mol dm−3), CR = 0.05 % arsenazo III in 0.1 mol dm−3 HCl, E and CR flow rate = 3.2 mL min−1
In order to investigate the optimum time for the metal–CR complex formation, the effect of the varying reaction coil lengths (45 to125 cm) after the confluence point to the flow through cell of the spectrophotometer was studied. The results showed that the peak absorbance gradually decreased as the reaction coil length increased. This can be attributed to the increased dispersion of the sample zone, particularly when the reaction rate of complex formation is fast (Takeshi et al. 2004). Therefore the minimum possible Teflon tube length (45 cm) was used between the confluence point and the flow cell of the spectrophotometer.
The preconcentration time has a remarkable influence on the sensitivity of the system. Thus; the influence of the preconcentration time on the analytical signal yielded by 100 μg L−1 U(VI) solution was studied by varying it from 10 to 440 s. The analytical signal increased with the preconcentration time up to 400 s without increasing the back pressure in the manifold. The signal became constant at preconcentration time >400 s that might be due to the saturation of active sites of the complexing agent (Lemos and Gama 2010). Preconcentration time of 60 s was chosen for subsequent experiments; however, longer preconcentration times can be used for samples having low metal concentration.
The flow system was operated while employing two different preconcentration times (60 and 300 s). For 60-s preconcentration time, 15.2 mL sample was used. Under this condition, the system showed linearity for concentration of uranium between 25 and 150 μg L−1,which can be represented by the equation: A = 0.0009C − 0.0006 with correlation coefficient 0.9992, for n = 15 (no. of readings), where A is peak absorbance, C is concentration of U(VI) in micrograms per liter. All the statistical calculations were based on average of triplicate readings for each standard solution in the given range. The detection limit (DL), calculated as three times the standard deviation of blank values, was 1.1 μg L−1. The enrichment factor (EF) was calculated as the ratio of the slopes of the calibration curves obtained with and without preconcentration and was found to be 30. The relative standard deviation (RSD) was calculated by taking ten measurements of 100 μg L−1 solution and was found to be 0.81 %.The analytical throughput achieved under the optimized experimental condition was 42 h−1.
In case of longer preconcentration time (300 s), 76 mL sample volume was used. Calibration graph was constructed from 10 to 30 μg L−1, the characteristic equation being A = 0.0045C + 0.0008, with a correlation coefficient 0.9994 for n = 15. The DL, calculated as described above, was 0.22 μg L−1. The RSD also assessed by ten measurements of a 10 μg L−1 solution was 1.32 %. In this case, the EF was 150 and sample throughput was 11 h−1.
The DL (0.22 μg L−1) of the proposed method is lowest among our already reported spectrophotometric methods for the determination of uranium such as arsenazo III (DL = 1.6 μg L−1) (Khan et al. 2006) and arsenazo III+ N-Cetyl-N,N,N-tri-methylammonium bromide surfactant method (DL = 0.5 μg L-1) (Khan et al. 2011) and online preconcentration method using XAD-4 resin impregnated with dibenzoylmethane (DL = 0.3 μg L−1) (Shahida et al. 2011). The DL of the proposed method is the lowest among the other FI-SPM methods reported so far in the literature, e.g., chromazurol S (Hirano et al. 2003), arsenazo III (Pavon et al. 1989, 1990), chlorophosphonazo-mN (Sun et al. 1994), 2-(5-bromo-2-pyridylazo)-5-diethylaminophenol (Lindh et al. 1984; Jones 1985), and PAR (Grudpan et al. 1995b) methods and online preconcentration methods using Duolite resin (Grudpan et al. 1995a, 1998), Levextrel CL-5209 resin (Wu and Qi 1988), Dowex 50-X8 resin (Pavon et al. 1992), and XAD-4 resin functionalized with β-nitroso-α-naphthol (Lemos and Gama 2010) whose detection limits lie in the range (0.5–500 μg L−1).
The EF calculated for the proposed method (150) is higher than the already reported methods such as online preconcentration methods using Levextrel CL-5209 resin (EF = 10) (Wu and Qi 1988), XAD-4 resin functionalized with β-nitroso-α-naphthol (EF = 10) (Lemos and Gama 2010), and impregnated with dibenzoylmethane (EF = 143) (Shahida et al. 2011). The EF of the proposed method is even greater than the FI method reported by Dadfarnia containing microcolumn of activated alumina coupled with ICP whose EF was 40 (Dadfarnia and McLeod 1994).
Features of the proposed system under the optimum conditions for the spectrophotometric determination of U(VI) are given in Table 2. To evaluate the HNA impregnated XAD-4 resin stability, almost 1,000 loading and elution column operations were carried out. The sorption capacity of the resin was reproducible after so many preconcentration–elution cycles. This showed an excellent stability of impregnated XAD-4 resin with HNA and no leakage of the HNA from the column was observed in this study. The reason may be the insolubility of HNA in aqueous solutions.
Interference
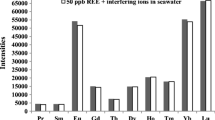
The effect of various cations as their chloride or nitrate salts and anions as their sodium or potassium salts has been studied on the preconcentration of U(VI) (100 μg L−1) using 60 s preconcentration time. The tolerance limits (±5.0 % variation in the peak height) are given in Table 3. From the table, it is clear that most of the cations do not interfere with the determination of U(VI) by the proposed method. Cations like Fe(III), Fe(II), Ni(II), Cd(II), Co(II), Mn(II), Zn(II), Pb(II), Al(III), and Cu(II) can be tolerated up to 1,000 times higher mass ratio of U(VI). Metals like rare earth elements can be tolerated up to 250 times higher mass ratio of U(VI) after adding 50 μg L−1 EDTA as masking agent, while only 35–50-fold higher mass ratio of these metals can be tolerated in FI-SPM systems such as PAR (Grudpan et al. 1995a, b, 1998) and arsenazo III method (Pavon et al. 1992). The tolerance limit of Th(IV) was increased up to 50 times higher mass ratio of U(IV) after adding 50 μg L−1 EDTA as masking agent in the proposed method as compared to other FI methods such as arsenazo III (Pavon et al. 1990, 1992) and chromazurol (Hirano et al. 2003) methods whose tolerance limits lie in the range of 6–16 μg L−1. This shows that the reported method is more selective than the other reported spectrophotometric methods.
It was observed that the anions like I−, Br−, Cl−, HPO −24 , ClO −4 , ClO −3 , and S2O −23 can be tolerated up to 1,000 times higher mass ratio of U(VI), while EDTA, SO -24 , and F− can be tolerated up to 500 times higher mass ratio of U(VI). The anions like CO −23 , HCO −3 , and C2O −24 interfered up to 15 times higher mass ratio of U(VI).
Analysis of spiked water and biological CRM samples
In order to assess the applicability of the method to real samples with different matrices containing varying amounts of a variety of diverse ions, it was applied to the separation and recovery of U(VI) from biological and three different water samples that were spiked with known amount of U(VI) using 300 s as preconcentration time and a washing step of 10 s was added to remove the matrix effect. The results of water samples are tabulated in Table 4 which shows the quantitative recovery of U(VI).
Analysis of uranium ore mock solution
The applicability of the proposed method to ore samples was tested by analyzing uranium ore (CANMET-DH-1a Canada Center for Mineral and Energy Technology) mock solution. The analytical results are shown in Table 5. According to the table, the U(VI) was quantitatively recovered with good precision.
Comparison with other FI–spectrophotometric methods
The figure of merits of this work and other reported FI–spectrophotometric methods is summarized in Table 6. The EF of the proposed method is higher than other reported methods. Comparison of the DL of the proposed method with other reported methods shows the lowest DL of the proposed method. The low matrix effect, as evident from the application on geological, biological, and water samples, is the additional advantage of the present method. Only the RSD of arsenazo III methods is better than the proposed method. Therefore, the proposed method using XAD-4-HNA as sorbent consists of simpler apparatus with easier manipulation having much lower equipment and running cost.
Conclusion
Amberlite XAD-4 resin impregnated with HNA (ingredient of a cheap and easily available drug, Negram) was successfully applied to the online preconcentration and determination of uranium by a spectrophotometer. A good DL of 1.1 and 0.22 μg L−1, a sampling frequency of 42 and 11, and EF of 30 and 150 were obtained for 60 and 300 s, respectively. The analytical characteristics of the system were improved compared to many methods listed in the literature. The present study has demonstrated that the active ingredients of cheap pharmaceutical drugs can be successfully used for the preconcentration of metals ions prior to their determination by UV-visible spectrophotometry. This has opened a new class of organic reagents that can be used as selective complexing agents for various metal ions. Further research work is required to explore more drugs to find more selective and specific complexing reagents for metal ions.
References
Ali, A., Abbasi, Y. A., Khan, M. H., & Saeed, M. M. (2010). Liquid–liquid extraction of Nd(III) and Eu(III) using nalidixic acid in dichloromethane. Radiochimica Acta, 98(8), 513–517.
Ali, A., Shahida, S., Khan, M. H., & Saeed, M. M. (2011). Online thorium determination after preconcentration in a minicolumn having XAD-4 resin impregnated with N-benzoylphenylhydroxylamine by FI–spectrophotometry. Journal of Radioanalanytical and Nuclear Chemistry, 288(3), 735–743.
Ayata, S., Kaynak, I., & Merdivan, M. (2009). Solid phase extractive preconcentration of silver from aqueous samples. Environmental Monitoring and Assessment, 153(1–4), 333–338.
Aydin, F. A., & Soylak, M. (2007). Solid phase extraction and preconcentration of uranium(VI) and thorium(IV) on Duolite XAD761 prior to their inductively coupled plasma mass spectrometric determination. Talanta, 72, 187–192.
Barbosa, J., Bergés, R., Toro, I., & Nebot, V. S. (1997). Protonation equilibria of quinolone antibacterials in acetonitrile–water mobile phases used in LC. Talanta, 44(7), 1271–1283.
Camel, V. (2003). Solid phase extraction of trace elements. Spectrochimica Acta Part B, 58(12), 1177–1233.
Dadfarnia, S., & McLeod, C. W. (1994). Online trace enrichment and determination of uranium in waters by flow injection inductively coupled plasma mass spectrometry. Applied Spectroscopy, 48(11), 1331–1336.
Demirel, N., Merdivan, M., Pirinccioglu, N., & Hamamci, C. (2003). Thorium(IV) and uranium(VI) sorption studies on octacarboxymethyl-C-methylcalix[4] resorcinarene impregnated on a polymeric support. Analytica Chimica Acta, 485(2), 213–219.
Grudpan, K., Laiwraungrath, S., & Sooksamiti, P. (1995a). Flow injection spectrophotometric determination of uranium with in-valve ion-exchange column preconcentration and separation. Analyst, 120(8), 2107–2110.
Grudpan, K., Sooksamiti, P., & Laiwraungrath, S. (1995b). Determination of uranium in tin tailings using 4-(2-pyridylazo) resorcinol by flow injection analyses. Analytica Chimica Acta, 314(1–2), 51–55.
Grudpan, K., Jakmunee, J., & Sooksamiti, P. (1998). Spectrophotometric determination of uranium by flow injection analysis using U/TEVA.Spec™ chromatographic resin. Journal of Radioanalytical and Nuclear Chemistry, 229(1–2), 179–181.
Hirano, Y., Ogawa, Y., & Oguma, K. (2003). Simultaneous spectrophotometric determination of uranium and thorium by flow injection analysis using selective masking. Analytical Science, 19(2), 303–307.
Issopoulos, P. B. (1989). Nalidixic acid, complex formation with iron(III) and its analytical applications. Acta Pharmaceutica Jugoslavia, 39(4), 267–274.
Jain, V. K., Handa, A., Sait, S. S., Shrivastav, P., & Agrawal, Y. K. (2001). Pre-concentration, separation and trace determination of lanthanum(III), cerium(III), thorium(IV) and uranium(VI) on polymer supported o-vanillinsemicarbazone. Analytica Chimica Acta, 429(2), 237–246.
Jones, E. A. (1985). Determination of uranium(VI) with 2-(5-bromo-2-pyridylazo)-5-diethylaminophenol in a flow injection system. Analytica Chimica Acta, 169, 109–115.
Khan, M. H., Warwick, P., & Evans, N. (2006). Spectrophotometric determination of uranium with arsenazo-III in perchloric acid. Chemosphere, 63(7), 1165–1169.
Khan, M. H., Bukhari, S. M. H., Ali, A., Liaqat, K., & Fazal, S. (2011). Spectrophotometric determination of uranium with arsenazo-III in the presence of N-acetyl-N, N, N- trimethylammonium bromide as surfactant. Journal of Radioanalytical and Nuclear Chemistry, 289(1), 113–119.
Lemos, V. A., & Gama, E. M. (2010). An online preconcentration system for the determination of uranium in water and effluent samples. Environmental Monitoring and Assessment, 171(1–4), 163–169.
Lemos, V. A., Novaes, G. S., Carvalho, A. L., Gama, E. M., & Santos, A. G. (2009). Determination of copper in biological samples by flame atomic absorption spectrometry after precipitation with Me-BTAP. Environmental Monitoring and Assessment, 148(1–4), 245–253.
Lin, C. E., Deng, Y. J., Liao, W. S., Sun, S. W., Lin, W. Y., & Chen, C. C. (2004). Electrophoretic behavior and pK a determination of quinolones with a piperazinyl substituent by capillary zone electrophoresis. Journal of Chromatography. A, 1051(1–2), 283–290.
Lindh, C. S., Nord, L., Danielsson, L. G., & Ingman, F. (1984). The analysis of aqueous solutions with ethanol soluble reagents in a flow injection system. Spectrophotometric determination of uranium. Analytica Chimica Acta, 160, 11–19.
Lozano, E. J., Marques, I., Barron, D., Beltrán, J. L., & Barbosa, J. (2002). Determination of pK a values of quinolones from mobility and spectroscopic data obtained by capillary electrophoresis and a diode array detector. Analytica Chimica Acta, 464(1), 37–45.
Lyman, G., & Felming, R. H. (1940). Composition of sea water. Journal of Marine Research, 3, 134–136.
Martindale, W. (2002). The extra pharmacopia. London: Royl Pharmaceutical Society.
Meshkova, S. B., Litvinenko, A. V., Nazarenko, N. A., Topilova, Z. M., & Efryushina, N. P. (2004). Improving the selectivity of the luminescence determination of europium(III) with the use of a zirconium phosphate solid support. Journal of Analytical Chemistry, 59(3), 246–249.
Metilda, P., Gladis, J. M., & Rao, T. P. (2004). Synthesis of malonic acid-functionalized Amberlite XAD-4 and its use in solid phase extraction/preconcentrative separation of thorium (IV). Radiochimica Acta, 92(12), 931–937.
Metilda, P., Sanghamitra, K., Gladis, J. M., Naidu, G. R. K., & Rao, T. P. (2005). Amberlite XAD-4 functionalized with succinic acid for the solid phase extractive preconcentration and separation of uranium(VI). Talanta, 65(1), 192–200.
Pathak, R., & Rao, G. N. (1996). Synthesis and metal sorption studies of p-tert butylcalix[8]arene chemically bound to polymeric support. Analytica Chimica Acta, 335(3), 283–290.
Pavon, J. L. P., Cordero, B. M., Mendez, J. H., & Agudo, R. M. I. (1989). Application of a nested-loop system for the simultaneous determination of thorium and uranium by flow injection analysis. Analytical Chemistry, 61(15), 1789–1791.
Pavon, J. L. P., Cordero, B. M., Garcia, E. R., & Mendez, H. (1990). Determination of uranium using a flow system with reagent injection. Application to the determination of uranium in ore leachates. Analytica Chimica Acta, 230, 217–220.
Pavon, J. L. P., Pinto, C. G., Garcia, E. R., & Cordero, B. M. (1992). Flow injection determination of thorium and uranium after on-line ion-exchange preconcentration on Dowex 50-X8. Analytica Chimica Acta, 264(2), 291–296.
Salim, E. F., & Shupe, I. S. (1966). Qualitative and quantitative tests for nalidixic acid. Journal of Pharmaceutical Sciences, 55(11), 1289–1290.
Shahida, S., Ali, A., Khan, M. H., & Saeed, M. M. (2011). Flow injection online determination of uranium after preconcentration on XAD-4 resin impregnated with dibenzoylmethane. Journal of Radioanalanytical and Nuclear Chemistry, 289(3), 929–938.
Soylak, M., & Akkaya, Y. (2003). Separation/preconcentration of xylenol orange metal complexes on Amberlite XAD-16 column for their determination by flame atomic absorption spectrometry. Journal of Trace and Microprobe Technology, 21(3), 455–466.
Soylak, M., Elci, L., & Dogen, M. (2001). Solid phase extraction of trace metal ions with Amberlite XAD resins prior to atomic absorption spectrometric analysis. Journal of Trace and Microprobe Technology, 19(3), 329–344.
Soylak, M., Narin, I., Bezerra, M. A., & Ferreira, S. L. C. (2005). Factorial design in the optimization of preconcentration procedure for lead determination by FAAS. Talanta, 65(4), 895–899.
Sun, J. Y., Chen, X. G., & Hu, Z. D. (1994). Spectrophotometric determination of uranium(VI) with chlorophosphonazo-mN by flow injection analysis. Analytical Letters, 27(10), 1989–1998.
Takeshi, Y., Hiroya, I., & Tatsuhiko, T. (2004). Simple, rapid and sensitive, determination of bismuth in iron and steel based on in-line preconcentration/separation directly coupled with spectrophotometric detection in a continuous flow system. ISIJ International, 44(4), 698–703.
Tokalioglu, S., Yilmaz, V., & Kartal, S. (2009). Solid phase extraction of Cu(II), Ni(II), Pb(II), Cd(II) and Mn(II) ions with 1-(2-thiazolylazo)-2-naphthol loaded Amberlite XAD-1180. Environmental Monitoring and Assessment, 152(1–4), 369–377.
Tzanavaras, P. D., & Themelis, D. G. (2007). Review of recent applications of flow injection spectrophotometry to pharmaceutical analysis. Analytica Chimica Acta, 588(1), 1–9.
Venkatesh, K., & Maiti, B. (2004). Preconcentration and separation of uranium from thorium by solid phase extraction with XAD–4 modified with organic reagents. Separation Science and Technology, 39(8), 1779–1789.
Wu, X., & Qi, W. (1988). Spectrophotometric determination of trace uranium in geological samples by flow injection analysis with online levextrel resin separation and preconcentration. Analytica Chimica Acta, 214, 279–288.
Xiangqi, J., Lanying, L., & Hanguo, H. (1996). Fluorometric determination of europium, terbium and dysprosium with nalidixic acid system. Fenxi Huaxue, 24(10), 1216–1218.
Acknowledgments
S. Shahida, one of the authors, is highly grateful to Higher Education Commission, Pakistan, for awarding a fellowship for Ph. D studies.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shahida, S., Ali, A., Khan, M.H. et al. Flow injection online spectrophotometric determination of uranium after preconcentration on XAD-4 resin impregnated with nalidixic acid. Environ Monit Assess 185, 1613–1626 (2013). https://doi.org/10.1007/s10661-012-2655-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10661-012-2655-4