
Abstract
Curvularia lunata causes Curvularia leaf spot disease of maize, resulting in periodically significant yield losses around the world. To understand the molecular mechanisms of fungal pathogenicity and virulence factors contributed to the host, here we report a knockout transformation system against target genes. The concentration of conidia, Agrobacterium cell density, and method of co-incubation were optimized for deletion of the gene encoding 1, 3, 8-trihydroxynaphthalene reductase (Brn1), a gene in the melanin biosynthesis pathway, as a test case. Transformants contained a single T-DNA copy. The Brn1 mutant strain was reduced in virulence compared with the wild type strain when inoculated on susceptible maize. Transformation efficiency was 130 ± 10 transformants per 1 × 105 germlings and homologous recombination efficiency was 60.0 to 100.0 %.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Curvularia lunata (anamorph Cochliobolus lunatus) is the causal agent of Curvularia leaf spot of maize, one of the most serious maize foliar diseases impacting productive yield in China (Dai et al. 1995). In 1996, the loss of grain yield was estimated as 8 million kilograms in Liaoning province in northern China (Dai et al. 1998). Subsequently, resistant varieties containing tropic and sub-tropic germplasms were identified and introduced into maize to generate a large number of resistant cultivars. Those cultivars had already been planted to control Curvularia leaf spot disease of maize (Li et al. 2002; Zhao et al. 2002). However, in recent years, the disease has reappeared and caused serious damage in some maize growing areas such as Liaoning, Anhui and Henan provinces. The main cause of the disease recurrence was expected to link to large areas of monoculture with the same or similar resistant germplasm, which became a matrix to induce pathogen virulence variation.
The genus Cochliobolus is an important group of species that includes pathogens of monocots such as Cochliobolus sativus (wheat/barley/cereals Common root rot), Cochliobolus victoriae (Oat victoria blight), Cochliobolus miyabeanus(Rice brown spot), and the maize pathogens Cochliobolus heterostrophus(Southern Corn Leaf Blight) and Cochliobolus carbonum(Northern Leaf Spot), all of which have been sequenced (http://genome.jgi.doe.gov/programs/fungi/index.jsf). Recently, C. lunata strain CX-3 was sequenced (http://ncbi.nlm.nih.gov/bioproject/182303) (Gao et al. 2014), which provides an opportunity for in-depth understanding of virulence requirements of the fungus to infect its maize host. However, there are no efficient targeted gene deletion protocols for this fungus that are needed to assess candidate genes for their involvement in mechanisms of pathogenicity to plants. This is especially critical for testing mutants of field isolates representing the virulence of the fungus on susceptible and resistant plants.
Protoplast-polyethylene glycol (PEG)-based transformation and Agrobacterium tumefaciens-mediated transformation (ATMT) are two major gene manipulation protocols for deleting genes by homologous recombination in filamentous ascomycetes. The protoplast-polyethylene glycol transformation approach has been used with many important plant-pathogenic fungi (e.g., Cooley et al. 1988; Henson et al. 1988; Kistler and Benny 1988; Parsons et al. 1987; Rodriguez and Yoder 1987; Turgeon et al. 1985). However, for some fungi, low yield and low viability of protoplasts coupled to low rates of homologous integration of exogenous DNA constructs are limiting (Bundock et al. 1995; Mullins and Kang 2001).
ATMT has been used successfully with many fungi as an alternative to using protoplasts (de Groot et al. 1998; Michielse et al. 2005; Xue et al. 2013). With ATMT, it is often possible to choose germinating conidia, fresh mycelia, or fruiting bodies, as starting materials for transformation (Sugui et al. 2005). Random insertion mutations have been reported for C. lunata using ATMT and protoplast-PEG-based transformation (Liu et al. 2010; Huang et al. 2010). However, high-efficiency transformation with targeted gene disruption has not been established to date.
The present paper focused on the evaluation of factors that influence the efficiency of gene knockout by ATMT. The 1,3,8-trihydroxynaphthalene reductase gene (Brn1), associated with the 1,8-dihydroxynaphthalene-type melanin pathway, was chosen as a target because mutants can be easily identified by an alteration in pigmentation (Thompson et al. 2000). Deletion of Brn1 altered virulence of the corresponding mutant on susceptible maize inbred line.
Materials and methods
Fungal strains, culture conditions, and harvesting conidia
Curvularia lunata strains CX-3, ND-108 and WS18, obtained from the molecular and physiological plant pathology laboratory at Shenyang Agriculture University, were tested for sensitivity to hygromycin B, pathogenicity and ability to produce conidia. Strain CX-3 was chosen initially for transformation attempts, based on hygromycin B sensitivity, pathogenicity and spore production capacity. Fungal strains were stored as single conidial cultures at −80 °C in Potato Dextrose (PD) medium [200 g of potato, 20 g of dextrose per liter] in 25 % glycerol. For each experiment, an aliquot was recovered from glycerol and transferred to PD agar (PDA) for 6 to 7 days at 25 °C under full darkness. To harvest conidia, sterile distilled H2O was added to PDA plates, the surface was rubbed with a flat knife to dislodge conidia, and the resulting conidial suspension was filtered through three layers of lens paper to remove mycelial debris.
PCR for target gene
Genomic DNA of the wild type and candidate transformants of C. lunata CX-3 grown on PDA plates was extracted from the mycelium using the Plant Genomic DNA Isolation kit (TIANGEN, Beijing, China). PCR were performed in a total volume of 20 μl containing 0.4 mM of each dNTP, 5 mM of each primer, 1 unit of easyTaq DNA polymerase (TaKaRa, Dalian, China), 2.0 μl of 10× reaction buffer, and 10 to 20 ng of genomic DNA. PCR products were purified by Axyprep DNA Gel extraction kit (Axygen Scientific Inc. USA) and sequenced (Sangon Biotech (Shanghai) Co., Ltd. Shanghai, China). Three primer pairs were used to obtain full length genomic Brn1.
Construction of a plasmid for Agrobacterium transformation
Fragments of the Brn1 gene, Brn1-5 F (524 bp) and Brn1-3R (453 bp), were amplified by PCR using primer combinations Brn1-5 F-F/R and Brn1-3R-F/R. PCR was performed using Phusion DNA polymerase (New England Biolabs, lpswich, MA). HindIII/SalI restriction enzyme recognition sites were added to the 5′ ends of the Brn1-5 F-F/R primers, and KpnI/SacI restriction enzyme recognition sites were added to the 5′ ends of Brn1-3R-F/R primers, respectively. These two PCR products were then constructed into the pMD19-T vector (TaKaRa, Dalian, China), giving rise to plasmids pMD19-T-Brn1-5 F and pMD19-T-Brn1-3R. The Brn1-5F fragment was cut from pMD19-T-Brn1-5 F with HindIII/SalI and ligated into pPZP100H (Hajdukiewicz et al. 1994), precut with HindIII/SalI, to generate pPZP100HClBrn1-5 F. The Brn1-3R fragment was cut from pMD19-T-Brn1-3R using KpnI/SacI and ligated into pPZP100HClBrn1-5 F,precut with KpnI/SacI. The final plasmid was named pPZP100HClBrn1 (Fig. 3). pPZP100HClBrn1 was transformed into A. tumefaciens strain AGL-1 by the electroporation method (Sambrook and Russell 2001). Insertion orientation was determined by PCR using primers PtrpC/Brn1-5 F-F and Brn1-3R-R/TtrpCend. All primer sequences are shown in Table 1.
A second C. lunata gene, FTR1, was disrupted using the same approach to construct the transformation plasmid, which was transformed into AGL-1 by electroporation. The primers for this experiment are listed in Supplementary Table 1.
Transformation of C. lunata
A. tumefaciens strain AGL-1, containing pPZP100HClBrn1 or the other plasmid, was grown at 28 °C for 2 days in Luria broth (LB) (5 mL), supplemented with chloramphenicol (100 μg/mL) and ampicillin (100 μg/mL). The culture was diluted with induction medium (IM) to an optical density at 600 nm (OD600) = 0.25 in a final volume of 5 ml. IM (in 1 l) is 10.5 g of K2HPO4, 4.5 g of KH2PO4, 1.0 g of NH4SO4, 0.5 g of Na3citrate • 2H2O, 0.2 g of MgSO4 • 7H2O, 1.0 mg of thiamine-HCl, 2.0 g of glucose, 40 mM 2- (4-Morpholino) ethanesulfonic acid, and 0.5 % glycerol, pH 5.3 (containing the same concentrations of antibiotics as LB), plus acetosyringone (AS) (200 μg/mL). Cultures were grown to OD600 = 0.4 to 0.7. CX-3 conidial suspension (1 × 105/mL to 1 × 107/mL) was germinated in IM (not containing AS) in a 250 mL flask, for 6 h before mixing with an equal volume of induced AGL-1 cells in a 50 mL falcon centrifuge tube. The mixture (500 μL) was placed on a cellulose nitrate membrane (Whatman, 0.45 μm) on top of IM agar (15 g/liter) and incubated at 25 °C for 36 or 48 h. Membranes were transferred onto Czapek yeast extract agar (CYA) plates containing cefotaxime (300 μg/mL) and hygromycin B (200 μg/mL) to kill Agrobacterium cells and select for candidate transformants, respectively. CYA (in 1 l) is 3.0 g of NaNO3, 1.0 g of K2HPO4, 0.5 g of KCl, 0.5 g of MgSO4 · 7H2O, 0.01 g of FeSO4 · 7H2O, 5.0 g of yeast, 30 g of sucrose, and 14 g agar. Plates were incubated at 25 °C for 5 to 7 days until conidiogenesis. Candidate transformants were purified by isolating single conidia and reselecting on CYA medium containing the same concentrations of hygromycin B and cefotaxime. Purified hygromycin B resistant candidate transformants were stored in CY in 25 % glycerol at −80 °C.
PCR confirmation of gene deletion
To verify insertion of the HPH gene at the native Brn1 locus, three primer pairs were used: PtrpC/Brn1-F and TtrpCend/Brn1-R to confirm correct insertion into the 5′ and 3′ flanks of Brn1, respectively; and Brn1-F/Brn1-R to confirm expected size of insertion. All primer sequences in this paper are listed in Table 1.
For confirmation of deletion of an additional gene in C. lunata, FTR1 encoding an iron permease, the same PCR primer strategies described above for Brn1 were used (Supplemental Table 1).
DNA extraction and southern blot hybridization
Southern blot analysis of DNA from wild type and three of five confirmed Brn1 mutants was performed. Genomic DNA of mutant and wild type strains was isolated from lyophilized mycelia as described previously (Xue et al. 2013). For blotting, genomic DNA was digested with XbaI and EcoRI overnight at 37 °C, separated by electrophoresis in an agarose gel, and transferred to a nitrocellulose membrane using a standard protocol (Sambrook and Russell 2001). A digoxigenin (DIG)-labeled probe was generated by PCR using primers Hph F1/Hph R1 to amplify the HPH gene and the membrane hybridized to the labeled DNA. Hybridizing bands were detected using the DIG High Prime DNA Labeling and Detection Starter Kit II Direct Labeling and Detection System according to the manufacturer’s instructions (Roche, Mannheim, Germany).
Virulence assays
Five-week-old ‘78599’ and ‘Huangzaosi’ maize inbred line plants were used to evaluate the virulence of brn1 mutants, as previously described (Xue et al. 2008). A spore suspension (2 mL of 5 × 104 spores/mL) was spray inoculated on each plant. Diseased leaves were photographed at 7 days post-inoculation.
Results
Strain selection
Field strain CX-3 was chosen from three candidate wild type strains because it conidiated well, had high pathogenicity and was sensitive to hygromycin B. The level of sensitivity to hygromycin B was tested by growing strains on PDA plates supplemented with different concentrations of the antibiotic (0, 50, 100, 150, 200, 300, 400, 500 μg/mL). Growth of CX-3 was inhibited partly at 100 μg/mL and completely at 200 μg/mL. Other strains showed less sensitivity to hygromycin B. For selection of candidate transformants, 200 μg/mL was chosen.
Target gene ClBrn1 analysis
Sequencing revealed that C. lunata CX-3 Brn1 (912 bp, JQ698339) possesses two introns at positions 62 bp (55 bp long) and 594 bp (53 bp long) and is similar to the structure of the C. lunata m118 Brn1 gene (Fig. 1). A putative open reading frame of 801 bp was identified that was predicted to code for a protein of 267 amino acids, with a calculated molecular mass of 28.65 kDa and pI of 6.60 (AFJ97108). The amino acid sequence of Brn1 was found to be highly similar to that of other fungal Brn1 or 3HNRs (Figs. 2 and 3). There was more than 95 % identity between the amino acid sequences of C. lunata Brn1 and those of C. heterostrophus, C. carbonum, C. sativus, C. miyabeanus, Pyrenophora tritici-repentis, Setosphaeria turcica, Alternaria alternata, Leptosphaeria maculans, Pleospora tarda, Phaeosphaeria nodorum and Paradendryphiella salina.

Sequence comparison of Brn1 genes from two C. lunata isolates (CX-3 and m118) and positions of oligonucleotide primers. First, second and three round PCR amplifications were performed to obtain 549, 430 and 300 bp by Brn1-F1/Brn1-R1, Brn1-F2/Brn1-R2 and Brn1-F3/Brn1-R3. Three fragments were assembled as full length ClBrn1 (912 bp). Nucleotides with red represent the open reading frame (ORF), blue nucleotides represent introns

An alignment of the amino acid sequences of C. lunata Brn1 and 11 homologs. Green line: Glucose/ribitol dehydrogenase, Red line: Short-chain dehydrogenase/reductase SDR; blue line: Short-chain dehydrogenase/reductase, conserved site

Agrobacterium tumefaciens-mediated transformation plasmid for 1,3,8-trihydroxynaphthalene reductase (Brn1) gene knockout. Restriction map of pPZP100HClBrn1, a binary vector for deletion of the C. lunata Brn1 gene. The XbaI site was used for insertion of the hygromycin B resistance cassette (HPH). Brn1-5 F (upstream flanking sequence) was inserted as a Hind III/Sal I fragment between the HPH cassette and the T-DNA right border (RB), while Brn1-3R (downstream flanking sequence) was inserted as KpnI/SacI fragment between the HPH cassette and the T-DNA left border (LB)
Factors influencing ATMT efficiency in C. lunata
Conidial concentration was tested in three different levels, 1 × 105/mL, 1 × 106/mL, and 1 × 107/mL, and transformants were obtained the most with 1 × 105/mL conidia even though a number of parameters, including varying the Agrobacterium cell density and co-cultivation conditions, were tried as discussed below.
Agrobacterium cell density
Cell concentration (OD600 value) of AGL-1 after induction was also a key factor. Transformation rate was the highest when OD value reaches about 0.6, no gene knockouts were obtained when the OD value was <0.4, although it was evident that HPH was randomly inserted into the genome at lower cell densities (0.2 to 0.4 OD).
Plating conditions
The mixture of Agrobacterium cells and fungal conidia was plated on a cellulose nitrate membrane. Varying the time of co-culture (36 or 48 h) made an obvious difference to transformation efficiency, with more transformants being obtained at the longer incubation time. Nevertheless incubation time should not be more than 48 h to avoid integration of multiple copies of the T-DNA molecules. Putative transformants grew faster than background growth on selection medium (Fig. 4). Overall transformation efficiency was 130±10 transformants per 1 × 105 starting conidia.

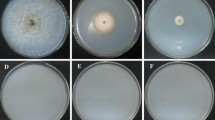
Transformation plates. a, Control wild type strain CX-3 and AGL-1 cells without plasmid pPZP100HClBrn1 on hygromycin B (200 μg/ml) selection medium plus cefotaxime (300 μM). Note background growth. b, Example of a typical original transformation plate containing CX-3 and AGL-1 cells carrying pPZP100HClBrn1 plasmid on the same selection medium as in A, 3 days after plating. Note the large, light brown colony is a candidate brn1 mutant
Verification of transformation and targeted integration
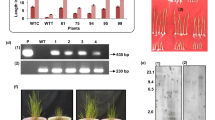
The Brn1 gene in the melanin biosynthesis pathway was chosen as the target for deletion because mutants were expected to be brown and, thus, readily distinguishable from black wild type colonies (Fig. 5). Hygromycin-B-resistant colonies obtained with ATMT were screened by PCR using primer pairs PtrpC/Brn1-F and TtrpCend/Brn1-R, which were expected to amplify products of 1.1 and 0.8 Kb, respectively, if the Brn1 gene was deleted (Fig. 6a). These two bands should be absent in the wild type. Primer pair Brn1-F/Brn1-R should amplify a product of 3.6 Kb in deletion strains and 1.5 Kb in the wild type. According to PCR assays, 9 of 15 transformants screened had the Brn1 gene deleted (Fig. 6b). Thus, under these conditions, homologous recombination efficiency was 60 %.

Phenotype of a 1,3,8-trihydroxynaphthalene reductase (brn1) mutant compared with the wild type (WT). a, WT and mutant on lactose casein (CY) agar and b, in CY liquid. The mutant is light brown, indicating a block in the melanin synthesis pathway

Polymerase chain reaction (PCR) and Southern blot analysis of brn1 mutants of Curvularia lunata. a, Schematic representation of the DNA used for transformation. Brn1-5 F = 5′ flanking sequence of Brn1, Brn1-3R = 3′ flanking sequence of Brn1, PtrpC = Aspergillus nidulans trpC promoter; HPH is the hygromycin B resistance gene and TtrpC is the A. nidulans trpC terminator. Brn1-F, PtrpC, TtrpCend, and Brn1-R, are primers used for screening brn1 mutants (Table 1). b, PCR confirmation of brn1 mutants from two transformants using primers Brn1-F/Brn1-R (lanes 1 to 4), Brn1-F2/PtrpC (lanes 5 to 8) and TtrpCend/Brn1-R2 (lanes 9 to 12). Lane M, TaKaRa 5 kb DNA ladder marker; lanes 1, 5, and 9, H2O; lanes 2, 6 and 10, DNA from wild type; lanes 3, 4, 7, 8, 11 and 12, DNA from transformants. Size of PCR product from the mutants matches the predicted size of Brn1-5 F + HPH + Brn1-3R, indicating replacement of the Brn1 gene by the hygromycin B resistance gene at the Brn1 locus. C, Southern blot analysis of wild type and brn1 mutants. Genomic DNA was cut with XbaI and EcoRI, two restriction enzymes that do not cut in HPH, and separated on a 1.0 % agarose gel by electrophoresis. DNAs were transferred to a nitrocellulose membrane and hybridized using a digoxigenin-labeled 1.1 kb fragment of HPH. Lane CK is DNA from the wild type strain, and lanes 1 to 3 DNA from transformants. When homologous integration has occurred, a hybridizing band of ≈ 2.1 kb (XbaI digested) was observed due to integration of the selectable marker, and a band of ≈ 3.5 kb was also observed in the mutants (EcoRI digested), indicating that only a copy of HPH integrated the Brn1 locus. The wild type lane does not have any hybridization signal due to lack of the HPH gene
Southern blot analysis of three independent transformants, determined by PCR to be brn1 mutants, confirmed that the HPH gene had integrated at a single site in all cases. In the three transformants, a single XbaI hybridizing band of 2.1 kb and a single EcoRI hybridizing band of 3.5 Kb were detected (Fig. 6c).
Gene replacement of the FTR1 gene
The deletion of an additional gene was tested and verified by diagnostic PCR. Since strain CX-3 is highly homologous to strain C. lunata m118 isolated from sorghum, longer flanking sequences were readily available for constructions of gene deletions. Increasing the flanking region to more than 800 bp improved the efficiency of targeted gene replacements within the transformants analyzed to 100 % (Table 2; Supplemental Table 1, Supplemental Figure 1).
Virulence phenotypes on susceptible maize
The brn1 mutant, ftr1 mutant and wild type strain CX-3 were inoculated on resistant and susceptible maize inbred lines ‘78599’ and ‘Huangzaosi’. Virulence phenotypes of the ftr1 mutant were similar to those of the wild type. Inoculation with the brn1 mutant caused a significant reduction in virulence to maize. At 7 days post-inoculation, the wild type strain produced semi-round brown eye lesions; in contrast, inoculation with the mutants did not elicit this type of lesion, but instead produced tiny green back flecks and the number of scab decreased visually (Fig. 7). This matches results demonstrating the role of this gene in disease development of other plant-pathogenic fungi (e.g., Setosphaeria turcica, Magnaporthe oryzae and Cochliobolus heterostrophus: Xue et al. 2013; Thompson et al. 2000; Oide et al. 2006).

Virulence phenotypes of Curvularia lunata wild type (WT) and a brn1 mutant on resistant and susceptible maize. a, Virulence of WT strain CX-3 and the brn1 mutant on resistant maize ‘78599’. WT caused small faded green spots, comparing to the no spots on the mutant brn1. b, Virulence of the wild type strain CX-3 and the brn1 mutant on maize ‘Huangzaosi’. Wild type produced semi-round brown eye lesions, whereas the brn1 mutant strain produced only small chlorotic flecks
Discussion
In this study, we describe the construction of binary vectors for gene deletion and the conditions of ATMT that affect the transformation efficiency in C. lunata. In previous studies, transformation of C. lunata was achieved with a vector containing the hygromycin B selection marker (Liu et al. 2010). However, that study had only constructed a library of random insertional mutants. In this work, we constructed a system of targeted gene replacement in C. lunata by ATMT. This system of gene deletion for C. lunata can contribute to understand the mechanisms of its pathogenicity in future studies.
The recommended protocol for transformation is as follows. The growing condition for AGL-1 containing vector in LB is at 28 °C for 2 days. The culture is diluted with IM to OD = 0.25 and grown overnight to OD600 ≈ 0.6. A CX-3 conidial suspension (1 × 105/mL) is germinated 6 h in darkness, before mixing with an equal volume of induced A. tumefaciens. The mixture (500 μL) is spread on a cellulose nitrate membrane (Whatman 0.45 μm) on top of IM agar in the presence of AS (200 μM) and incubated at 23 °C for 48 h. Membranes are transferred onto CYA plates containing cefotaxime (300 μg/ml) and hygromycin B (200 μg/ml). Plates are incubated at 28 °C for 5 to 7 days until conidiogenesis. Candidate transformants are purified by picking single conidia and reselected on CYA medium containing the same concentrations of hygromycin B and cefotaxime.
The gene targeting efficiency varies greatly between different fungal species. As examples, a gene targeting efficiency of 95 % is reported for Saccharomyces cerevisiae, 5–75 % for Aspergillus nidulans and 1–30 % for Neurospora crassa (Amberg et al. 1995; Bird and Bradshaw 1997; Aronson et al. 1994). Comparisons between species are hampered by the fact that different lengths of homologous regions are used for transformation experiments. The efficiency of the gene targeting depends on the length of the homologous region present on the vector. The standard gene targeting procedures use a replacement vector, with the selection marker cloned within the region of homology. To obtain high efficiency of targeted integration in C. lunata, flanking sequences that are longer than 800 bp are suggested. This protocol appears robust, based on trials using the same transformation parameters and protocols with two different genes and phenotypic screens, including virulence assays, for alterations from wild type.
References
Amberg, D. C., Botstein, D., & Beasley, E. M. (1995). Precise gene disruption in Saccharomyces cerevisiae by double fusion polymerase chain reaction. Yeast, 11, 1275–1280.
Aronson, B. D., Lindgren, K. M., Dunlap, J. C., & Loros, J. J. (1994). An efficient method for gene disruption in Neurospora crassa. Molecular and General Genetics, 242, 490–494.
Bird, D., & Bradshaw, R. (1997). Gene targeting is locus dependent in the filamentous fungus Aspergillus nidulans. Molecular and General Genetics, 255, 219–225.
Bundock, P., Den Dulk Ras, A., Beijersbergen, A., & Hooykaas, P. J. J. (1995). Trans-kingdom T-DNA transfer from Agrobacterium tumefaciens to Saccharomyces cerevisiae. EMBO Journal, 14, 3206–3214.
Cooley, R., Shaw, R., Franklin, F., & Caten, C. (1988). Transformation of the phytopathogenic fungus Septoria nodorum to hygromycin B resistance. Current Genetics, 13, 383–389.
Dai, F. C., Gao, W. D., Wu, R. J., & Jin, X. H. (1995). A noticeable corn disease: Curvularia leaf spot. ACTA Phytopathologica Sinica, 25, 330.
Dai, F. C., Wang, X. M., Zhu, Z. D., Gao, W. D., & Huo, N. X. (1998). Curvularia leaf spot of maize: pathogens and varietal resistance. ACTA Phytopathologica Sinica, 2, 123–129.
de Groot, M. J., Bundock, P., Hooykaas, P. J., & Beijersbergen, A. G. (1998). Agrobacterium tumefaciens-mediated transformation of filamentous fungi. Nature Biotechnology, 16, 839–842.
Gao, S. G., Li, Y. Q., Gao, J. X., Suo, Y. J., & Fu, K. H. (2014). Genome sequence and virulence variation-related transcriptome profiles of Curvularia lunata, an important maize pathogenic fungus. BMC Genomics, 15, 627–645.
Henson, J., Blake, N., & Pilgeram, A. (1988). Transformation of Gaeumannomyces graminis to benomyl resistance. Current Genetics, 14, 113–118.
Huang, W. D., Xue, C. S., Zhao, Z. W., & Chen, J. (2010). Agrobacterium tumefaciens-mediated transformation of Curvularia lunata. Hubei Agricultural Sciences, 49, 1537–1539.
Kistler, H., & Benny, U. (1988). Genetic transformation of the fungal plant wilt pathogen Fusarium oxysporum. Current Genetics, 13, 145–150.
Li, Y., Dai, F. C., Jing, R. L., Wang, T. Y., Du, J. Y., & Jia, J. Z. (2002). QTL analysis of resistance to Curvularia lunata in maize. Scientia Agricultura Sinica, 35, 1221–1227.
Liu, T., Liu, L. X., Jiang, X., Hou, J. M., Fu, K. H., Zhou, F. H., & Chen, J. (2010). Agrobacterium-mediated transformation as a useful tool for the molecular genetic study of the phytopathogen Curvularia lunata. European Journal of Plant Pathology, 126, 363–371.
Michielse, C. B., Arentshorst, M., Ram, A. F., & Van Den Hondel, C. A. (2005). Agrobacterium-mediated transformation leads to improved gene replacement efficiency in Aspergillus awamori. Fungal Genetics and Biology, 42, 9–19.
Mullins, E. D., & Kang, S. (2001). Transformation: a tool for studying fungal pathogens of plants. Cellular and Molecular Life Sciences, 58, 2043–2052.
Oide, S., Moeder, W., Krasnoff, S., Gibson, D., Haas, H., Yoshioka, K., & Turgeon, B. G. (2006). NPS6, encoding a nonribosomal peptide synthetase involved in siderophore-mediated iron metabolism, is a conserved virulence determinant of plant pathogenic Ascomycetes. Plant Cell, 18, 2836–2853.
Parsons, K. A., Chumley, F. G., & Valent, B. (1987). Genetic transformation of the fungal pathogen responsible for rice blast disease. Proceedings of the National Academy of Sciences USA, 84, 4161–4165.
Rodriguez, R. J., & Yoder, O. C. (1987). Selectable genes for transformation of the fungal plant pathogen Glomerella cingulata f. sp. phaseoli (Colletotrichum lindemuthianum). Gene, 54, 73–81.
Sambrook, J., & Russell, D. W. (Eds.). (2001). Molecular cloning: A laboratory manual. Cold Spring Harbor: Cold Spring Harbor Laboratory Press.
Sugui, J. A., Chang, Y. C., & Kwon-Chung, K. J. (2005). Agrobacterium tumefaciens-mediated transformation of Aspergillus fumigatus: an efficient tool for insertional mutagenesis and targeted gene disruption. Applied and Environment Microbiology, 71, 1798–1802.
Thompson, J. E., Fahnestock, S., Farrall, L., Liao, D. I., Valent, B., & Jordan, D. B. (2000). The second naphthol reductase of fungal melanin biosynthesis in Magnaporthe grisea: tetrahydroxynaphthalene reductase. Journal of Biological Chemistry, 275, 34867–34872.
Turgeon, B. G., Garber, R. C., & Yoder, O. C. (1985). Transformation of the fungal maize pathogen Cochliobolus heterostrophus using the Aspergillus nidulans amdS gene. Molecular Genetics and Genomics, 201, 450–453.
Xue, C. S., Xiao, S. Q., Zhai, Y. H., Gao, Y., Gao, Z. G., & Chen, J. (2008). Pathogenicity differentiation of Curvularia lunata. Acta Phytopathologica Sinica, 38, 6–12.
Xue, C. S., Wu, D. L., Condon, B. J., Bi, Q., Wang, W. W., & Turgeon, B. G. (2013). Efficient gene knockout in the maize pathogen Setosphaeria turcica using Agrobacterium tumefaciens-mediated transformation. Phytopathology, 103, 641–647.
Zhao, J., Wang, G. Y., Hu, J., Zhang, X. H., & Dai, J. R. (2002). Genetic analysis of maize resistance to Curvularia leaf spot by ADAA model. Acta Agronomica Sinica, 28, 127–130.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (grant number 31271992). We gratefully thank Dr. Gillian Turgeon, Department of Plant Pathology & Plant-Microbe Biology, Cornell University, for providing the plasmid pPZP100.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
(DOC 35 kb)
Supplementary Figure 1
(DOC 193 kb)
Rights and permissions
About this article

Cite this article
Zhang, L., Li, H., Xiao, S. et al. Efficient Agrobacterium tumefaciens-mediated target gene disruption in the maize pathogen Curvularia lunata . Eur J Plant Pathol 145, 155–165 (2016). https://doi.org/10.1007/s10658-015-0825-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10658-015-0825-2