
Abstract
Austrolebias wolterstorffi is a critically endangered annual fish, occurring in temporary ponds in a restricted area of Southern Brazil and Uruguay. Here, we evaluate the levels of genetic diversity and morphometric differentiation presented by A. wolterstorffi, attempting to reconstruct the spatiotemporal scenario by which this species reached their current distribution. Part of the mitochondrial cytochrome b and nuclear rhodopsin genes were characterized and analysed for a set of 122 and 110 specimens, respectively, collected along the entire distribution range of the species. Additionally, shape variations were evaluated for 92 individuals (43 males and 49 females) through geometric morphometric methods. Our analyses demonstrated several cases of significantly high levels of genetic differentiation among individual populations, in an isolation-by-distance pattern of divergence, with at least six different population groups along the Patos-Mirim lagoon. These groups differed by a minimum of 0.9% and a maximum of 2.6% of corrected cyt b nucleotide distances and did not share any mitochondrial haplotype. Such a pattern, added to the slight morphometric differentiation detected for most of the groups, suggests the occurrence of incipient speciation as consequence of allopatric fragmentation. The chronophylogenetic tree performed with the concatenated dataset supported independent oriental and occidental colonization routes, with the population located in the northwest part of the Rio Grande do Sul coastal plain presenting the most ancient divergence. In general, the recovered biogeographic patterns are highly consistent with the records of Quaternary climatic changes and depositional events that have occurred along the area inhabited by the studied species. This allowed us to establish a molecular clock calibration system for Neotropical annual fish. Thus, although the taxonomic status of each of the detected population units needs further study, it is clear that independent conservation strategies must be taken in each of the major areas covered by this study, most of which are located in Brazil.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Freshwater annual fish (Cyprinodontiformes: Aplocheiloidei) possess uncommon developmental, ecological, and physiological adaptations. They have an unusually short life cycle of less than one year, which is entirely correlated to the seasonal ponds they inhabit (Loureiro and de Sá 2015). Their non-overlapping generation times and habitats make annual fish an excellent model for studies of evolution. Also, their patchy distribution provides ideal conditions for studying the underlying evolutionary mechanisms (e.g., genetic drift, gene flow, selection) that usually result in rapid divergence between populations or even allopatric speciation with or without morphological differentiation (de Sá et al. 2015).
Among Aplocheiloidei, Aplocheilidae is one of the most diverse families, with about 350 described species living in South and Central America and in the southern United States (Costa 2008). Austrolebias currently encompasses 45 annual species, distributed along the Paraná-La Plata, Amazonas and Patos-Mirim basins (Costa 2006; Nielsen and Pillet 2015). This genus is particularly diverse in southern Brazil and Uruguay, especially in the Patos-Mirim lagoon system (Costa 2006). Nevertheless, several of these species are endangered because of their frequently restricted and patchy distributions, their characteristic low vagility, and the loss and fragmentation of their habitats (ICMBio 2013; Volcan et al. 2015).
Austrolebias wolterstorffi is one of the largest species of annual fish and has one of the widest distribution ranges within the genus (~50,000 km2), occurring in temporary ponds from the north of the Patos Lagoon to the south of the Mirim Lagoon, along Southern Brazil and in Uruguay (Loureiro et al. 2015). It is considered a critically endangered (CR) species (Reis et al. 2003; ICMBio 2013) because of a combination of its peculiar evolutionary properties and the loss and degradation of its habitats. Nevertheless, despite the incipient incentive of studies aiming to enhance the knowledge of annual fish (ICMBio 2013), even the phylogenetic position of A. wolterstorffi remains controversial. In this sense, although classical studies (Costa 2006, 2010) tend to allocate this species as a member of the A. elongatus group, a recent analysis (García et al. 2014) recovered it as an early offshoot within its genus.
Although the relatively wider geographical distribution of A. wolterstorffi suggests it might recover more easily than other congeners annual fish, the combination of high levels of genetic drift, frequent bottlenecks, inbreeding, and low levels of gene flow (de Sá et al. 2015) might have led to population differentiation or unrecognized cryptic speciation, which could affect its long-term persistence. Therefore, we aimed to assess the levels of diversity and the genetic and morphometric structure within and among populations of A. wolterstorffi, attempting to help in the reconstruction of its evolutionary history and in the establishment of management and conservation strategies.
Materials and methods
Study area
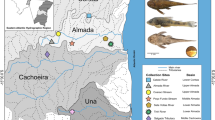
This study includes molecular and/or morphometric data from a total of 134 individuals of A. wolterstorffi collected between 2014 and 2015 in 22 sampling locations distributed in the entire known distribution range of the species (Loureiro et al. 2015; Volcan et al. 2015), which comprises the Patos-Mirim lagoon system, in the southernmost Brazilian state of Rio Grande do Sul and Uruguay (Fig. 1; Supporting Information Table 1S and 2S).

Median-joining networks of the 39 cyt b haplotypes (a) and of the four RHO alleles (b). The size of each circle is proportional to haplotype frequencies, and colours refer to the main population groups recovered in this study (see Results) in relation to their geographic distribution (presented in the map)
With the exception of Eldorado do Sul, all of the sampled sites in the Rio Grande do Sul Brazilian state are within Quaternary sedimentary deposits of the coastal plain. This region was intensely reworked during the paleoclimatic alternations of Quaternary, which caused variations in sea level, thereby opening and closing areas of communication with the Atlantic Ocean, and building a system referred to as the Multiple Barrier (Villwock and Tomazelli 2007). This system is formed by four major depositional events (Barrier I–IV) that extended from 400 to 5 thousand years ago (kyr) and led to the formation of the Patos, Mirim, and Mangueira lagoons. The locality of Eldorado do Sul is inserted in the western portion of the Permian-Triassic sedimentary deposits, locally named the Peripheral Depression, which is part of the Paraná geological basin. Sampled localities in Uruguay are distributed along the Precambrian Shield, Paraná basin (Permian sedimentary and Mesozoic volcanic formation), and Quaternary sedimentary deposits (Rocha department) (Bossi and Navarro 1988).
Samplings
The fishes were collected in temporary ponds with the help of hand nets, euthanized with an overdose of 3000 mg/L of eugenol anesthetic and then fixed in 95% ethanol. For the molecular procedures, a piece of the caudal peduncle was dissected from a set of 83 and 39 Brazilian and Uruguayan specimens, respectively (Table 2S). For geometric morphometry, 43 males and 49 females were photographed on their left side with a digital câmera (Olympus VG-120, supermacro mode).
DNA manipulation
Total DNA was extracted from each individual from approximately 30 mg of muscular tissue, using a phenol/chloroform protocol (Sambrook et al. 1989). Approximately 800 bp of the mitochondrial cytochrome b (cyt b) gene and 820 bp of the nuclear rhodopsin (RHO) were amplified from each sample using the primers L14735 (5′-AAAAACCACCGTTGTTATTCAACTA-3′) and CB3-H (5′-GGCAAATAGGAARTATCATTC-3′) (Palumbi et al. 1991; Wolf et al. 1999) and Rh193 (5′-CNTATGAATAYCCTCAGTACTACC-3′) and Rh1039r (5′-TGCTTGTTCATGCAGATGTAGA-3′) (Chen et al. 2003), respectively. PCR reactions were carried out using 100 ng of DNA in 25 μL reactions, containing 1× buffer, 0.5–1 μM of each primer, 0.25 mM of each dNTP, 2.5–3 mM of MgCl2 and 1–1.5 U of Taq DNA polymerase, with the aid of 5% Dimethyl Sulfoxide in the amplification of RHO. PCR conditions for cyt b consisted of an initial stage of denaturation at 94 °C for 5 min, followed by 35 cycles of denaturation at 94 °C for 45 s, annealing at 55 °C for 45 s and extension at 72 °C for 60 s, and a final extension stage at 72 °C for 10 min; for RHO, cycling consisted of denaturation at 94 °C for 30 s, annealing at 58 °C for 30 s and extension at 72 °C for 30 s. To check whether the amplification was successful, 5 μL of the PCR product were separated by agarose (0.8%) gel electrophoresis and stained with GelRed (Biotium). The amplified fragments were then purified with a solution of 7.5 M ammonium acetate (C2H7NO2) and directly sequenced. Sequencing was performed in a Perkin-Elmer ABI Prism 377 Automated Sequencer (MACROGEN, Seoul, Korea) using the same amplification primers.
Data analysis
First, electropherograms were assembled and edited in the Gap4 software of the Staden package (Staden 1996). The consensus sequences thus obtained had their identity confirmed using BLASTN (NCBI website) and were then aligned using the ClustalW algorithm, as implemented in Mega 6 software (Tamura et al. 2013). Each polymorphic site encountered along this alignment was individually checked and manually corrected, if necessary. For RHO, heterozygous sites were coded according to the nucleotide degeneracy/redundancy table and after unphased in DnaSP 5.10 software (Librado and Rozas 2009). This software was also employed to measure the minimum number of recombination events that best explains the diploid dataset.
The DnaSP 5.10 software was used to calculate the levels of genetic diversity within populations or population groups individually for each gene, as estimated from the average number of different haplotypes (H) or alleles (A) (Nei 1987), the average number of nucleotide differences between haplotypes or alleles (k) (Tajima 1983), the haplotype diversity (Hd) or expected heterozigosity (He) (Nei 1987), and the nucleotide (π) diversities (Nei 1987). The neutrality tests Tajima’s (1989), and Fu’s (1997) were performed in Arlequin 3.5 (Excoffier and Lischer 2010), with significance measured through 10,000 random permutations for both genes.
The relationships between haplotypes or alleles were inferred for both genes individually from networks generated by median-joining in the Network v.4.510 software (Bandelt et al. 1999). The levels of genetic differentiation among populations were measured for cyt b and RHO by the fixation index (FST) in Arlequin 3.5 using pairwise differences with 10,000 random permutations. A Mantel test was finally performed using these measures to check the correlation between the genetic (FST) and geographic distances.
For the concatenated dataset, different hypotheses of population groupings were assessed through spatial analysis of molecular variance (SAMOVA) (Dupanloup et al. 2002), in order to define the number and structure of the groups that are geographically homogeneous and maximally differentiated from each other. In this case, different hypotheses were evaluated through hierarchical analysis of molecular variance (AMOVA), as performed in Arlequin, with 10,000 permutations. The population structure and the most likely number of clusters of individuals were also assessed using the Bayesian Population Structure analysis performed in BAPS 6 (Corander et al. 2008).
The supermatrix phylogenetic tree was reconstructed using Bayesian analysis and maximum likelihood estimates in MrBayes 3.2.6 (Ronquist et al. 2012) and RaxMLHPC (Stamatakis 2014), respectively, under the partitioning scheme and substitution models suggested by PartitionFinder 1.1.1 (Lanfear et al. 2012). The Markov Chain Monte Carlo (MCMC) of the BA was run for 10,000,000 generations, sampling trees every 1000 generations, and burning 25% of the initial results. The ML search was performed under the new rapid hill-climbing algorithm, with 1000 bootstrap replicates. These analyses were performed without the use of outgroups, and the phylogenetic trees were further visualized and edited in FigTree 1.4.3 (Rambaut and Drummond 2009) as radial phylograms.
Additionally, a chronophylogenetic tree was reconstructed under a Bayesian approach (BA) in Beast 1.5.4 (Drummond and Rambaut 2007), using cyt b and RHO sequences with unlinked substitution and clock models. In this case, an uncorrelated lognormal relaxed molecular clock analysis was performed with a mean rate of 2% per million years (SD = 0.5%) for cyt b, as adjusted for ectotherms mitochondrial genomes (Brown et al. 1979). This prior was further complemented by the dating of 325 kyr (SD = 12.5 kyr) to the major split between oriental and occidental lineages, as recovered in the first Bayesian search conducted with MrBayes (see results). The analysis encompassed two independent runs of 20,000,000 generations, with trees and parameters sampled every 2000 iterations, and a burn-in of 25%. Results of each run were visualized in Tracer 1.7 (Rambaut et al. 2018) to ensure that stationarity was achieved and that convergence was reached. Posterior probabilities and the maximum credibility tree were inferred using TreeAnnotator 1.5.4 (Drummond and Rambaut 2007) and further visualized and edited in FigTree 1.4.3.
Finally, to estimate the effective number of females of each population group, a Bayesian analysis based on coalescence was implemented in LAMARC 2.1.10 (Kuhner 2006) under the GTR nucleotide substitution model, with effective population sizes set to 1 and 4, and relative mutation rates set 1 and 0.5 for the mitochondrial and nuclear partitions, respectively. Runs consisted of four simultaneous searches, each with 100 initial and four final chains, with a minimum of 1000 and 10,000 recorded parameter sets, respectively, and sampling every 20 generations after a burn-in of 1000 genealogies. Only population groups with more than 10 sampled individuals were used in this analysis.
Morphometry analysis
A set of 14 landmarks were defined and digitalized using TpsDig2 software ver. 2.26 (Rohlf 2016) (Supporting Information Fig. 1S). The coordinates were aligned using a Generalized Procrustes Analysis (GPA), where non-shape variability was removed after minimizing the differences in translation, scaling and rotation. Due to sexual dimorphism, the analyses were performed independently for males and females.
Shape variations among individuals of the different groups were first evaluated through an exploratory Principal Component Analysis (PCA), followed by MANOVA and pairwise MANOVA. Afterwards, a Canonical Variate Analysis (CVA) was employed to find the linear combination of predictors that best discriminates among groups. Cross validation test was used to measure the accuracy of correct classifications. These analyses were permormed in R (R Core Team 2017), using Geomorph (Adams and Otarola-Castillo 2013) and Vegan packages (Oksanen et al. 2017).
Results
In this study, sequences spanning 798 bp of the mitochondrial cyt b gene and 821 bp of the nuclear RHO gene were characterized, respectively, for 122 and 110 individuals of A. wolterstorffi collected at 22 different localities (Fig. 1) (Supporting Information Tables 1S and 2S). The intraspecific matrix of cyt b encompassed 39 different haplotypes presenting 65 variable sites, of which 48 were parsimoniously informative. For RHO, only four alleles were detected, and variability was restricted to three sites.
Molecular analyses
Cyt b dataset
Concerning cyt b diversity estimates, the number of haplotypes per sampling locality ranged from 1 (Sal, 33-INIA, 33-Pas, SJN-4, Buj, Pel-S, PN-RG and TAIM) to 6 (Tav and IL-RG), with minimum Hd and π of 0 (Sal, 33-INIA, 33-Pas, SJN-4, Buj, Pel-S, PN-RG and TAIM), and maximum Hd and π values of 0.889 (IL-RG) and 0.006 (SJN-3), respectively (Table 3S). None locality presented significant deviations from neutrality (Table 3S). A total of 111 cases of significant genetic structure were detected in the pairwise comparisons between populations, and these Fst values varied from 0.20 (in the comparison between IL-RG and Pel-P) to 1.00 (in the comparison between SJN-4 and Sal, 33-INIA, Buj and between Buj and 33-INIA) (Table 4S). The Mantel test indicated a significant correlation between genetic and geographic distances (r = 0.52; p < 0.000).
The haplotype network reconstructed with cyt b sequences revealed the presence of six independent haplogroups (Fig. 1a): (1) Eldorado, with haplotypes sampled at the municipality of Eldorado do Sul, RS, Brazil; (2) Pelotas, clustering haplotypes sampled in seven localities of Pelotas and Rio Grande, RS, Brazil; (3) Uruguay, grouping together the haplotypes sampled in the nine localities of Uruguay; (4) Tavares, with haplotypes sampled at Tavares, RS, Brazil; (5) São José do Norte (SJN), with haplotypes sampled at Cachoeirinha, RS, Brazil and in three localities of SJN, RS, Brazil; and (6) São José do Norte 2, with haplotypes sampled at one locality of SJN, where haplogroup 5 was also collected. No haplotype was shared between these haplogroups, and a star-like pattern of ramification could be evidenced for Uruguay (Fig. 1a).
RHO dataset
The pattern detected for RHO is less informative, since this marker revealed to be highly conserved in A. wolterstorffi. In this case, more than a single allele was detected only for SJN-3, SJN-4, Buj, Tav, Pel-P, PN-RG, IL-RG, IL2-RG and MP-RG, and the higher values of He and π were 0.644 (SJN-4) and 0.0009 (SJN-4), respectively (Table 5S). None locality presented significant deviations from neutrality (Table 5S), and significant Fst values were restricted to the comparisons involving some Uruguayan and Brazilian localities (Table 6S), which resulted in a significant correlation between genetic and geographic distances in the Mantel Test (r = 0.20; p < 0.018). Even so, in the Network, none clear subdivision could be detected, and a starlike pattern was recovered for the species considered as a whole (Fig. 1b).
Concatenated dataset
Despite the differences in variability levels, as cyt b and RHO presented similar divergence patterns in the target species, they were jointly considered in the remaining analyses. The population structure suggested by SAMOVA for the supermatrix dataset lead to a plateau in FCT values when k was set to six, which subdivided A. wolterstorffi into the same population groups previously recovered in the cyt b Network: Pelotas, Uruguay, Tavares, São José do Norte, Eldorado and São José do Norte 2. In fact, the AMOVA performed with this structure revealed that the six groups of populations were able to explain 75.95% of the variation encountered in the concatenated dataset (Table 1). Bayesian clustering approaches implemented through the spatial BAPS model also supported the same structure.
Accordingly, the topology of the radial phylogram also recovered a similar structure, and all the population groups were recovered as reciprocally monophyletic in both BA and ML trees, with support values higher than 0.9 and 63, respectively (Fig. 2). These lineages grouped into two main clusters encompassing populations inhabiting drainages at the western (population groups of Pelotas, Uruguay, and Eldorado) and eastern (Tavares, São José do Norte and São José do Norte 2) margins of the Patos Lagoon. After setting this divergence to a time prior of 325 kyr (SD = 12.5 kyr) in a chronophylogenetic analysis, diversification of these clades was dated to approximately 222 and 186 kyr, respectively (Fig. 3). In this case, the Eldorado and Tavares population groups seem to have encompassed the most ancient divergences, respectively, although support for this position was tenuous, especially for the last. Diversifications of each of the six population groups were set between 104 and 7 kyr (Fig. 3).

Radial phylogram of the concatenated sequences, with internal node labels representing support values, given by posterior probabilities and bootstrap values in BA and ML evaluations, respectively. Branch lengths are proportional to the number of substitutions per site, and colours refer to population groups (see Fig. 1)

Bayesian chronophylogenetic tree based on cyt b and RHO sequences sampled for A. wolterstorffi. Values above internal branches represent support values, given by posterior probabilities values in BA analysis. Values below nodes indicate divergence time estimates, with the highest posterior density (HPD) interval containing 95% of the sampled values presented within brackets for some of the detached clades. Groups of populations were represented by their respective names and colours (see Fig. 1). This tree was rooted with cyt b and RHO sequences of A. nigrofasciatus, A. minuano, and A. adloffi
Morphometric analyses
Applying the subdivision in six population groups, significant differences were found in the shape patterns for both, males and females (p < 0.01). Among females, most pairwise comparisons involving the population groups of Eldorado and Uruguay evidenced significant differentiation, although for males such a result was restricted to two comparisons involving Uruguayan specimens (Table 7S). In this sense, the PCA for males and females also suggested the existence of some differences between groups, especially in regard to the insertion of the caudal fin and to the position of the dorsal fin, respectively (Fig. 4a, b). For males, the PC1 explained 29.5% of shape variation, whereas PC2 explained 27.1%. For females, PC1 and PC2 explained 22 and 19.2% of shape variation, respectively. Nevertheless, when a CVA was used with the four population groups that had at least six individuals sampled, morphological differentiation between the different population groups became more evident (Fig. 4c, d). In this case, the overall classification accuracy reached 59.5 and 60.9% for males and females, respectively.

Plots of Principal Component Analysis (PCA) (a and b) and Canonical Variate Analysis (c and d) showing the distribution of variation in A. wolterstorffi morphometric patterns: (a and c) males, (b and d) females. For the PCA plots, extreme PC values are represented by fishes’ warped drawings on grid deformations. Colours refer to the different population groups (see Fig. 1)
Diversity and differentiation evaluations in face of the new subdivision
As the subdivision among six population groups was consistently defined by the network of cyt b, and by SAMOVA, spatial BAPS, and the phylogenetic analyses of the concatenated dataset, this structure was further employed in new neutrality, diversity, and genetic differentiation tests. In general, at least two haplotypes were sampled for cyt b in each of the six population groups, which always presented haplotype diversity (Hd) values higher than 0.60 for this marker (Table 2). For rhodopsin, the population groups of Eldorado, SJN2 and Uruguay were fixed for a single allele, and the highest value of He was 0.54 for Tavares (Table 2). Conversely, the nucleotide diversity values (π) were generally moderate to low, ranging from 0.010 to 0.058 and 0.0004 to 0.0007 for cyt b and RHO, respectively. Regarding the neutrality tests, significant negative results were only obtained by cyt b for the populations of Pelotas and Uruguay (Table 2).
When testing genetic differentiation levels for cyt b, most comparisons between population groups presented significant genetic differences (Table 3, below the diagonal). The sole comparison that did not present a significant FST value was between Eldorado and SJN 2, which is probably an outcome of reduced sampling size. In all the significant comparisons, FST values between groups were higher than 0.7, indicating the presence of high levels of genetic differentiation (Table 3, below the diagonal). For RHO, significant genetic differences were only detected between the Uruguayan and Brazilian groups (Table 4, below the diagonal). The net Tamura 3-parameters distances also supported such a pattern, and the six lineages differed by a minimum of 0.9% (as seen between Tavares and SJN) and a maximum of 2.6% (as seen between groups Pelotas and SJN 2) for cyt b (Table 3, above the diagonal). Nevertheless, net distances between groups were zero when only RHO sequences were analysed (Table 4, above the diagonal).
Population parameters inferred for the concatenated dataset in LAMARC revealed theta values ~12.4-fold higher for Pelotas than for SJN, whereas the populations of Uruguay and Tavares presented similar intermediate theta values (Table 5). Even so, Bayesian analysis recovered effective numbers larger than 11,000 for all populations (Table 5).
Discussion
It is generally assumed that annual fish are subject to the concomitant action of a set of environmental, demographic, and metabolic conditions commonly associated with high evolutionary rates (Whitlock 2000; Loureiro 2004), which can lead to a great variability among populations. In agreement with this, our analyses have shown that, in addition to several cases of significantly high differentiation levels between individual populations, in an isolation-by-distance divergence pattern, A. wolterstorffi is subdivided into at least six different groups of populations. Such a structure was evidenced by the Network of cyt b, and by SAMOVA, BAPS, and the phylogenetic analyses of the concatenated dataset, and further supported by shape variation. So, despite the differences in evolutionary rates presented by the mitochondrial and nuclear markers, which can be attributed to distinct mutation rates, modes of inheritance and population sizes (Avise 2004; Templeton 2006), this subdivision was able to explain more than 75% of the variation detected for this species in both of the sampled loci. Moreover, cross validation tests performed with discriminant CVA resulted in overall classifications accuracies ranging 60%, evidencing an incipient morphometric differentiation between at least some of the evaluated groups. This suggests that vicariance has played an important role in the diversification of A. wolterstorffi, as also reported for several other Aplocheilidae species (Jowers et al. 2008; García et al. 2009, 2012, 2015; Bartáková et al. 2013; Ponce de León et al. 2014; Loureiro et al. 2015).
Although allopatric fragmentation seems to be an ongoing process within A. wolterstorffi, some level of gene flow seems to occur, especially at short distances, encompassing mainly populations located within each of the population groups. In fact, all FST values recovered in pairwise comparisons involving the six groups were significant and high, and they differed by a minimum of 0.9% corrected cyt b distances (with a maximum of 2.6%). These results, added by the shape divergence patterns and the evidence of recent diversification (see below), lead us to conclude that at least some of these groups may constitute incipient species. In fact, distances as small as 1.4% were previously reported for cyt b between different species of Austrolebias (García et al. 2000), with even lower distance ranges (0 to 1.8%) being recently reported within the A. bellottii species complex (García et al. 2015). Considering that DNA-based approaches previously unravelled several unrecognized lineages of Neotropical annual fishes (Costa and Amorim 2011; Costa 2013; Costa et al. 2014, 2016, 2017), we argue here that A. wolterstorffi may in fact constitute a species complex. Nevertheless, it is likely that this complex presents a continuum of differentiation, in which at least some of the lineages might have yet reached a speciation endpoint, given by establishment of complete reproductive isolation.
Independent of the taxonomic status assigned to each of the six evolutionary units detected here, it is important that independent conservation strategies are applied to each these major sampling areas, four of which are located in Brazil. Although the levels of genetic diversity encountered for A. wolterstorffi as a whole (or for each of the individual population groups) were relatively high, this is likely explained by small-scale events of gene flow within population groups or even by the putative higher mutational rates previously assigned to mitochondrial genes in Austrolebias (García et al. 2015). In fact, similar levels of intrapopulation diversity were previously reported for several other species of annual fishes inhabiting different regions (García et al. 2000, 2015; Bartáková et al. 2013).
Nevertheless, the fact that each of these areas embraces an independent genetic stock calls attention to the need for rapid interventions, principally in the face of the rapid fragmentation and degradation of wetlands within the Brazilian territory (Volcan et al. 2015). This situation might decrease effective population sizes and gene flow, enhancing the action of random genetic drift and inbreeding and increasing the risk of mutational meltdown and inbreeding depression, respectively (Frankham et al. 2013). Likewise, the periodic flooding of rivers and lagoons within the distribution range of A. wolterstorffi might also threaten the persistence of each of these independent evolutionary units. In this sense, signs of population admixture were encountered for the population of São José do Norte, which presents two different haplogroups differing by a minimum of 11 mutational steps and assigned to different population groups. This result led us to hypothesize that the SJN2 individuals might represent immigrants from a distinct unknown population. Secondary colonisations might also be invoked to explain the presence of individuals from the SJN haplogroup in Cachoeirinha, a municipality located northwest of the Patos Lagoon. So, although current estimates of the effective population sizes do not suggest an incipient risk of extinction for any of the sampled population groups (see Frankham et al. 2004), and none of the populations presented significant signs of genetic bottlenecks, this could change rapidly and it is important that interventions are performed before the evolutionary potential of the populations is lost.
Finally, regarding the spatio-temporal evolutionary scenario, it is possible to infer that the six groups of populations encompass two distinct lineages, whose distribution coincides with the eastern and western margins of the Patos Lagoon, and suggest a north-south colonization route. Using the onset of the formation of the Patos Lagoon, related to the occurrence of the second Pleistocene Lagoon Barrier Depositional System and dated to approximately 325 kyr (Villwock and Tomazelli 2007) as prior to the divergence time between Oriental and Occidental lineages, together with the mean mitochondrial ectothermic rate of 2% per million years (Brown et al. 1979; Avise 1994), it was possible to infer that the first divergence within these two clades occurred around 222 (western) and 186 (eastern) kyr, compatible with range expansions enabled by the second Pleistocene Lagoon Barrier Depositional System related to the paleogeographical evolution of the South American Coastal Plain (Montaña and Bossi 1995; Tomazelli and Villwock 2005; Villwock and Tomazelli 2007). Interestingly, for the western clade, the lineage with the further north distribution encompasses the early offshoot, which is also in agreement with the paleogeographic history of the region (Villwock and Tomazelli 2007). Furthermore, the diversification of each of the six population groups seem to have occurred around 104 kyr (Pelotas population group) and 7 kyr (SJN2), and it is tempting to speculate that at least some of these events were related to the third or fourth Lagoon Barrier Depositional Systems, dated to approximate 120 kyr and 18 kyr to near the present, respectively (Villwock and Tomazelli 2007). The Uruguayan population group, in particular, inhabits a geologically older formation, which seems to have been recently colonized (about 63 kyr). In agreement with this, this lineage presents significant signs of population expansion, as given by the Fu’s FS test and by the star-like network pattern recovered with the cyt b dataset, and by the lower diversity values revealed by RHO. More subtle signals of demographic or spatial expansions were also detected for the species complex taken as a whole (as given by the star-like pattern recovered with RHO), and for the Pelotas population group (as given the Tajima’s D test performed with cyt b).
The well-registered paleogeoclimatic history of the South American Coastal Plain (Montaña and Bossi 1995; Tomazelli and Villwock 2000, 2005; Villwock and Tomazelli 2007) and the clear vicariance between Oriental and Occidental lineages of A. wolterstorffi also allowed us to use this divergence to calibrate our relaxed molecular clock analysis and to establish a new calibration system to the group. Until now, the most widely used method to date divergence events in the group employed the mean substitution rate for mitochondrial genes that was established by Brown et al. (1979) and adjusted by Avise (1994) for ectothermal species taken as a whole (García et al. 2012, 2014, 2015). As molecular clock substitution rates need to be calibrated for each gene in each studied lineage, the use of this molecular clock calibration system will allow the estimation of substitution rates for several other genes within the genus, enabling refinement of several questions related to the spatio-temporal evolutionary history of this and several other species of annual fish.
So, although this study clearly helped to enhance knowledge about the evolution of A. wolterstorffi, evidencing some hidden taxonomic and conservation problems, it should be followed by additional studies and initiatives aimed at the conservation of annual fish, which encompass one of the most threatened vertebrate groups in Brazil (ICMBio 2013; Volcan et al. 2015). It is important that the knowledge generated here be used to help in the promotion and implementation of environmental education programs and to encourage the creation of protected areas. Only the adoption of a whole set of actions can help in the conservation of this fascinating and biologically unique group of species.
References
Adams DC, Otarola-Castillo E (2013) Geomorph: an R package for the collection and analysis of geometric morphometric shape data. Methods Ecol Evol 4:393–399
Avise JC (1994) Molecular markers, natural history and evolution. Chapman and Hall, New York, 511 p
Avise JC (2004) Molecular markers, natural history and evolution. 2nd edn. Sinauer Associates, Sunderland
Bandelt HJ, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48
Bartáková V, Reichard M, Janko K et al (2013) Strong population genetic structuring in an annual fish, Nothobranchius furzeri, suggests multiple savannah refugia in southern Mozambique. BMC Evol Biol 13:196
Bossi J, Navarro R (1988) Geología del Uruguay. Universidad de la República, Montevideo, 966 p
Brown WM, Jr George M, Wilson AC (1979) Rapid evolution of animal mitochondrial DNA. Proc Natl Acad Sci U S A 76:1967–1971
Chen WJ, Bonillo C, Lecointre G (2003) Repeatability of clades as a criterion of reliability: a case study for molecular phylogeny of Acanthomorpha (Teleostei) with larger number of taxa. Mol Phylogenet Evol 26:262–288
Corander J, Sirén J, Arjas E (2008) Bayesian spatial modeling of genetic population structure. Comput Stat 23:111–129
Costa WJEM (2006) The South American annual killifish genus Austrolebias (Teleostei: Cyprinodontiformes: Rivulidae): phylogenetic relationships, descriptive, morphology and taxonomic revision. Zootaxa 1213:1–162
Costa WJEM (2008) Catalog of Aplocheiloid killifishes of the world. UFRJ, Rio de Janeiro
Costa WJEM (2010) Historical biogeography of Cynolebiasine annual killifishes inferred from dispersal-vicariance analysis. J Biogeogr 37:1995–2004
Costa WJEM (2013) Historical biogeography of aplocheiloid killifishes (Teleostei: Cyprinodontiformes). Vertebr Zool 63:139–154
Costa WJEM, Amorim PF (2011) A new annual killifish species of the Hypsolebias flavicaudatus complex from the São Francisco River basin, Brazilian Caatinga (Cyprinodontiformes: Rivulidae). Vertebr Zool 61:99–104
Costa WJEM, Amorim PF, Aranha GM (2014) Species limits and DNA barcodes in Nematolebias, a genus of seasonal killifishes threatened with extinction from the Atlantic Forest of South-Eastern Brazil, with description of a new species (Teleostei: Rivulidae). Ichthyol Explor Fres 24:225–236
Costa WJEM, Amorim PF, Mattos JLO (2016) A new species of inseminating seasonal killifish of the Cynopoecilus melanotaenia complex from southern Brazil (Cyprinodontiformes: Rivulidae). Biodivers Data J 4:e6888–17
Costa WJEM, Cheffe MM, Amorim PF (2017) Two new seasonal killifishes of the Austrolebias adloffi group from the Lagoa dos Patos basin, southern Brazil (Cyprinodontiformes: Aplocheilidae). Vertebr Zool 67:139–149
de Sá RO, Berois N, García G (2015) Overview, future challenges, and evolution of Annualism. In: Berois N, García G, de Sá RO (eds) Annual fishes: life history strategy, diversity, and evolution, 1st edn. Press Taylor & Francis group, Boca Raton, pp 309–318
Dupanloup I, Schneider S, Excoffier L (2002) A simulated annealing approach to define the genetic structure of populations. Mol Ecol 11:2571–2581
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and windows. Mol Ecol Resour 10:564–567
Frankham R, Ballou JD, Briscoe DA (2004) A primer of conservation genetics. Cambridge University Press, Cambridge
Frankham R, Brook BW, Bradshaw CJA, Traill LW, Spielman D (2013) 50/500 rule and minimum viable populations: response to Jamieson and Allendorf. Trends Ecol Evol 28:187–188
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
García G, Wlasiuk G, Lessa E (2000) High levels of mitochondrial cytochrome b divergence in annual killifishes of the genus Cynolebias (Cyprinodontiformes, Rivulidae). Zool J Linn Soc-Lond 129:93–110
García G, Loureiro M, Berois N et al (2009) Pattern of differentiation in the annual killifish genus Austrolebias (Cyprinodontiformes; Rivulidae) from a biosphere reserve site in South America: a multidisciplinary approach. Biol J Linn Soc 98:620–635
García G, Gutiérrez V, Vergara J, Calviño P, Duarte A, Loureiro M (2012) Patterns of population differentiation in annual killifishes from the Paraná-Uruguay-La Plata basin: the role of vicariance and dispersal. J Biogeogr 39:1707–1719
García G, Gutiérrez V, Ríos N, Turner B, Santiñaque F, López-Carro B, Folle G (2014) Burst speciation processes and genomic expansion in the neotropical annual killifish genus Austrolebias (Cyprinodontiformes, Rivulidae). Genetica 142:87–98
García G, Gutiérrez V, Ríos N, de Sá RO (2015) Comparative Phylogeographic patterns in Austrolebias from different south American basins. In: Berois N, García G, de Sá RO (eds) Annual fishes: life history strategy, diversity, and evolution, 1st edn. Press Taylor & Francis group, Boca Raton, pp 259–279
ICMBio, Instituto Chico Mendes de Conservação da Biodiversidade (2013) Sumário executivo do plano de ação nacional para a conservação dos peixes Rivulídeos ameaçados de extinção. Brasília. http://www.icmbio.gov.br/portal/biodiversidade/fauna-brasileira/plano-de-acao/2833-plano-de-acao-nacional-para-a-conservacao-dos-rivulideos.html. Accessed 16 November 2016
Jowers MJ, Cohen BL, Downie JR (2008) The cyprinodont fish Rivulus (Aplocheiloidei: Rivulidae) in Trinidad and Tobago: molecular evidence for marine dispersal, genetic isolation and local differentiation. J Zool Syst Evol Res 46:48–55
Kuhner MK (2006) LAMARC 2.0: maximum likelihood and Bayesian estimation of population parameters. Bioinformatics 22:768–770
Lanfear R, Calcott B, Ho SY, Guindon S (2012) PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol 29:1695–1701
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Loureiro M (2004) Sistemática y biogeografía de los peces anuales de la subtribu Cynolebiatina (Cyprinodontiformes: Rivulidae: Cynolebiatinae). PhD thesis, Montevideo
Loureiro M, Borthagaray A, Hernández D, Duarte A, Pinelli V, Arim M (2015) Austrolebias in space: scaling from ponds to biogeographical regions. In: Berois N, García G, de Sá RO (eds) Annual fishes: life history strategy, diversity, and evolution, 1st edn. Press Taylor & Francis group, Boca Raton, pp 111–132
Loureiro M, de Sá RO (2015) Diversity of Aplocheiloidei. In: Berois N, García G, de Sá RO (ed) Annual fishes: life history strategy, diversity, and evolution, 1st edn. Press Taylor & Francis group, Boca Raton, pp 3–31
Montaña JR, Bossi J (1995) Geomorfología de los humedales de la cuenca de la Laguna Merín en el departamento de Rocha. UDELAR. Revista Facultad de Agronomía. Montevideo, Uruguay 2:1–32
Nei M (1987) Molecular evolutionary genetics. Columbia Univ. Press, New York
Nielsen DTB, Pillet D (2015) Austrolebias accorsii, a new annual fish (Cyprinodontiformes: Rivulidae: Cynolebiatinae) from the upper río Grande basin, Amazon basin. Bolivia Aqua 21:172–179
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Henry M, Stevens H, Szoecs E, Wagner H (2017) Vegan: community ecology package. R Package Version 2:4–3
Palumbi S, Martin A, Romano S, McMillan WO, Stice L, Grabowski G (1991) The simple fool’s guide to PCR. University of Hawaii, Honolulu
Ponce de León JL, León G, Rodríguez R et al (2014) Phylogeography of Cuban Rivulus: evidence for allopatric speciation and secondary dispersal across a marine barrier. Mol Phylogenet Evol 79:404–414
R Core Team (2017) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Rambaut A, Drummond AJ (2009) FigTree, v1.4.3. http://tree.bio.ed.ac.uk/software/figtree/. Accessed 18 July 2017
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA (2018) Tracer v1.7. http://tree.bio.ed.ac.uk/software/tracer/. Accessed 6 July 2016
Reis RE, Lucena ZMS, Lucena CAS, Malabarba LR 2003. Peixes. In: Fontana CS, Bencke GA, Reis RE (ed) Livro vermelho da fauna ameaçada de extinção no Rio Grande do Sul, Edipucrs, Porto Alegre, pp 632
Rohlf FJ TpsDig Version 2.26 (2016) Department of ecology and evolution, stony brook 565 University, New York
Ronquist F, Teslenko M, Van Der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:1–4
Sambrook J, Fritschi EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, New York, p 1120
Staden R (1996) The Staden sequence analysis package. Mol Biotechnol 5:233–241
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313
Tajima F (1983) Evolutionary relationship of DNA sequences in finite populations. Genetics 105:437–460
Tajima F (1989) Statistical methods to test for nucleotide mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Templeton AR (2006) Population genetics and microevolutionary theory. Hoboken, New Jersey: John Wiley & Sons. In: Inc
Tomazelli LJ, Villwock JA (2000) O Cenozóico no Rio Grande do Sul: geologia da Planície Costeira. In: Holz M, De Ros LF (ed) Geologia do Rio Grande do Sul, CIGO/UFRGS, Porto Alegre, pp 375–406
Tomazelli LJ, Villwock JA (2005) Mapeamento Geológico de Planícies Costeiras: o Exemplo da Costa do Rio Grande do Sul. Gravel 3:109–115
Villwock JA, Tomazelli LJ (2007) Planície Costeira do Rio Grande do Sul: gênese e paisagem atual. In: Becker FG, Ramos RA, Moura LA (ed) Biodiversidade. Regiões da Lagoa do Casamento e dos Butiazais de Tapes, planície costeira do Rio Grande do Sul, Ministério do Meio Ambiente/SBF, pp 388
Volcan MV, Lanés LEK, Gonçalves AC, Guadagnin DL (2015) Annual fishes (Rivulidae) from Southern Brazil: a broad-scale assessment of their diversity and conservation. In: Berois N, García G, de Sá RO (ed) Annual fishes: life history strategy, diversity, and evolution, 1st edn. Press Taylor & Francis group, Boca Raton, pp 185–203
Whitlock MC (2000) Fixation of new alleles and the extinction of small populations: drift load, beneficial alleles, and sexual selection. Evolution 54:1855–1861
Wolf C, Rentsch J, Hübner P (1999) PCR-RFLP analysis of mitochondrial DNA: a reliable method for species identification. J Agr Food Chem 47:1350–1355
Acknowledgements
This study was funded by Fundação Grupo Boticário de Proteção à Natureza as part of the “Peixes Anuais do Pampa” and the “Padrões micro e macroevolutivos em peixes anuais de Cynopoecilus e Austrolebias (Cyprinodontiformes: Rivulidae) ao longo do Sistema de Drenagens Patos-Mirim: um enfoque comparativo com aplicações para a conservação – 1090_20171” projects. We thank CAPES and CNPq for providing fellowships to DKG and CB, respectively. All the collections were authorized by the Brazilian Ministério do Meio Ambiente (MMA), in the form of the Sistema de Autorização e Informação em Biodiversidade (SISBIO) (process number 55651-1). This study was also approved by the Ethics Comittee on Animal Use of the Universidade Federal do Rio Grande (process number 23116.008163/2015-23).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(DOCX 1141 kb)
Rights and permissions
About this article

Cite this article
Garcez, D.K., Barbosa, C., Loureiro, M. et al. Phylogeography of the critically endangered neotropical annual fish, Austrolebias wolterstorffi (Cyprinodontiformes: Aplocheilidae): genetic and morphometric evidence of a new species complex. Environ Biol Fish 101, 1503–1515 (2018). https://doi.org/10.1007/s10641-018-0795-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10641-018-0795-2