
Abstract
Background
Glycolysis and tumor immunity were interrelated. In present study, we aimed to construct a prognostic model based on glycolysis-immune-related genes (GIGs) of osteosarcoma (OS) patients.
Methods
The mRNA expression data of OS patients were downloaded from GEO and TARGET databases. The hub genes were screened from 305 differentially expressed genes by univariate cox regression analysis and used to further establish a prognostic Risk Score. The independence of the Risk Score prognostic prediction model based on five genes was tested by multivariate Cox regression analysis. Finally, CIBERSORT and LM22 feature matrix were used to estimate the differences in immune infiltration of OS patients.
Results
A total of 141 OS patients’ mRNA expression data and 296 glycolysis-associated genes were analyzed. Based on these 296 genes, all patients could be divided into two clusters: high glycolysis state and low glycolysis state. In the group with high glycolysis status, patients had low immune scores, indicating that glycolysis status was negatively correlated with immune function. The OS patients with high glycolysis and low immunity had the worst prognosis. Next, the Risk Score was constructed by 5 GIGs, including RAI14, MAF, CLEC5A, TIAL1 and CENPJ. Moreover, the Risk Score was shown to be an independent prognostic model, and high Risk Score patients had a greater risk of death.
Conclusions
The Risk Score based on GIG could predict the prognosis of OS patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Osteosarcoma (OS) is an invasive malignant tumor in the skeletal system, with rapid progression and poor prognosis, and has become a major fatal disease in children and adolescents [1]. OS often occurred in long epiphysis of extremities, such as the proximal tibia and distal femur. It grows rapidly, causes large bone defect and motor function limitation, and leads to pulmonary or skeletal metastasis, resulting in a significant reduction in the 5-year overall survival rate [2]. Despite the great advances have been made in surgery and chemotherapy in recent years [3], patients with metastatic and recurrent OS was still very depressed, the survival rate of 5-year was less than 25% due to the lack of prognosis assessment and therapeutic strategies efficacy [4]. Therefore, an urgent need exists to investigate the pathogenic mechanisms of OS and establish a reliable predictive model for the prognosis of OS patients.
Dysregulation of cellular energy or metabolic reprogramming was a striking feature of solid tumor. Normal cells proliferated at a limited rate, while tumor cells proliferated rapidly and without restriction, so tumor cells needed to produce more energy to meet their proliferation needs. Whether oxygen content was sufficient or not, tumor cells tended to go through anaerobic glycolysis rather than aerobic oxidation capacity, which was known as the Warburg effect [5]. Based on the clinical observations, the sugar uptake of tumor tissues was significantly increased relative to adjacent normal tissues [6], so glycolysis was considered as a potential prognostic indicator of tumors [7]. Recently, glycolysis has been proved to be closely related to the occurrence and development of OS [8]. Han et al. showed that the effect of TUG1 on the viability of OS cells was affected by glycolysis [9]. Additional evidence indicated that restriction of glycolysis could inhibit the metastasis of OS in vivo and in vitro [10]. Moreover, tumor glycolysis and the tumor microenvironment (TME) or immune evasion were interdependent [11, 12]. Obviously, we could conclude that if tumor cells consumed a large amount of glucose for glycolysis, the immune activity in the TME would be limited by the glucose competition between immune cells and tumor cells [13]. Currently, there are few studies on the relationship between glycolysis and immune response in OS. Therefore, a more complete understanding of the interaction between glycolysis and immune response in OS was necessary, which would open up new avenues for OS treatment and prognostic strategies.
Accumulating literatures have reported OS prognosis prediction methods. For example, Yang et al. established glycolysis-related risk signature based on the three genes P4HA1, ABCB6, and STC, which could predict the prognosis of OS patients and provided us with new ideas [14]. However, the prognostic model of OS based on glycolysis-immune comprehensive evaluation has not been reported. In present study, we focused on constructing a prognostic model based on GIG, which could be used to predict the prognosis of OS patients.
Materials and methods
Study objects
We downloaded mRNA data of 53 patients (GSE21257) and 88 patients from Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) and Therapeutically Applicable Research to Generate Effective Treatments (TARGET, https://ocg.cancer.gov/programs/target) databases respectively. The clinical information of 53 patients was shown in Table 1, and 84 of the 88 patients had complete survival information.
Differentially expressed genes (DEGs) analysis
The limma package of R software (version4.1.0) [15] was used to screen DEGs from patient samples with the |log2FC| > 0.5 and adj. p.val < 0.05 (the corrected p value wasrepresented by adj. p.val).
Functional enrichment analysis
GO (including Molecular Function, Cellular Component, and Biological Process) and KEGG enrichment analyses were conducted on the DEGs by clusterProfiler package in R software (p < 0.05 was considered significantly enriched).
Immune Score
The R software was used to cluster OS patients with complete clinical survival information based on their mRNA expression profile data via “K-means” method. And the “ESTIMATE” package was used to calculate the Immune Score of OS samples.
LASSO Cox regression analysis
The univariate Cox regression analysis was performed on gene expression values of OS samples, and p value < 0.05 was used as the threshold to screen genes significantly associated with OS prognosis. Then, LASSO Cox regression was conducted with GLMNET package of R software [16] to further optimize the genes related to OS prognosis. The screened genes were used to created the Risk Score of each sample by the following formula:
Coefi was the risk coefficient of each gene calculated by the LASSO-Cox model, and Xi was the expression value of each gene. Next, the most optimal threshold of Risk Score was determined by R package survival, survminer, and bilateral log rank test. The patients were divided into low and high risk group according to the median.
Survival analysis
The overall survival rate of patients was estimated by R software survival package and survminer package based on Kaplan Meier (KM) method, and the significance of survival rate difference between different groups was tested by log rank or breslow. Multivariate Cox regression model was used to analyze whether risk score can predict the survival of OS patients independently of other factors.
Evaluation of immune cell infiltration (ICI)
The relative proportion of 22 immune cells in each cancer sample was calculated by software CIBERSORT [17]. According to the gene expression matrix, CICERSORT executed deconvolution algorithm to characterize the composition of immune infiltrating cells with 547 preset barcode genes. The sum of the proportion of all estimated immune cell types in each sample was equal to one.
Establishment of Nomogram prognostic model
Nomogram has been widely used to predict cancer prognosis. In order to predict the 1-, 3- and 5-year survival probabilities of patients, all independent prognostic factors determined by multivariate Cox regression analysis were used to establish Nomogram with R software “RMS” package [18].
Results
Glycolysis-immune-related analysis could define different prognosis
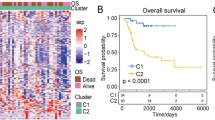
In this study, a total of 296 glycolysis related genes (Table S1) were obtained from GSEA. Based on these genes, we used the “K-means” method in R software to cluster the OS samples in GSE21257. According to the sum of squares errors (Fig. 1A), we selected the number of clusters k = 2 and clustered the samples into two clusters (Fig. 1B). Furthermore, the KM carves showed that there was significant difference in overall survival between the two clusters of samples (Fig. 1C), and the prognosis of cluster 2 was better. Therefore, we define cluster 2 as the low glycolysis group, on the contrary, cluster 1 as the high glycolysis group.

glycolysis-immunoassay defines different prognosis. (A) The “Elbow diagram” of the optimal number of clustering. The horizontal and vertical axes represented the number of clusters K and sum of the squared errors respectively; the point where the decline tends to be gentle was the optimal number of clusters K = 2. (B) Sample clustering diagram, different colors represented different clusters. (C) KM survival curve defined by glycolysis. p value was obtained based on log-rank test. (D-E) The best cutoff values and KM survival curve defined by Immune Score. (F) Kaplan Meier survival curve defined by glycoly-immune score
For further survival analysis, the “Estimate” function package was conducted to evaluate the Immune Score of samples and these samples were grouped subsequently. According to the results, the optimal cutoff value divided the samples into two groups (Fig. 1D), and the prognosis of the samples in the high Immune Score group was significantly higher than that in the low Immune Score group (Fig. 1E).
According to the glycolysis status and Immune Score, we further divided the samples into low glycolysis-high immunity group (Low/High), high glycolysis-low immunity group (High/Low) and other mixed groups (Mix). We noted that the prognosis of Low/High group was the best among three groups, the prognosis of High/Low group was the worst, and mix group crossed between these two groups, there were significant differences among the groups (Fig. 1F).
Identification of candidate GIGs
We also analyzed the DEGs in each group. Compared with the high glycolysis group, the low glycolysis group had 1960 DEGs (Fig. 2A), the low immunity group had 659 DEGs compared with the high immunity group (Fig. 2B), 2517 DEGs between the Low/High group and the High/Low group (Fig. 2 C), and 305 overlap genes among the three groups (Table S2, Fig. 2D). Further enrichment analysis of GO and KEGG indicated that these 305 genes were significantly enriched in GO terms such as extracellular matrix organization and KEGG Pathway such as Nicotine addiction, respectively. The top 10 GO terms with the most significant enrichment were shown in Fig. 2E, as well as the top 10 KEGG terms in Fig. 2F. The detailed results of enrichment analysis as listed in Table S3.

DEGs and functional enrichment. (A-C) Volcano map of DEGs, blue dots were down-regulated genes, red dots were up-regulated genes. (D) Venn’s diagram of DEGs. (E-F) The top 10 items of GO and KEGG enrichment analysis
Construction and validation of prognostic model
The prognostic model was established and verified in this study. Firstly, we used the GSE21257 data set sample to conduct univariate Cox regression analysis with the expression values of 305 overlap genes as the continuous variables, and calculated the Hazard ratio (HR). The first 8 genes were screened with p < 0.01 as the threshold (Fig. 3A). Next, LASSO Cox regression analysis was performed on the 8 genes to screen the most closely related genes. According to the lambda values corresponding to the number of different genes, the optimal number of genes was determined to be 5 (Fig. 3B, lambda value was the minimum). And the five genes were RAI14, MAF, CLEC5A, TIAL1 and CENPJ. In order to obtain a uniform critical value, we divided patients into high risk group and low risk group, and weighted the expression values of 5 genes in the GSE21257 dataset with LASSO Cox regression coefficient to establish a Risk Score model for predicting patient survival. Risk Score=(-0.13626991*RAI14)+(-0.13438968*MAF)+(-0.30713757*CLEC5A)+ (0.09256004*TIAL1)+(0.05059066*CENPJ).

Construction of OS prognostic model. (A) Forest map of univariate analysis of 8 genes significantly associated with OS prognosis. HR: the Hazard ratio; 95% CI: the 95% confidence interval. (B) Optimization parameter Lambda by the LASSO regression model. The lambda value at the minimum was the best. (C-D) The predicted Risk Score for patients. The vertical dotted lines represented the median Risk Score, which distinguished between high-risk (red) and low-risk (green) populations in terms of population proportion, duration of survival (life/death), and gene expression of interest (genes involved in model construction). (E-F) KM survival curve in GEO dataset and TARGET dataset, respectively. p value was obtained based on log-rank test
After that, we calculated the Risk Score for each patient and divided the GSE21257 dataset and the TARGET validation sample into the high-risk and low-risk groups by median. Thus, we obtained the relationship between patients ranked by predicted value of risk and time to survival. As shown in Fig. 3C-D, the survival time of the low-risk group was slightly higher than that of the high-risk group. Clearly, the number of deaths was higher in the high-risk group than in the low-risk group. In addition, survival analysis showed that high-risk OS samples had poor overall survival compared with low-risk samples in the GSE21257 data set (Fig. 3E), as well as in the TARGET data set. Overall, these results suggested that the Risk Score can successfully distinguish the prognosis of OS patients.
Risk Score was an independent prognostic marker of OS
To determine whether Risk Score was an independent prognostic indicator, age, gender, tumor stage and Risk Score were included in this study for multivariate Cox regression analysis. The results showed that Risk Score was significantly correlated with overall survival, and the sample with high Risk Score had a greater risk of death and was a poor prognostic factor (HR = 5.35, 95% CI: 1.547–18.5, p < 0.008).
In addition, the prognostic value of Risk Score in OS samples with different clinicopathological factors (including age and sex) was further evaluated. We regrouped OS patients according to these factors and performed KM survival analysis. As the analysis results showed, in both female and male samples (Fig. 4B-C), the overall survival rate of male samples in the high-risk group was significantly lower than that in the low-risk group, while the p value of KM survival curve of female samples was greater than 0.05, which may be caused by too few samples. Meanwhile, overall survival was significantly lower in the high-risk group than in the low-risk group in both samples under 16 years old (inclusive) and over 16 years of age (Fig. 4D-E). These results indicated that Risk Score can be used as an independent indicator to predict the prognosis of OS patients.

Risk score was an independent prognostic marker of OS. (A) Forest map by multivariate Cox regression analysis. Compared with the reference sample, samples with a Hazard ratio greater than 1 had a higher risk of death, and those with a Hazard ratio less than 1 had a lower risk of death. (B-C) KM survival curves of OS samples of different genders. (D-E) KM survival curves of OS samples in different age groups
Nomogram model predicts the prognosis and survival of OS patients
A Nomogram was an effective model for pictorial representation of a complex mathematical formula and now was widely used in oncology to assist with prediction of prognosis [19]. In this study, we constructed a Nomogram model by Risk Score to further verify the accuracy and rationality of Risk Score (Fig. 5A). As shown in Fig. 5B-D, the corrected curve in the calibration diagram was closed to the ideal curve (the 45-degree line with slope 1 through the origin of the coordinate axis), indicating that the prediction was in good agreement with the actual results.

Nomogram predicted survival of OS patients. (A) Nomogram model was used to predict the probability of 1-, 3-, and 5-year overall survival of OS patients. (B-D) Calibration curves for Nomogram to predict the probabilities of 1-, 3-, and 5-year overall survival of OS patients. The X-axis and Y-axis represented the survival rate predicted by Nomogram and actual survival rate, respectively
Characterisation of the ICI in OS patients
We also used the CIBERSORT method combined with the LM22 feature matrix to evaluate the differences in immune infiltration of 22 immune cells in OS patients. In the analysis of ICI results of 53 OS patients (Fig. 6 A), obviously, there were differences in the infiltrating proportion of various immune cells in the high and low risk groups (Fig. 6B). the changes in the proportion of tumor infiltrating immune cells in different patients represented the internal characteristics of individual differences. Among them, plasma cells, T cells CD8, T cells CD4 memory resting, T cells follicular helper, NK cells activated, Monocytes and MacrophagesM0 had significant differences in the proportion of immune cell infiltration (Fig. 6C). Further PCA analysis showed that the 7 types of immune cells with significant differences could be clustered and divided into two groups (Fig. 6E).

Immune infiltration of OS patients in high and low risk groups. (A) Relative proportion of immune-infiltrating cells in patients. (B-C) Violin diagram of different immune cells in the high and low risk groups. p value was calculated by the Wilcoxn method, the significance level label ns: p > 0.05, *: p ≤ 0.05, **: p ≤ 0.01, ***: p ≤ 0.001, ****: p ≤ 0.0001. (D) Correlation matrix of the proportion of 22 immune cells. Orange represented positive correlation, and blue represented negative correlation. The darker the color, the greater the correlation. (E) PCA three-dimensional clustering map
Relationship between Risk Score and immune checkpoint genes
The expression of immune checkpoint has become a biomarker for OS patients to choose immunotherapy. In this study, we investigated whether there was an association between patient Risk Score and key immune checkpoints (CTLA4, PDL1, LAG3, TDO2). As expected, the Risk Score was associated with all of them (Fig. 7 A). Furthermore, there were significant differences in the expression of CTLA4 and LAG3 of OS patients in the high- and low-risk groups (Fig. 7B C).

Relationship between four immune checkpoints, tumor purity and Risk Score. (A) Chord diagrams expressed by Risk Score and four prominent immune checkpoints, the wider the line between them the stronger the correlation between them. (B) Violin plot of immune checkpoint with significant difference in expression levels in the high and low risk groups, different colors represented the high and low risk groups, the vertical axis represented expression levels, and p value was calculated by Wilcoxon method
Discussion
OS progresses rapidly and the prognosis of patients in the middle and late stages was often unsatisfactory [20]. Therefore, it is urgent to accurately evaluate the recurrence risk and provide appropriate monitoring and treatment for patients at high risk. Metabolic reprogramming in TME was involved in the survival, growth, and proliferation of tumor cells [21]. Recent studies have focused on the glycolysis process in the proliferation, invasion and drug resistance of OS cells [9, 22]. For example, Wen et al. [23] showed that lncRNA-SARCC-mediated cisplatin sensitivity may be via glycolysis in the miR-143-inhibited OS cells. Although prognostic signatures of patients with OS have been studied, the prognostic significance of glycoly-related genes was still unclear. In this study, we established a comprehensive evaluation model of glycolysis status combined with Immune Score, which could more clearly distinguish the difference in prognosis of patients. According to this unique model, we could easily group patients. Patients in different groups showed different glycolysis degrees and immune status, and the prognosis was also significantly different.
To numerically formulate this model, we analyzed 296 glycolsis-related genes and calculated Immune Score of OS patients to establish a GIG Risk Score. Next, we verified that the novel established model was an independent prognostic indicator of OS patients by using multivariate Cox analysis. Five prognostic genes related to glycolysis and immunity of OS were screened from 305 genes by univariate Cox regression analysis, including RAI14, MAF, CLEC5A, TIAL1 and CENPJ. Most of these genes have been studied in various tumors. RAI14 was highly expressed in gastric cancer, and its expression level was related to poor prognosis of patients [24]. MAF was a transcription factor and was considered as a tumor suppressor or oncogene, depending on the cell type [25]. For instance, MAF overexpression was a common tumorigenic event in multiple myeloma, triggering pathological bone marrow stromal cell interaction and promoting proliferation [26]. A study showed that the expression level of CLEC5A was significantly increased in ovarian cancer cells, and the higher the expression, the lower the survival rate of patients [27]. And CENPJ has been also verified to be tightly associated with prognosis of breast cancer [28]. Moreover, the stable silencing of TIAL1 expression in HeLa cells promoted cell proliferation, anchoring-dependent and -independent growth and invasion [29]. However, almost none of the 5 prognostic genes has been studied in OS. In our present work, the Risk Score constructed based on these five genes could satisfactorily predict the prognosis of OS patients.
Lactic acid produced by glycolysis of tumor cells subsequently accumulated in extracellular environment, which reduced pH of the TME and inhibited the innate immune response of the tumor, resulting in a barrier to treatment [30]. At the present, the immune Risk Score model based on tumor infiltrating immune cells has been reported to predict the prognosis of OS. For example, Chen et al. [31] constructed an immune Risk Score model based on tumor infiltrating immune cell types selected by forward stepwise approach to provide more valuable ideas for further research on the prognosis of OS. In this study, our results demonstrated that the prognosis of the low Immune Score group was significantly lower than the high Immune Score group, suggested that the low Immune Score group has a greater risk of death. Meanwhile, in the high and low risk group defined by Risk Score, there were differences in 22 types of immune cells, among which 7 immune cells, including plasma cells, T cells CD8, T cells CD4 memory resting, T cells follicular helper, NK cells activated, monocytes, and macrophages M0 were significantly different. These findings indicated that glycolysis affected the composition of the immune cell population of OS. Hence, it is more reasonable for our prognostic model to take into account both tumor glycolysis and immunity.
Compared with traditional methods, our GIG Risk Score model can predict the prognosis of OS patients more conveniently and accurately. In clinical observation, we can easily obtain the expression levels of these five genes to calculate the patient’s Risk Score and predict their survival rate. Our research also suffers from some limitations. The target sample size obtained from the GEO and TARGET datasets was small, which may affect the reproducibility of the results and lack verification of molecular mechanisms. These issues may be the focus of further research.
Conclusions
In conclusion, our study revealed an association between clinical outcomes of osteosarcoma and glycolysis and tumor immunity. The Risk Score model based on 5 glycolysis-immume-related genes was reliable in predicting the prognosis of osteosarcoma.
Availability of data and materials:
The datasets generated and analyzed during the current study are available in the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) repository and the Therapeutically Applicable Research to Generate Effective Treatments (TARGET, https://ocg.cancer.gov/programs/target) repository.
References
Ren C, Liu J, Zheng B, Yan P, Sun Y, Yue B (2019) The circular RNA circ-ITCH acts as a tumour suppressor in osteosarcoma via regulating miR-22. Artif Cells Nanomed Biotechnol 47:3359–3367. http://doi.org.10.1080/21691401.2019.1649273
He M, Shen P, Qiu C, Wang J (2019) miR-627-3p inhibits osteosarcoma cell proliferation and metastasis by targeting PTN. Aging (Albany NY) 11:5744–5756. http://doi.org.10.18632/aging.102157
Huang C, Wang Q, Ma S, Sun Y, Vadamootoo AS, Jin C (2019) A four serum-miRNA panel serves as a potential diagnostic biomarker of osteosarcoma. Int J Clin Oncol 24:976–982. http://doi.org.10.1007/s10147-019-01433-x
Lv S, Guan M (2018) miRNA-1284, a regulator of HMGB1, inhibits cell proliferation and migration in osteosarcoma. Biosci Rep 38:http://doi.org.10.1042/BSR20171675
Warburg O (1956) On the origin of cancer cells. Science 123:309–314. http://doi.org.10.1126/science.123.3191.309
Teicher BA, Linehan WM, Helman LJ (2012) Targeting cancer metabolism. Clin Cancer Res 18:5537–5545. http://doi.org.10.1158/1078-0432.CCR-12-2587
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033. http://doi.org.10.1126/science.1160809
Wan J, Liu Y, Long F, Tian J, Zhang C (2021) circPVT1 promotes osteosarcoma glycolysis and metastasis by sponging miR-423-5p to activate Wnt5a/Ror2 signaling. Cancer Sci 112:1707–1722. http://doi.org.10.1111/cas.14787
Han X, Yang Y, Sun Y, Qin L, Yang Y (2018) LncRNA TUG1 affects cell viability by regulating glycolysis in osteosarcoma cells. Gene 674:87–92. http://doi.org.10.1016/j.gene.2018.06.085
Sottnik JL, Lori JC, Rose BJ, Thamm DH (2011) Glycolysis inhibition by 2-deoxy-D-glucose reverts the metastatic phenotype in vitro and in vivo. Clin Exp Metastasis 28:865–875. http://doi.org.10.1007/s10585-011-9417-5
Jiang Z, Liu Z, Li M, Chen C, Wang X (2019) Increased glycolysis correlates with elevated immune activity in tumor immune microenvironment. EBioMedicine 42:431–442. http://doi.org.10.1016/j.ebiom.2019.03.068
Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD et al (2015) Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 162:1229–1241. http://doi.org.10.1016/j.cell.2015.08.016
Bi J, Bi F, Pan X, Yang Q (2021) Establishment of a novel glycolysis-related prognostic gene signature for ovarian cancer and its relationships with immune infiltration of the tumor microenvironment. J Transl Med 19:382. http://doi.org.10.1186/s12967-021-03057-0
Yang M, Ma X, Wang Z, Zhang T, Hua Y, Cai Z (2021) Identification of a novel glycolysis-related gene signature for predicting the prognosis of osteosarcoma patients. Aging (Albany NY) 13:12896–12918. http://doi.org.10.18632/aging.202958
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W et al (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43:e47. http://doi.org.10.1093/nar/gkv007
Friedman J, Hastie T, Tibshirani R (2010) Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw 33:1–22
Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y et al (2015) Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12:453–457. http://doi.org.10.1038/nmeth.3337
Zhao W, Wu L, Zhao A, Zhang M, Tian Q, Shen Y et al (2020) A nomogram for predicting survival in patients with de novo metastatic breast cancer: a population-based study. BMC Cancer 20:982. http://doi.org.10.1186/s12885-020-07449-1
Sun Y, Deng C, Zhang Z, Ma X, Zhou F, Liu X (2021) Novel nomogram for predicting the progression of osteoarthritis based on 3D-MRI bone shape: data from the FNIH OA biomarkers consortium. BMC Musculoskelet Disord 22:782. http://doi.org.10.1186/s12891-021-04620-y
Mirabello L, Zhu B, Koster R, Karlins E, Dean M, Yeager M et al (2020) Frequency of Pathogenic Germline Variants in Cancer-Susceptibility Genes in Patients With Osteosarcoma. JAMA Oncol 6:724–734. http://doi.org.10.1001/jamaoncol.2020.0197
Yang E, Wang X, Gong Z, Yu M, Wu H, Zhang D (2020) Exosome-mediated metabolic reprogramming: the emerging role in tumor microenvironment remodeling and its influence on cancer progression. Signal Transduct Target Ther 5:242. http://doi.org.10.1038/s41392-020-00359-5
Zheng XM, Xu CW, Wang F (2018) MiR-33b inhibits osteosarcoma cell proliferation through suppression of glycolysis by targeting Lactate Dehydrogenase A (LDHA). Cell Mol Biol. (Noisy-le-grand) 64:31–35
Wen JF, Jiang YQ, Li C, Dai XK, Wu T, Yin WZ (2020) LncRNA-SARCC sensitizes osteosarcoma to cisplatin through the miR-143-mediated glycolysis inhibition by targeting Hexokinase 2. Cancer Biomark 28:231–246. http://doi.org.10.3233/CBM-191181
Xiao Y, Zhang H, Du G, Meng X, Wu T, Zhou Q et al (2020) RAI14 Is a Prognostic Biomarker and Correlated With Immune Cell Infiltrates in Gastric Cancer. Technol Cancer Res Treat 19:1533033820970684. http://doi.org.10.1177/1533033820970684
Janiszewska J, Bodnar M, Paczkowska J, Ustaszewski A, Smialek MJ, Szylberg L et al (2021) Loss of the MAF Transcription Factor in Laryngeal Squamous Cell Carcinoma. Biomolecules 11:http://doi.org.10.3390/biom11071035
Hurt EM, Wiestner A, Rosenwald A, Shaffer AL, Campo E, Grogan T et al (2004) Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell 5:191–199. http://doi.org.10.1016/s1535-6108(04)00019-4
Wang Q, Shi M, Sun S, Zhou Q, Ding L, Jiang C et al (2020) CLEC5A promotes the proliferation of gastric cancer cells by activating the PI3K/AKT/mTOR pathway. Biochem Biophys Res Commun 524:656–662. http://doi.org.10.1016/j.bbrc.2019.10.122
Li Y, Wang X, Vural S, Mishra NK, Cowan KH, Guda C (2015) Exome analysis reveals differentially mutated gene signatures of stage, grade and subtype in breast cancers. PLoS One 10:e0119383. http://doi.org.10.1371/journal.pone.0119383
Izquierdo JM, Alcalde J, Carrascoso I, Reyes R, Ludena MD (2011) Knockdown of T-cell intracellular antigens triggers cell proliferation, invasion and tumour growth. Biochem J 435:337–344. http://doi.org.10.1042/BJ20101030
Hayes C, Donohoe CL, Davern M, Donlon NE (2021) The oncogenic and clinical implications of lactate induced immunosuppression in the tumour microenvironment. Cancer Lett 500:75–86. http://doi.org.10.1016/j.canlet.2020.12.021
Chen Y, Zhao B, Wang X (2020) Tumor infiltrating immune cells (TIICs) as a biomarker for prognosis benefits in patients with osteosarcoma. BMC Cancer 20:1022. http://doi.org.10.1186/s12885-020-07536-3
Acknowledgements
Not applicable.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
Kangsong Tian and Wei Qi contributed to the study conception and design. Material preparation, data collection and analysis were performed by Kangsong Tian, Wei Qi, Qian Yan and Ming Lv. The first draft of the manuscript was written by Kangsong Tian, Wei Qi and Delei Song and Delei Song commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Disclosure of potential conflicts of interest
Not applicable.
Research involving human participants and/or animals
Not applicable.
Informed consent
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Kangsong Tian and Wei Qi contributed equally to this study.
Rights and permissions
About this article

Cite this article
Tian, K., Qi, W., Yan, Q. et al. Signature constructed by glycolysis-immune-related genes can predict the prognosis of osteosarcoma patients. Invest New Drugs 40, 818–830 (2022). https://doi.org/10.1007/s10637-022-01228-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-022-01228-4