
Summary
Objective The problem of drug resistance to BRAF-targeted therapy often occurs in melanoma treatment. Activation of PI3K/AKT/mTOR signaling pathway is one of the mechanisms of acquired resistance and a potential target for treatment. In the current research, we investigated that dual inhibition of mTOR and MEK synergistically reduced the viability of melanoma cells in vitro. Methods A combination of rapamycin (a macrolide immunosuppressant, mTOR inhibitor) and binimetinib (an anti-cancer small molecule, selective inhibitor of MEK) was studied using a panel of melanoma cell lines, including patient-derived cells. Results It was found, that combinatorial therapy of rapamycin (250 nM) and binimetinib (2 μM) resulted in 25% of cell viability compared to either rapamycin (85%) or binimetinib alone (50%) for A375 and vemurafenib-resistant Mel IL/R cells. The suppressed activation of mTOR and MEK by combined rapamycin and binimetinib treatment was confirmed using Western blot assay. Cell death occured via the apoptosis pathway; however, the combination treatment significantly increased the apoptosis only for Mel IL/R cells. The enhanced cytotoxic effect was also associated with enhanced cell cycle arrest in the G0/G1 phase. Conclusion In general, we provide the evidence that dual inhibition of mTOR and MEK could be promising for further preclinical investigations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Treatment of advanced melanoma has changed dramatically within the last decade. The target inhibition of mutated BRAF with vemurafenib and dabrafenib have shown marked downregulation of BRAF signaling and significant clinical activity [1]. However, the effectiveness of existing therapies remains questionable due to development of drug resistance. This acquired resistance is commonly associated with MAPK pathway reactivation and PI3K/AKT/mTOR pathway activation, NRAS activating mutations, or hyperactivation of receptor tyrosine kinases (RTKs) [2, 3].
Both PI3K/AKT/mTOR and MAPK signaling pathways play a central role in the transmission of oncogenic signals [4, 5]. These pathways can be constitutively activated or mutated in melanoma cells by hyperactivation of the upstream RTKs or somatic mutations in specific signaling components leading to activation of cell proliferation and survival signals [6].
Despite the prevalence of persistent MAPK signaling in resistant metastases, inhibition downstream of BRAF using the MEK inhibitor trametinib as a single agent had minimal clinical activity in melanoma patients that had relapsed on BRAF inhibitor (BRAFi) therapy [7]. Another MEK inhibitor, binimetinib (MEK162), has led to a partial response in a phase II study in patients with melanoma containing BRAFV600 [8]. A Phase III trial has also shown significantly improved progression-free survival compared with dacarbazine in NRAS-mutated melanoma [9]. In June 2018 FDA approved a combination of binimetinib and BRAF inhibitor encorafenib for unresectable or metastatic melanoma with BRAF mutations [11].
However, little information is known about its efficacy on BRAF-resistant melanomas. The search for new combinations with this group of drugs is continuing to increase the tumor-specific cytotoxic effects and increase patient survival.
Although PI3K/AKT/mTOR is a potential drug target, its inhibition alone does not provide sufficient and sustained results in multiple studies of malignancies [10, 11]. Whereas inhibiting both mTOR and MEK in parental cells makes sense due to the genetic alterations in melanoma, the rationale of co-targeting mTOR and MEK in resistant cells is a challenge. Thus, combination therapy with PI3K/AKT/mTOR and MAPK pathway inhibitors might be an effective therapeutic strategy [12]. Indeed, several preclinical studies of concurrent PI3K/AKT/mTOR and MAPK signaling pathway inhibition showed more pronounced antitumor activity in various models of malignant tumors such as basal breast cancer, BRAF mutant skin melanoma, and renal cell carcinoma [13].
To date, most efforts to dual target the PI3K/AKT/mTOR and MAPK pathways have used a combination of PI3K and MEK inhibitors [12, 14,15,16] but not concurrent inhibition of mTOR and MEK in melanoma cells. Here, mTOR is a serine/threonine kinase that plays a crucial role in both normal physiological development and carcinogenic processes, including cell growth, proliferation, apoptosis, autophagy, nutrient starvation, and survival [17]. Allosteric MEK inhibitors are highly specific for their targets [18], while the current RAF inhibitors are significantly less effective in inhibiting downstream signaling in tumors with wildtype RAF. Recently, Carlino et al., showed that the combination of ERK and PI3K/mTOR inhibition was effective at promoting cell death in all resistant melanoma cell models, and was substantially more potent than the MEK/PI3K/mTOR inhibitor combination [19, 20].
The aim of the current research was to investigate the ability of rapamycin to increase the sensitivity to MEKi binimetinib in BRAF-mutant melanoma cells in vitro.
Materials and methods
Drugs
Rapamycin was purchased from Sigma Aldrich (St. Louis, MO, USA) and binimetinib was obtained from Selleck Chemicals (Cambridge, UK). Stock concentrations of rapamycin and binimetinib were prepared in DMSO and diluted to working concentrations in culture medium. Stock concentrations of rapamycin were stored in aliquots at −20 °C and binimetinib at −80 °C according to the manufacture’s recommendations.
Cell lines
All experiments were performed on three melanoma cell lines: Mel IL was derived from a patient at N.N. Blokhin National medical research center for oncology. Resistant Mel IL/R subline was obtained via long-term cultivation of maternity Mel IL with increasing doses of vemurafenib. The A375 cell line was purchased from ATCC. Mel IL and A375 cell lines were established as vemurafenib-sensitive and Mel IL/R cell line was vemurafebin-resistant. We used keratinocytes HaCaT and the hFB-hTERT6 skin fibroblasts (obtained via a lentiviral transduction of full-length TERT gene under a CMV promoter (generated in Engelhardt Institute of Molecular Biology; gift of E. Dashinimaev) [21] as normal cells. Both Mel IL and Mel IL/R cells harbored a BRAFV600K and TP53 c.326 T > C p.F109S mutations [22], while A375 harbored oncogenic BRAFV600E mutation. HaCaT, Mel IL and Mel IL/R cells were cultured in RPMI-1640 (Gibco, Gaithersburg, MD, USA) and hFB-hTERT6 and A375 cells were grown in DMEM (Gibco, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (HyClone, South Logan, Utah, USA), 2 mM L-glutamine (Sigma, St. Louis, MO, USA), 10 U/mL penicillin, and 10 mg/mL streptomycin (Sigma, St. Louis, MO, USA) at 37 °C in a 5% CO2 humidified atmosphere.
Cell viability assay and CI determination
Cells were plated in 96-well plates at 7000 cells per well and incubated overnight. The cells were then treated with binimetinib (0.1–10 μM) and rapamycin (10–500 nM), and were further incubated for 48 h. The cytotoxicity was assessed by incubating cells with 20 μl of MTT reagent (3- [4,5-dimetiltriazol-2-yl] -2,5 diphenyl tetrazolium bromide, M2128, Sigma) for 4 h followed by DMSO elution and measuring the absorbance at 570 nm on a microplate analyzer Multiscan FC (Thermo Scientific, Waltham, MA, USA). The IC50 values were determined as the dose at which cell viability was decreased by 50% compared to control in three independent experiments. Combination Index (CI) was calculated by the Chou-Talalay method [23], and synergy assessment was performed using CalcuSyn software (Biosoft, Cambridge, UK). CI values were interpreted as follows: < 1 is synergism, = 1 is additivity, and > 1 is antagonism.
Colony-formation assay
Mel IL, Mel IL/R, and A375 melanoma cells were seeded in 6-well plates at 2000 cells per well. After complete cell adherence, the cells were exposed to 2 μM binimetinib, 250 nM rapamycin, or both (in triplicates). Cells were cultivated for 12 days (37 °C, 5% CO2), and the medium with drugs was changed every 3 days. At the end of the experiment, colonies were fixed in 1% formalin for 15 min, stained with 0.5% crystal violet, and counted using Image J software (NIH, Bethesda, Maryland, USA). Three independent experiments were done.
The entrapment of spheroids in Matrigel
Tumor spheroid formation on agarose-coated plates was performed as previously described [24]. Briefly, 1.5% wt of agarose in PBS was heated on water bath for 15 min. Then 50 μL of agarose gel was added to each well of a flat-bottom 96-well plate under sterile conditions. Plates with agarose were cooled down to room temperature for 30 min. Mel IL, Mel IL/R sand A375 cells were seeded on agarose-coated plates (10,000 cells/well, 100 μL of media in each well) and incubated at 37 °C, 5% CO2 for 2–3 days. Then the spheroids were collected in a 15 ml centrifuge tube for 10 min for spheroids sedimentation. The supernatant was carefully removed and 1 ml of Matrigel (BD Bioscience, Belgium) was added to spheroids pellet. The spheroids were gently resuspended in Matrigel, and 50 μl of suspension was transferred to the wells of pre-cooled 96-well plate. The plate was incubated at 37 °C for 10 min for Matrigel polymerization, and 100 μl of full media containing rapamycin (250 nM) and binimetinib (2 μM) or both was added to the each well. Cell spreading into the Matrigel was assessed within 24 h using an inverted light microscope. The quantification of spheroids spreading area was performed using ImageJ software (wound healing tool macros). For these, the cell-free surface was accessed for each microphotography, and the total spreading area was calculated. Three independent experiments were done.
Apoptosis measurement and caspase 3/7 activity
Mel IL, Mel IL/R, and A375 melanoma cells were seeded in 6-cm2 Petri’s dishes (300,000 cells per dish). Next day cells were treated with 250 nM rapamycin, 2 μM binimetinib, or both for 24 or 48 h. Then cells were collected to trypsinised and centrifuged. Apoptosis was quantified via combined staining of annexin V and propidium iodide (PI) using the Annexin V–FITC Kit according to the manufacturer’s protocol (Molecular probes, Waltham, MA, USA). Caspase 3 and caspase 7 activities were measured within the Mel IL, Mel IL/R, and A375 cells 4 h after drug treatment with 250 nM rapamycin, 2 μM binimetinib, or both. After treatment, cells were trypsinized, centrifuged, and permeabilized in 0.1% Triton X-100-citrate buffer. They were then incubated for 30 min at 4 °C with mouse anti-caspase 3 (1:100, Santa Cruz, Dallas, Texas, USA) or anti-caspase 7 antibodies (1:100, Santa Cruz, Dallas, Texas, USA). After incubation, the cells were washed and incubated with anti-mouse antibody AlexaFluor® 488 nm (1:2000, Life Technologies, Waltham, MA, USA), washed, and fixed in 1% formalin followed by analysis. Apoptosis and caspase 3/7 activities were analyzed on a NovoCyte 2000R flow cytometer (ACEA Biosciences, USA) using NovoExpress v.1.2.4 software. All experiments were performed three times for each experimental condition. The results were presented as the percent increase in the control.
Cell cycle analysis
Mel IL, Mel IL/R, and A375 cells were seeded in 6-cm2 Petri’s dishes (300,000 cells per dish) and were treated with 250 nM rapamycin, 2 μM binimetinib, or both for 24 h. Then they were trypsinized, washed with PBS, and the cell pellets were resuspended in 1 ml of 50 μg/ml solution of PI in buffer (Becton Dickinson, Franklin Lakes, NJ, USA) and incubated in the dark at room temperature for 15 min. The PI fluorescence was measured using NovoCyte 2000R flow cytometer (ACEA Biosciences, USA), and the cell cycle distribution was analyzed using NovoExpress v.1.2.4 software. All experiments were performed three times for each experimental condition.
TUNEL assay
Melanoma cells A375, Mel IL, and Mel IL/R were seeded in 8-well culture slides at 20,000 cells per well (SPL Lifesciences, Gyeonggi-do, Korea) and incubated overnight. The next day, the cells were treated with rapamycin (250 nM), binimetinib (2 μM), or both for 24 h. Apoptotic cells were determined by TUNEL assay using the InSitu cell death detection kit (Roche, Basel, Switzerland) according to the manufacturer’s protocol. Briefly, slides with cells were washed in PBS once, dried, and fixed with 4% paraformaldehyde (Sigma-Aldrich, St. Louis, MO, USA) for 1 h at 4 °C. The fixed cells were incubated with blocking solution (3% H2O2 in methanol). They were then rinsed in PBS and incubated with 0.1% Triton X-100 (MP Biochemicals, Leicester, UK) in 0.1% sodium citrate for 2 min on ice (Sigma-Aldrich, St. Louis, MO, USA). The cells were then washed twice with PBS and incubated with the deoxynucleotide transferase recombinant (rTdT)-catalyzed reaction and nucleotide mixture for 1 h at 37 °C. Cells were washed twice in PBS and stained with Hoechst 33258 dye for 10 min in the dark (Sigma-Aldrich, USA). Cells were analyzed with InCell Analyzer 6000 (GE Healthcare, Chicago, Illinois, USA) and InCell developer toolbox software (GE Healthcare, Chicago, Illinois, USA) to calculate the percentage of apoptotic cells.
Western blot analysis
Mel IL, Mel IL/R, and A375 cells were seeded in 6-cm2 Petri’s dishes (300,000 cells per dish) and were treated with 250 nM rapamycin, 2 μM binimetinib, or both and cultivated for 24 and 48 h. Cells were trypsinized, centrifuged and lysed with cell lysis buffer (Cell Signaling, Leiden, The Netherlands) containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM b-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin, 1 mМ PMSF, 10 μl/ml inhibition cocktail, and 100 μM DTT for 40 min at +4°С. The cells were then centrifuged at 13,500 g for 15 min at +4°С. The total protein was analyzed using Quant-IT Protein assay kit according to manufacturer’s protocol (Invitrogen, Waltham, MA, USA) on a Quibit 2.0 (Invitrogen, Waltham, MA, USA).
An equal amount of protein (60 μg) from each group was separated using 10% SDS-polyacrylamide gel electrophoresis (PAGE) followed by transfer to nitrocellulose membrane. Membranes were incubated with a 5% BSA (A1391, Applichem, Darmstadt, Germany) in TBS/0.1% Tween-20 for 1.5 h and then incubated with the following primary antibodies at 4 °C overnight: p-AKTSer473 (1:1000; Cell Signaling, Leiden, The Netherlands), pan-AKT (1:500; Abcam, Cambridge, UK), p-ERK1/2Thr202/Tyr204 (1:1000; Leiden, The Netherlands), MEK1/2 (1:1000; Cell Signaling, Leiden, The Netherlands), mTOR (1:1000; Cell Signaling, Leiden, The Netherlands), p-mTOR Ser2448 (1:1000; Cell Signaling, Leiden, The Netherlands) and β-actin (1:4000; Sigma Aldrich, St. Louis, MO, USA). Membranes were washed in TBS/0.1% Tween-20 and incubated with HRP-conjugated anti-mouse (Amersham, Little Chalfont, UK) or anti-rabbit (Amersham, Little Chalfont, UK) antibodies for 1.5 h at room temperature. The detection of protein-antibody complexes used the enhanced chemiluminescence reagent Clarity ECL (BioRad, Hercules, California, USA). The density of bands was determined on a ChemiDoc Touch Imaging System (BioRad, Hercules, California, USA) and quantification of immunoreactive signals used ImageJ software (NIH, Bethesda, MD, USA). The relative ratio of activated protein to loading control (β-actin) was calculated for each experiment.
Ethics statement
The protocol to obtain Mel IL cells was initially approved by the N.N. Blokhin National Medical Research Center ethics committee in 1999 with written consent. Since then, the cell line was patented (#RU 2287577 C1) and used for many melanoma studies [22, 25].
Statistical analysis
All experiments were performed in triplicate unless otherwise noted, and each experiment was repeated 3 times independently. Data are expressed as mean ± SD. The results of cell viability, colony formation and TUNEL assays were analyzed using one-way analysis of variance (ANOVA), factor Rap/Bin/Rap+Bin followed by the post hoc Duncan’s test. The data were additionally evaluated with ANOVA multiple comparison mode (Rap vs. Rap+Bin and Bin vs. Rap+Bin). The results of Western blot, apoptosis and cell cycle distribution assays were analyzed using two-way ANOVA followed by the post hoc Duncan’s test, since we evaluated two time points. Assumption of normal distribution of differences was verified with the use of Shapiro–Wilk test. For cell cycle distribution: factor I - Rap, factor II - Bin, and for the interaction between factors I and II: Rap/Bin× Rap/Bin/Rap+Bin. For apoptosis and Western blot assays: factor I - 24/48 h, factor II - Rap/Bin/Rap+Bin, and for the interaction between factors I and II: 24/48 h × Rap/Bin/Rap+Bin. Growth assay of 3D tumor spheroids treated with rapamycin and/or binimetinib in Matrigel for 24 h. was analyzed using Mann-Whitney test (median values with quartiles). The results were considered as statistically significant when p < 0.05. All statistical analyses were performed with GraphPad Prism 8.0 software (GraphPad Software, La Jolla, San Diego, CA, USA).
Results
Effects of combined therapy on melanoma viability
We first evaluated the effects of rapamycin alone on cell viability. The cytotoxicity of binimetinib alone has been demonstrated previously [26]. We observed a dose-dependent decrease in cell viability, and the IC50 values of rapamycin were 489 ± 14 nM in A375 and Mel IL/R; it was 267 ± 15 nM in Mel IL (Fig. 1a). Mel IL cells were more sensitive to rapamycin than A375 and Mel IL/R (A375 F2, 6 = 193.6, p < 0.0001; Mel IL/R F2, 6 = 602.4, p < 0.0001; Mel IL F2,6 = 401.3, p < 0.0001).

Rapamycin and binimetinib dose-dependently decrease melanoma cell proliferation. (a) Cell viability after rapamycin treatment (50–500 nM) was determined in A375, Mel IL and Mel IL/R cells by MTT assay. Data are expressed as mean ± SD of at least three independent experiments done in triplicate. (b) Combined treatment with rapamycin (250 nM) and binimetinib (2 μM) for 48 h synergistically decreased cell viability in melanoma cells compared to rapamycin or binimetinib alone. One-way ANOVA. Duncan’s test: statistical significance is shown as * p < 0.001
The Chou-Talalay method was used to define the nature of drug interaction. To evaluate the CI index we conducted experiments with several series of drug doses at a consistent ratio of binimetinib and rapamycin. According to CI results for further investigation, we chose 2 mkM binimetinib and 250 nM rapamycin as lower concentrations of rapamycin and binimetinib did not result in synergetic effect. In Mel IL cells, the CI of binimetinib (2 μM) and rapamycin (250 nM) was found to be 0.97. The CI with combined binimetinib and rapamycin in both Mel IL/R and A375 was 0.45. These results indicate that the combination of binimetinib and rapamycin is synergistic in three cell lines (suppl. File 1).
In A375 and Mel IL/R cells, combinatorial therapy with 250 nM rapamycin and 2 μM binimetinib significantly reduced cell viability (25%) compared to either rapamycin (85%) or binimetinib alone (50%). There was a more obvious decrease in cell viability in Mel IL: 19% cell viability in a combined treatment group compared to monotherapy with rapamycin (61%) or binimetinib (40%) (Fig. 1b).
The results were confirmed in a series of colony-forming assays. Melanoma cells were cultivated in the presence or absence of rapamycin and binimetinib for 12 days. Rapamycin as a monotherapy inhibited the viability of A375, Mel IL, and Mel IL/R cells by 60–70%. Binimetinib inhibited cell growth in 50% of A375 cells, in 70% of Mel IL cells, and in 75% of Mel IL/R cells versus control. The number of colonies in melanoma cells decreased by an additional 15% with combined treatment compared to binimetinib alone. The combined rapamycin and binimetinib reduced the amount of colonies compared to control by 60% in A375, 75% in Mel IL/R, and 90% in Mel IL (Fig. 2, Table 1).

Combined treatment with rapamycin and binimetinib decrease colony-formation activity. Melanoma cells were seeded in 6-well plates and were treated with 2 μM binimetinib, 250 nM rapamycin, or a combination. The numbers of longterm surviving Mel IL/R, A375, and Mel IL cells that were able to form colonies were determined 12 days later. Data are expressed as mean ± SD of at least three independent experiments done in triplicate. The outcome of one-way ANOVA with multiple comparison mode is shown in Table 1. Results are shown as the means ± SD. Duncan’s test: statistical significance is shown as *p < 0.05, **p < 0.01 compared to control
To evaluate the effect of drug combination on normal cells we conducted experiments on keratinocytes HaCat and the hFB-hTERT6 skin fibroblasts cell lines. Our data indicated that 250 nM rapamycin and 2 μM binimetinib combination results in moderate toxicity on fibroblasts, but keratinocytes were not sensitive to either drugs alone or their combo (Fig. S2).
Rapamycin and binimetinib inhibit cell invasion
We examined the rapamycin and binimetinib capacity to inhibit cell invasion using 3D spheroids embedded in Matrigel. For this, Mel IL spheroids were treated with rapamycin (250 nM) and binimetinib (2 μM) for 48 h. It was found that treatment with rapamycin (250 nM), binimetinib (2 μM), or their combination resulted in significant reduction of Mel IL spheroids spreading area compared to the control. Despite the measurement of total spreading area does not allow demonstrating differences between three experimental conditions, these differences can be confirmed with single spreading cells observation. Thus, we have a lot of spreading cells in control, a few spreading cells in rapamycin (250 nM) and binimetinib (2 μM) samples, and no spreading cells for drug combination (Fig. 3).

Effects of rapamycin and binimetinib on melanoma migration in Mel IL cells. 3D spheroid growth assay in Matrigel using Mel IL melanoma cells treated with rapamycin (250 nM) and/or binimetinib (2 μM) for 24 h. Microphotographs of cell spreading (a) and quantification of spheroids spreading area (b). The arrows indicate spreading cells. The total spreading area was measured using ImageJ software, median with interquartile range. Statistical analysis was performed using non-parametric Mann-Whitney test, *p < 0.05. Scale bar is 200 μm
Combined mTOR and MEK/ERK pathways inhibition effects intracellular signaling in melanoma cell lines
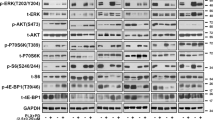
We next investigated whether combined rapamycin and binimetinib treatment indeed inhibits both PI3K/AKT/mTOR and MEK/ERK pathways. To understand the molecular effects of rapamycin, binimetinib, and their combination, we measured the phosphorylated levels of AKT, mTOR, and ERK in Mel IL, Mel IL/R, and A375 cell lines by Western blot after 24 and 48 h treatment to determine whether this drug combination effectively suppressed activation of the two indicated signaling pathways. Rapamycin suppressed phosphorylation of mTOR in Mel IL and Mel IL/R cell lines, but there was no decrease in phosphorylation of mTOR in A375 after 24 h treatment. Phosphorylation of mTOR was completely stopped in all three cell lines after 48 h of rapamycin treatment. Interestingly, rapamycin alone significantly increased AKT phosphorylation by positive feedback in Mel IL/R cell line after 24 h therapy, but p-AKT decreased after an additional 24 h of rapamycin therapy (Fig. 4a, suppl. File 3). In A375 cells, the rapamycin monotherapy reduced phosphorylation of AKT by 24 h, but this effect was abrogated by 48 h.

Combined treatment with rapamycin and binimetinib decrease the expression of mTOR and MEK but not AKT. (a) Rapamycin (Rap) and binimetinib (Bin) in combination decrease mTOR and MEK but not AKT phosphorylation. Western blots show phosphorylation status of mTOR, AKT and MEK from A375, Mel IL, Mel IL/R cells treated for 24 or 48 h with 2 μM binimetinib and 250 nM rapamycin or both. (b) Densitometric analysis was performed using ImageJ for each cell line, and protein expression levels of p-mTOR, p-AKT and p-MEK were normalized to β-actin loading control. Representative western blots and summaries (mean ± SD, western bands represents one of three repeated experiments). The outcome of two-way ANOVA is shown in Table 2. Results are shown as the means ± SD. Duncan’s test: statistical significance is shown as *p < 0.05, **p < 0.01, ***p < 0.001 compared to control
We further evaluated MEK/ERK pathway activation using binimetinib monotherapy by phosphorylation of its downstream effector ERK. Binimetinib inhibited the activation of ERK in Mel IL and Mel IL/R after 24 h incubation with the drug but did not affect A375. However, the ERK phosphorylation was completely reduced in Mel IL/R and A375 cells after an additional 24 h of treatment but not Mel IL. Moreover, binimetinib resulted in increased AKT phosphorylation in all cell lines studied here.
Combined rapamycin and binimetinib therapy suppressed mTOR and ERK phosphorylation to levels seen with binimetinib or rapamycin alone in Mel IL, Mel IL/R, and A375 cell lines. Moreover, mTOR and ERK phosphorylation was totally reduced after 48 h of combined treatment.
On the other hand, rapamycin, and binimetinib incompletely inhibited AKT—the combined therapy reduced AKT phosphorylation within 24 h but this effect was overcome in an additional 24 h in Mel IL and A375 cell lines. This was absolutely reduced in Mel IL/R (Fig. 4b). Тwo factors were taken into account in the two-way ANOVA: factor I (24/48 h) and factor II (binimetinib/rapamycin/ rapamycin+binimetinib). Statistically significant values of F and p derived from the two-way ANOVA analysis are shown in Table 2. All the p values of Duncan’s test are presented in the figure captions. Thus, inhibition of components in these proliferative pathways cannot fully explain the synergistic effects of enhanced cell death.
Effects of combined therapy is dependent on apoptosis
To investigate the mechanism underlying the enhanced cytotoxic effect of combined rapamycin and binimetinib treatment in melanoma cells, we investigated activation of apoptosis by Annexin V/PI double staining. Here, A375, Mel IL, and Mel IL/R cell lines were treated with 250 nM rapamycin, 2 μM binimetinib, or both for 24 and 48 h. Rapamycin alone did not induce apoptosis in any cell line at 24 h (Fig. 5). After 48 h, rapamycin activated apoptosis only in Mel IL cells (12%). Binimetinib led to apoptosis activation in 35% of A375 cells, 23.4% of Mel IL/R cells, and 18.5% of Mel IL cells after 24 h incubation. Further treatment with binimetinib increased apoptosis only in Mel IL cells (18.5% vs 51%) (Fig. 5, Fig. S4). However, combined treatment with rapamycin and binimetinib led to a dramatic increase in apoptosis in these three cell lines compared to monotherapy with either rapamycin or binimetinib — the combined therapy resulted in 41.5% and 66.4% apoptosis cells in Mel IL after 24 or 48 h treatment, respectively. This was 39.1% and 53.3% in Mel IL/R and 56.5% and 48.8% in A375. Тwo factors were taken into account in the two-way ANOVA: factor I (24/48 h) and factor II (binimetinib/rapamycin/rapamycin+binimetinib). Statistically significant values of F and p derived from the two-way ANOVA analysis are shown in Table 3. All the p values of Duncan’s test are presented in the figure captions.

The enhancement of apoptosis by combined rapamycin and binimetinib in melanoma cells. Apoptosis was measured by annexin V/Propidium iodate staining in Mel IL, Mel IL/R and A375 cells after 24 h and 48 h treatment with rapamycin (250 nM), binimetinib (2 μM) or both. Values are percentage increase to untreated control. The histogram data represents 3 individual experiments. The outcome of two-way ANOVA is shown in Table 3. Results are shown as the means ± SD. Duncan’s test: statistical significance is shown as **p < 0.01 compared to control
Next, we determined the apoptosis by the TUNEL assay to detect apoptotic cells that undergo extensive DNA degradation after 24 h treatment with rapamycin (250 nM), binimetinib (2 μM), or both (Fig. 6a). Rapamycin alone did not induce apoptosis compared to the DMSO-treated control. Binimetinib resulted in an increased number of apoptotic cells in A375, Mel IL, and Mel IL/R cells. Combination treatment significantly increased the amount of apoptotic cells only in Mel IL/R (43% vs 62%) (Fig. 6b). Statistically significant values of F and p derived from the one-way ANOVA analysis with multiple comparison mode are shown in Table 4. All the p values of Duncan’s test are presented in the figure captions.

Morphological assessment of binimetinib and rapamycin induced apoptosis via TUNEL assay. (a) Cells were treated for 24 h with DMSO solution as a vehicle control, 2 μM binimetinib (Bin) and 250 nM rapamycin (Rap) in combination or alone, respectively. Treatment-induced apoptosis was visualized by TUNEL (green fluorescence) and DAPI (blue fluorescence) staining. (b) The percentage of TUNEL positive cells in each group was counted and the cumulative data from three independent experiments was shown here as mean ± SD compared to control. Scale bar 50 μm. The outcome of one-way ANOVA with multiple comparison mode is shown in Table 4. Results are shown as the means ± SD. Duncan’s test: statistical significance is shown as** p < 0.01,*** p < 0.001, # not significant compared to control
We evaluated whether apoptosis is mediated by caspases by measuring caspase 3 and caspase 7 activation. We treated melanoma cell lines as described above for 4 h and examined caspase 3 and caspase 7 activity by flow cytometry. Indeed, rapamycin and binimetinib increased caspase 3 and caspase 7 activation compared to control cells in A375 and Mel IL/R cell lines. Moreover, caspase activation was more obvious in a combination treatment group than rapamycin or binimetinib alone (Table 5, Fig. S5). In Mel IL cells, drugs dramatically increased the caspase-mediated apoptosis versus the control, but there were no differences in caspase 3 and caspase 7 activation between combined treatment and rapamycin or binimetinib alone. Тwo factors were taken into account in the two-way ANOVA: factor I (caspase 3/7) and factor II (binimetinib/rapamycin/rapamycin+binimetinib). Statistically significant values of F and p derived from the two-way ANOVA analysis are shown in Table 6. All the p values of Duncan’s test are presented in the figure captions. Therefore, the induction of apoptosis is a significant contributor to the synergistic effect of rapamycin and binimetinib in melanoma cell survival.
Enhanced cytotoxic effect of mTOR and MEK/ERK inhibition-induced cell cycle arrest
We further analyzed cell cycle distribution to investigate the effects of combined mTOR and MEK signaling pathway inhibition therapy on cytotoxic mechanisms other than caspase-mediated apoptosis. Cells were treated with rapamycin (250 nM), binimetinib (2 μM), or both for 24 h. Cell cycle was analyzed by flow cytometry. Rapamycin-induced cell cycle arrest at the G0/G1 phase in Mel IL (64.2% vs 52.4%) and Mel IL/R (74.8% vs 63%) cells compared to control cells, but did not influence A375 (64.5 vs 61.7 in control). There were no changes in the G2/M phase arrest in these cells after rapamycin treatment. The binimetinib treatment led to a more significant increase in the G0/G1 population in A375 (80% vs 64.5%), Mel IL (82.3% vs 52.4%), and Mel IL/R (81.7% vs 63%). The inhibition of S-phase in drug combination groups has been demonstrated for all studied cell lines, and for Mel IL/R cells the difference was statistically significant in comparison to both binimetinib and rapamycin monotherapy. Combined rapamycin and binimetinib did not significantly induce cell cycle arrest compared to binimetinib alone but did lead to a more obvious reduction in the number of cells undergoing S phase (Fig. 7, Fig. S6). Тwo factors were taken into account in the two-way ANOVA: factor I rapamycin and factor II and the interaction between them (binimetinib/rapamycin/rapamycin+binimetinib). Statistically significant values of F and p derived from the two-way ANOVA analysis are shown in Table 7. All the p values of Duncan’s test are presented in the figure captions. Thus, binimetinib resulted in a significant increase in G0/G1 arrest and decreased S phase; however, rapamycin did not influence the cell cycle distribution in tandem with binimetinib.

Cell cycle distribution under rapamycin and binimetinib treatment. Mel IL, Mel IL/R, and A375 cells were treated with binimetinib (2 μM), rapamycin (250 nM), or their combination for 24 h followed by propidium iodide staining and flow cytometry analysis. The histogram represents the data from three independent experiments, CV 5%. The outcome of two-way ANOVA is shown in Table 7. Results are shown as the means ± SD. Duncan’s test: statistical significance is shown as ***p < 0.0001 compared to control
Discussion
The major factor limiting the efficacy of targeted therapies in melanoma patients is drug resistance. Acquired resistance to BRAFi often involves MAPK re-activation or activation of alternative signaling pathways such as PI3K/AKT/mTOR [3]. The MEK inhibitor trametinib showed minimal clinical activity in melanoma patients who had progressed on BRAFi therapy [20]. Another MEK inhibitor, binimetinib, resulted in significantly improved progression-free survival compared with dacarbazine or vemurafenib in NRAS-mutated melanoma, but it was less effective in patients with BRAF-mutated melanoma [9]. Moreover, resistance to both BRAF and MEK inhibitors develops in most patients and is a major challenge in melanoma clinical practice [27]. The inhibition of ERK relieves the ERK-dependent negative feedback, reactivates RAS and PI3K/AKT/mTOR signaling, and increases the survival of growth-arrested melanoma cells.
In many MAPK resistant models, compensatory PI3K survival networks are active and often induced in response to MAPK inhibition.
Recently, Kircher et al. demonstrated the role of PI3K/AKT pathway in melanoma initiation [28]. Furthermore, the PI3K/AKT/mTOR and MAPK pathways are often concurrently dysregulated in human tumors because growth factors can initiate both signaling pathways through RTKs [20]. These two important pathways also converge downstream to regulate cell survival, growth, and proliferation.
However, clinical trials with mTOR inhibitors in monotherapy in patients with melanoma have not been successful [29], our results revealed that a dual inhibition of two signaling pathways with rapamycin (mTOR) and binimetinib (MEK) in melanoma cell lines sensitive or resistant to BRAFi has greater therapeutic efficacy than monotherapy in vitro.
Recently, Kiessling et al. showed that the combination of mTOR and MEK inhibitors resulted in synergistic growth inhibition and induced apoptosis in NRAS mutant neuroblastoma [30]. The combination of BYL719 (PI3Kα inhibitor) and binimetinib (MEK inhibitor) was studied in patients with advanced solid tumors with RAS or BRAF mutations [31]. Previously we demonstrated that the combined metformin-binimetinib treatment had a synergistic potentiation of the anti-proliferative and pro-apoptotic activity in vitro in both 2D and 3D human melanoma cells [26]. The mechanism of metformin and binimetinib synergy was associated with p-AMPKα upregulation and p-ERK and p-mTOR downregulation.
The rapamycin and binimetinib combination synergistically reduced cell viability in A375, Mel IL, and resistant Mel IL/R cell lines compared to drugs alone. We demonstrated that the combined treatment reduced the ability of the cell to form colonies in a cell-dependent manner and capacity to invasion in Matrigel. The synergistic effects of both PI3K/AKT/mTOR and MAPK pathways inhibitors is shown in vitro and in vivo in multiple cancer types [32]. Accordingly to our data the combination of 250 nM rapamycin and 2 μM binimetinib resulted in moderate toxicity on fibroblasts, but keratinocytes were not sensitive to either drugs alone or their combo.
Next, we evaluated whether the enhanced cytotoxic effect of combined rapamycin and binimetinib treatment indeed suppressed activation of both signaling pathways PI3K/AKT/mTOR and MAPK. We analyzed the phosphorylated levels of AKT, mTOR, and ERK in Mel IL, Mel IL/R, and A375 cell lines by Western blot after 24 and 48 h treatment. Immunoblot analysis indicated that the synergistic action of rapamycin and binimetinib in melanoma cells was associated with molecularly targeted inhibition of their respective cell proliferation targets, i.e., mTOR and MEK. However, rapamycin alone failed to effectively suppress mTOR phosphorylation after 24 h. Several studies indicates that rapamycin could rapamycin may have independent of mTOR modification functions (e.g. through stabilization of the mitochondrial membrane with concomitant decreases in ROS production or a FKBP3/GCF2-dependent repression of TRIB3 promoter activity) [33, 34].
Binimetinib significantly inhibited ERK phosphorylation even after 24 h, and this inhibitory effect was more obvious by 48 h.
On the other hand, treatment with binimetinib increased activation of AKT, and the inhibition of AKT level in a combined rapamycin and binimetinib treatment group was cell-dependent. Several studies have also reported an increase in the activation of the PI3K/AKT signaling pathway with MEK inhibition [6, 35]. The PI3K/AKT signaling pathway is known to modulate cell survival in response to BRAFi [27], and mTOR plays an important role in modulating cellular response to PI3K/AKT and MAPK signaling. Rapamycin is known for induction of feedback activation of AKT through an IGF-1R-dependent mechanism in multiple cancer cells [36, 37]. However, this phenomenon was not observed in our study.
We showed that the synergistic inhibition of cell proliferation by combined mTOR and ERK inhibitors is mainly associated with the induction of caspase-mediated apoptosis from 24 to 48 h. Recently, similar results were obtained on colorectal cells [38]; the cell cycle distribution was also investigated in melanoma cells. Both rapamycin and binimetinib induced cell cycle arrest at the G0/G1 phase. The BRAFi-resistant Mel IL/R cells were more sensitive to rapamycin alone suggesting that PI3K/AKT/ mTOR pathway are upregulated in this cell line compared to BRAFi-sensitive Mel IL and A375. However, combined treatment with rapamycin and binimetinib did not significantly induce arrest in G0/G1 phase compared to binimetinib; however, the proportion of cells undergoing S phase dropped significantly.
Nevertheless, our study demonstrated that concurrent mTOR and MEK inhibition likely are effective in patients previously treated with a BRAFi. They could be effective against heterogeneous resistance mechanisms identified within BRAF mutant melanoma patients. Although MAPK signaling is critical for the proliferation of BRAFi-resistant melanoma cells [20], compensatory PI3K survival pathway are upregulated in many MAPK-resistant models. These often induce a response to MAPK inhibition. Several trials combining PI3K or AKT inhibitors and with BRAF and/or MEK inhibitors are underway [8, 39]. Thus, we suggest that dual rapamycin and binimetinib could be promising for further preclinical investigations.
Data availability
All data and results contained in this paper are available upon request.
References
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O'Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur G, BRIM-3 Study Group (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364:2507–2516. https://doi.org/10.1056/NEJMoa1103782
McCubrey JA, Steelman LS, Kempf CR et al (2011) Therapeutic resistance resulting from mutations in Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR signaling pathways. J Cell Physiol 226:2762–2781. https://doi.org/10.1002/jcp.22647
Calero R, Morchon E, Martinez-Argudo I, Serrano R (2017) Synergistic anti-tumor effect of 17AAG with the PI3K/mTOR inhibitor NVP-BEZ235 on human melanoma. Cancer Lett 406:1–11. https://doi.org/10.1016/j.canlet.2017.07.021
Chappell WH, Steelman LS, Long JM, et al (2011) Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget 2:135–164. https://doi.org/10.18632/oncotarget.240
Wang A-X, Qi X-Y (2013) Targeting RAS/RAF/MEK/ERK signaling in metastatic melanoma. IUBMB Life 65:748–758. https://doi.org/10.1002/iub.1193
Turke AB, Song Y, Costa C, Cook R, Arteaga CL, Asara JM, Engelman JA (2012) MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res 72:3228–3237. https://doi.org/10.1158/0008-5472.CAN-11-3747
Kim KB, Kefford R, Pavlick AC, Infante JR, Ribas A, Sosman JA, Fecher LA, Millward M, McArthur GA, Hwu P, Gonzalez R, Ott PA, Long GV, Gardner OS, Ouellet D, Xu Y, DeMarini DJ, le NT, Patel K, Lewis KD (2013) Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J Clin Oncol Off J Am Soc Clin Oncol 31:482–489. https://doi.org/10.1200/JCO.2012.43.5966
Ascierto PA, Schadendorf D, Berking C, Agarwala SS, van Herpen CML, Queirolo P, Blank CU, Hauschild A, Beck JT, St-Pierre A, Niazi F, Wandel S, Peters M, Zubel A, Dummer R (2013) MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol 14:249–256. https://doi.org/10.1016/S1470-2045(13)70024-X
Dummer R, Schadendorf D, Ascierto PA, Arance A, Dutriaux C, di Giacomo AM, Rutkowski P, del Vecchio M, Gutzmer R, Mandala M, Thomas L, Demidov L, Garbe C, Hogg D, Liszkay G, Queirolo P, Wasserman E, Ford J, Weill M, Sirulnik LA, Jehl V, Bozón V, Long GV, Flaherty K (2017) Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 18:435–445. https://doi.org/10.1016/S1470-2045(17)30180-8
Kopetz S, Desai J, Chan E, Hecht JR, O'Dwyer PJ, Maru D, Morris V, Janku F, Dasari A, Chung W, Issa JPJ, Gibbs P, James B, Powis G, Nolop KB, Bhattacharya S, Saltz L (2015) Phase II pilot study of Vemurafenib in patients with metastatic BRAF-mutated colorectal Cancer. J Clin Oncol 33:4032–4038. https://doi.org/10.1200/JCO.2015.63.2497
Matulonis U, Vergote I, Backes F, Martin LP, McMeekin S, Birrer M, Campana F, Xu Y, Egile C, Ghamande S (2015) Phase II study of the PI3K inhibitor pilaralisib (SAR245408; XL147) in patients with advanced or recurrent endometrial carcinoma. Gynecol Oncol 136:246–253. https://doi.org/10.1016/j.ygyno.2014.12.019
Jokinen E, Koivunen JP (2015) MEK and PI3K inhibition in solid tumors: rationale and evidence to date. Ther Adv Med Oncol 7:170–180. https://doi.org/10.1177/1758834015571111
Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, Papa A, Nardella C, Cantley LC, Baselga J, Pandolfi PP (2008) Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest 118:3065–3074. https://doi.org/10.1172/JCI34739
Sweetlove M, Wrightson E, Kolekar S, Rewcastle GW, Baguley BC, Shepherd PR, Jamieson SMF (2015) Inhibitors of pan-PI3K signaling synergize with BRAF or MEK inhibitors to prevent BRAF-mutant melanoma cell growth. Front Oncol 5. https://doi.org/10.3389/fonc.2015.00135
Rewcastle GW, Kolekar S, Buchanan CM, et al (2017) Biological characterization of SN32976, a selective inhibitor of PI3κ and mTOR with preferential activity to PI3κα, in comparison to established pan PI3κ inhibitors. Oncotarget. https://doi.org/10.18632/oncotarget.17730
Leung EY, Askarian-Amiri M, Finlay GJ, Rewcastle GW, Baguley BC (2015) Potentiation of growth inhibitory responses of the mtor inhibitor everolimus by dual mTORC1/2 inhibitors in cultured breast cancer cell lines. PLoS One 10. https://doi.org/10.1371/journal.pone.0131400
Seto B (2012) Rapamycin and mTOR: a serendipitous discovery and implications for breast cancer. Clin Transl Med 1:29. https://doi.org/10.1186/2001-1326-1-29
Weekes CD, Von Hoff DD, Adjei AA et al (2013) Multicenter phase I trial of the mitogen-activated protein kinase 1/2 inhibitor BAY 86-9766 in patients with advanced cancer. Clin Cancer Res An Off J Am Assoc Cancer Res 19:1232–1243. https://doi.org/10.1158/1078-0432.CCR-12-3529
Carlino MS, Gowrishankar K, Saunders CAB, Pupo GM, Snoyman S, Zhang XD, Saw R, Becker TM, Kefford RF, Long GV, Rizos H (2013) Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Mol Cancer Ther 12:1332–1342. https://doi.org/10.1158/1535-7163.MCT-13-0011
Carlino MS, Todd JR, Gowrishankar K, Mijatov B, Pupo GM, Fung C, Snoyman S, Hersey P, Long GV, Kefford RF, Rizos H (2014) Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Mol Oncol 8:544–554. https://doi.org/10.1016/j.molonc.2014.01.003
Egorov EE, Moldaver MV, Vishniakova KS et al (2007) Enhanced control of proliferation in telomerized cells. Ontogenez
Emelyanova M, Ghukasyan L, Abramov I, et al (2017) Detection of BRAF, NRAS, KIT, GNAQ, GNA11 and MAP2K1/2 mutations in Russian melanoma patients using LNA PCR clamp and biochip analysis. Oncotarget 8:52304–52320. https://doi.org/10.18632/oncotarget.17014
Chou TC, Talalay P (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzym Regul 22:27–55
Friedrich J, Seidel C, Ebner R, Kunz-Schughart LA (2009) Spheroid-based drug screen: considerations and practical approach. Nat Protoc 4:309–324. https://doi.org/10.1038/nprot.2008.226
Mikhaĭlova IN, Lukashina MI, Baryshnikov AI, et al (2005) [melanoma cell lines as the basis for antitumor vaccine preparation]. Vestn Ross Akad Meditsinskikh Nauk 37–40
Ryabaya O, Prokofieva A, Akasov R, Khochenkov D, Emelyanova M, Burov S, Markvicheva E, Inshakov A, Stepanova E (2019) Metformin increases antitumor activity of MEK inhibitor binimetinib in 2D and 3D models of human metastatic melanoma cells. Biomed Pharmacother 109:2548–2560. https://doi.org/10.1016/j.biopha.2018.11.109
Shi H, Kong X, Ribas A, Lo RS (2011) Combinatorial treatments that overcome PDGFRβ-driven resistance of melanoma cells to V600EB-RAF inhibition. Cancer Res 71:5067–5074. https://doi.org/10.1158/0008-5472.CAN-11-0140
Kircher DA, Silvis MR, Cho JH, Holmen SL (2016) Melanoma brain metastasis: mechanisms, models, and medicine. Int J Mol Sci 17. https://doi.org/10.3390/ijms17091468
Rao RD, Windschitl HE, Allred JB, Lowe VJ, Maples WJ, Gornet MK, Suman VJ, Creagan ET, Pitot HC, Markovic SN (2006) Phase II trial of the mTOR inhibitor everolimus (RAD-001) in metastatic melanoma. J Clin Oncol 24:8043–8043. https://doi.org/10.1200/jco.2006.24.18_suppl.8043
Kiessling MK, Curioni-Fontecedro A, Samaras P, Lang S, Scharl M, Aguzzi A, Oldrige DA, Maris JM, Rogler G (2016) Targeting the mTOR complex by Everolimus in NRAS mutant Neuroblastoma. PLoS One 11:e0147682. https://doi.org/10.1371/journal.pone.0147682
Juric D, Soria J-C, Sharma S, Banerji U, Azaro A, Desai J, Ringeisen FP, Kaag A, Radhakrishnan R, Hourcade-Potelleret F, Maacke H, Rodon Ahnert J (2014) A phase 1b dose-escalation study of BYL719 plus binimetinib (MEK162) in patients with selected advanced solid tumors. J Clin Oncol 32:9051–9051. https://doi.org/10.1200/jco.2014.32.15_suppl.9051
Haagensen EJ, Kyle S, Beale GS, Maxwell RJ, Newell DR (2012) The synergistic interaction of MEK and PI3K inhibitors is modulated by mTOR inhibition. Br J Cancer 106:1386–1394. https://doi.org/10.1038/bjc.2012.70
Zimmerman MA, Biggers CD, Li PA (2018) Rapamycin treatment increases hippocampal cell viability in an mTOR-independent manner during exposure to hypoxia mimetic, cobalt chloride. BMC Neurosci 19:82. https://doi.org/10.1186/s12868-018-0482-4
Stefanovska B, Vicier CE, Dayris T, Ogryzko V, Scott V, Bouakka I, Delaloge S, Rocca A, le Saux O, Trédan O, Bachelot T, André F, Fromigué O (2020) Rapalog-mediated repression of Tribbles Pseudokinase 3 regulates pre-mRNA splicing. Cancer Res 80:2190–2203. https://doi.org/10.1158/0008-5472.CAN-19-2366
Wu YL, Maachani UB, Schweitzer M, Singh R, Wang M, Chang R, Souweidane MM (2017) Dual inhibition of PI3K/AKT and MEK/ERK pathways induces synergistic antitumor effects in diffuse intrinsic Pontine Glioma cells. Transl Oncol 10:221–228. https://doi.org/10.1016/j.tranon.2016.12.008
Wan X, Harkavy B, Shen N, Grohar PHL (2007) Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 26:1932–1940. https://doi.org/10.1038/sj.onc.1209990
Shi Y (2005) Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptor/insulin receptor substrate-1/phosphatidylinositol 3-kinase cascade. Mol Cancer Ther 4:1533–1540. https://doi.org/10.1158/1535-7163.MCT-05-0068
Pitts TM, Newton TP, Bradshaw-Pierce EL, Addison R, Arcaroli JJ, Klauck PJ, Bagby SM, Hyatt SL, Purkey A, Tentler JJ, Tan AC, Messersmith WA, Eckhardt SG, Leong S (2014) Dual pharmacological targeting of the MAP kinase and PI3K/mTOR pathway in preclinical models of colorectal Cancer. PLoS One 9:e113037. https://doi.org/10.1371/journal.pone.0113037
Bedard PL, Tabernero J, Janku F, Wainberg ZA, Paz-Ares L, Vansteenkiste J, van Cutsem E, Pérez-García J, Stathis A, Britten CD, le N, Carter K, Demanse D, Csonka D, Peters M, Zubel A, Nauwelaerts H, Sessa C (2015) A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin Cancer Res An Off J Am Assoc Cancer Res 21:730–738. https://doi.org/10.1158/1078-0432.CCR-14-1814
Funding
This work was supported by the Russian Science Foundation under Grant 14–35–00107.
Author information
Authors and Affiliations
Contributions
OR, AR, AP, DK and AI performed experiments, acquired and analysed the data. OR and AP designed the study, OR and AR wrote the manuscript. NS revised the manuscript. IA and DK contributed to materials and tools. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
This article does not contain any studies with human participants or animals performed by any of the authors. The protocol to obtain Mel IL cells was initially approved by the N.N. Blokhin National Medical Research Center ethics committee in 1999 with written consent. Since then, the cell line was patented (#RU 2287577 C1).
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare that there are no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A.Supplementary Information
Supplementary file 1
CI index calculations at a consistent ratio of binimetinib (2 μM) and rapamycin (250 nM). The output of the Chou-Talalay method, including Dm, m, and r with CI at the ED50, ED75, and ED90 values. Supplementary file 2 The effect of rapamycin, binimetinib, and its combination on normal cell proliferation. Cell viability with rapamycin (250 nM) and binimetinib (2 μM) was determined in keratinocytes HaCaT and hFB-hTERT6 skin fibroblasts by MTT assay for 48 h. Data are expressed as mean ± SD of at least three independent experiments done in triplicate. One-way ANOVA followed by the post hoc Duncan’s test (hFB-hTERT6 F2,6 = 29.42, p = 0.0008; HaCaT F2,6 = 33.43, p = 0.0006). Supplementary data 3 Combined treatment with rapamycin and binimetinib decrease the expression of mTOR and MEK but not AKT. The unprocessed data for Figs. 3. Supplementary file 4 The enhancement of apoptosis by combined rapamycin and binimetinib in melanoma cells. Apoptosis was measured by annexin V/Propidium iodate staining in Mel IL, Mel IL/R and A375 cells after 24 h and 48 h treatment with rapamycin (250 nM), binimetinib (2 μM) or both. Values are percentage increase to untreated control. The data represents 3 individual experiments, p < 0.05 vs. Control. Supplementary file 5 The enhancement of apoptosis by combined rapamycin and binimetinib in melanoma cells (caspase 3/7 staining). Caspase 3 and caspase 7 activities were measured in Mel IL, Mel IL/R, and A375 cells with rapamycin (250 nM), binimetinib (2 μM), or their combination after 4 h treatment. Data are presented as mean amount of stained cells (%) to untreated control (non-stained cells). Left peak (light grey) means control stained cells to either caspase 3 or caspase 7, right peak (dark grey) means experimental group results for caspase 3 or caspase 7. Supplementary file 6 Cell cycle distribution under rapamycin and binimetinib treatment. Mel IL, Mel IL/R, and A375 cells were treated with binimetinib (2 μM), rapamycin (250 nM), or their combination for 24 h followed by propidium iodide staining and flow cytometry analysis. The histogram represents the data from three independent experiments, CV 5%. (PDF 1622 kb)
Rights and permissions
About this article

Cite this article
Ryabaya, O.O., Abramov, I.S., Khochenkov, D.A. et al. Rapamycin synergizes the cytotoxic effects of MEK inhibitor binimetinib and overcomes acquired resistance to therapy in melanoma cell lines in vitro. Invest New Drugs 39, 987–1000 (2021). https://doi.org/10.1007/s10637-021-01089-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-021-01089-3