
Summary
Despite the great advances in the understanding of the molecular basis of acute leukemia, very little of this knowledge has been translated into new therapies. Stathmin 1 (STMN1), a phosphoprotein that regulates microtubules dynamics, is highly expressed in acute leukemia cells and promotes cell cycle progression and proliferation. GDP366 has been described as a STMN1 and survivin inhibitor in solid tumors. This study identified structural GDP366 analogs and the cellular and molecular mechanisms underlying their suppressive effects on acute leukemia cellular models. STMN1 mRNA levels were higher in AML and ALL patients, independent of risk stratification (all p < 0.001). Cheminformatics analysis identified three structural GDP366 analogs, with AD80 more potent and effective than GSK2606414 and GW768505A. In acute leukemia cells, GDP366 and AD80 reduced cell viability and autonomous clonal growth in a dose- and/or time-dependent manner (p < 0.05) and induced apoptosis and cell cycle arrest (p < 0.05). At the molecular level, GDP366 and AD80 reduced Ki-67 (a proliferation marker) expression and S6 ribosomal protein (a PI3K/AKT/mTOR effector) phosphorylation, and induced PARP1 (an apoptosis marker) cleavage and γH2AX (a DNA damage marker) expression. GDP366 induced STMN1 phosphorylation and survivin expression, while AD80 reduced survivin and STMN1 expression. GDP366 and AD80 modulated 18 of the 84 cytoskeleton regulators-related genes. These results indicated that GDP366 and AD80 reduced the PI3K/STMN1 axis and had cytotoxic effects in acute leukemia cellular models. Our findings further highlight STMN1-mediated signaling as a putative anticancer target for acute leukemia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acute leukemia comprises a group of hematological neoplasms characterized by the expansion of blasts in the bone marrow and peripheral blood, mainly due to processes that impair normal hematopoiesis, such as dysregulation of cell proliferation, resistance to apoptosis, and loss of the ability to differentiate functional terminal cells [1]. Leukemia blasts can originate from a myeloid or lymphoid lineage, resulting in acute myeloid leukemia (AML) or acute lymphoblastic leukemia (ALL), respectively [2]. The mortality of adult acute leukemia patients is very high, making the identification of new molecular markers and signaling pathways imperative for defining prognosis, therapeutic response, and the development of new treatment options [1, 3, 4].
Recently, large-scale genomics studies have led to advances in the understanding of the molecular basis of acute leukemia, but very little of this new knowledge has been translated into novel therapies [5, 6]. In this respect, previous studies have shown that Stathmin 1 (STMN1) is highly expressed in hematological neoplasms, including AML and ALL [7, 8]. Further evidence supports a contribution of STMN1 to the malignant phenotype by the triggering of three important oncogenes involved in leukemogenesis: JAK2V617F, PML-RARα, and BCR-ABL1 [9,10,11]. The genetic inhibition of STMN1 by shRNA or antisense RNA has been shown to reduce cell proliferation and clonogenicity of AML and ALL cell lines [7, 12]. Notably, STMN1 expression is not detected in normal leukocytes [7, 8]. Taken together, these data suggest that STMN1 may be a selective target for therapy against acute leukemia.
STMN1 is a phosphoprotein that acts by sequestering dimers of α/β tubulin, either by reducing the polymerization of microtubules or by stimulating already formed microtubules to undergo catastrophe by promoting their destabilization [13]. These molecular events contribute to a malignant phenotype characterized by cell cycle progression and high proliferation rates [14]. STMN1 activity is regulated by phosphorylation at specific sites, with serine 16 and 63 being inhibitory sites [13].
Pharmacological inhibition of STMN1 remains a challenge, with only one potential selective inhibitor described thus far in the literature [15]. This compound, GDP366 (1-[4-(4-Aminoethylene[2,3-d]pyrimidine-5-yl)phenyl]-3-(3-methylphenyl)urea), has been described as a dual inhibitor of both STMN1 and BIRC5 (survivin), which are both interesting targets from the point of view of antineoplastic therapy [15]. For example, treatment of HCT116 colon cancer cells with GDP366 inhibited the gene and protein expression of both STMN1 and BIRC5, reduced clonogenicity, led to cell cycle arrest in the G2/M phase, promoted aneuploidy, and increased senescence [15]. The aim of the present study was to identify structural GDP366 analogs and the cellular and molecular mechanisms underlying their suppressive effects in acute leukemia cellular models.
Materials and methods
Gene expression data
STMN1 (probes 200783_s_at and 217714_s_at) mRNA expression data from samples of healthy donors (n = 17, lymphocytes [n = 12] and bone marrow cells [n = 5]), AML patients (favorable risk [n = 120], intermediate risk [n = 342], and adverse risk [n = 66]) [16], B-ALL patients (n = 82), and T-ALL patients (n = 54) were derived from a public access data portal of the Amazonia! database 2008 (http://amazonia.transcriptome.eu) [17]. Gene expression values were measured using Affymetrix HGU133 plus 2.0 arrays, and data sets were cross-referenced using tumor-specific identification numbers.
Identification of GDP366 structural analogs and chemicals
Cheminformatics analyses were based on the identification of compounds with a chemical structure similar to GDP366 using SMILES codes for compounds from the eMolecules database (www.emolecules.com). The protonation state was verified at pH 7.4 using fixpka software v. 1.7.0.2 (QUACPAC: OpenEye Scientific Software, Santa Fe, NM, USA). Conformations were calculated using the standard with OMEGA v. 3.0.0 [18], while the Tanimoto index was applied with the reference compound GDP366 structure based on the three-dimensional shape and color similarity scores in ROCS v. 3.2.2.2 (OpenEye Scientific Software, Santa Fe, NM, USA) [19]. GDP366 (CAS number 501698-03-9) was obtained from MCE (MedicalChemExpress, Monmouth Junction, NJ, USA). AD80 (CAS number 1384071-99-1) was obtained from TargetMol (Target Molecule Corp., Boston, MA, USA). GW768505A (CAS number 1337531-89-1) was obtained from AOBIOUS (AOBIOUS INC, Gloucester, MA, USA). GSK2606414 (CAS number 501693-25-0) was obtained from ApexBio (ApexBio Technology, Houston, TX, USA). All compounds were prepared as 25 mM stock solutions in dimethyl sulfoxide (Me2SO4; DMSO).
Cell culture
Jurkat, Namalwa, and U937 cells were obtained from ATCC (Philadelphia, PA, USA). NB4 cells were provided by Prof. Eduardo Magalhães Rego (Medical school, University of São Paulo). Cells were cultured in RPMI-1640 medium containing 10 % fetal bovine serum (FBS), glutamine, and penicillin/streptomycin, and maintained at 37 °C, 5 % CO2. Cells were authenticated by Short Tandem Repeat (STR) matching analysis using the PowerPlex® 16 HS system (Promega, Madison, WI, USA) and the ABI 3500 Sequence Detector System (Life Technologies, Foster City, CA, USA).
Cell viability assay
Cell viability was evaluated by the methylthiazoletetrazolium (MTT) assay. Briefly, 2 × 104 cells were seeded in a 96-well plate in RPMI-1640 medium with 10 % FBS in the presence of vehicle (Ø) or different concentrations of GDP366, AD80, GW768505A or GSK2606414 (0.0032, 0.016, 0.08, 0.4, 2, 10, and 50 µM) for 24, 48, and/or 72 h. Next, 10 µL MTT solution (5 mg/mL) was added and incubated at 37 °C, 5 % CO2 for 4 h. The reaction was stopped using 100 µL 0.1N HCl in anhydrous isopropanol. Cell viability was evaluated by measuring the absorbance at 570 nm. IC50 values were calculated using nonlinear regression analysis in GraphPad Prism 5 (GraphPad Software, Inc., San Diego, CA, USA).
Apoptosis assessment by annexin V staining
A total of 1 × 105 cells per well were seeded in a 24-well plate in RPMI-1640 medium with 10 % FBS in the presence of vehicle (Ø) or different concentrations of GDP366 (2, 10, and 50 µM) or AD80 (0.4, 2, and 10 µM) for 24 h. The cells were then washed with ice-cold phosphate buffered saline (PBS) and resuspended in a binding buffer containing 1 µg/mL 7AAD and 1 µg/mL fluorescein isothiocyanate (FITC)-labeled annexin V. All specimens were analyzed by flow cytometry (FACSCalibur; Becton Dickinson) after incubation for 15 min at room temperature in a light-protected area. Ten thousand events were acquired for each sample.
Colony formation assay
Colony formation assays were carried out in semisolid methylcellulose medium (2 × 103/mL; MethoCult 4230; StemCell Technologies Inc., Vancouver, BC, Canada) in the presence of vehicle (Ø), GDP366 (2, 10, and 50 µM), or AD80 (0.4, 2, and 10 µM). Colonies were detected after 10 days of culture by adding 150 µL (5 mg/mL) MTT reagent and scored using Image J quantification software (U.S. National Institutes of Health, Bethesda, MD, USA).
Cell cycle analysis
A total of 6 × 105 cells per well were seeded in 6-well plates in the presence of vehicle (Ø), GDP366 (2, 10, and 50 µM) or AD80 (0.4, 2, and 10 µM) for 24 h, fixed with 70 % ethanol and stored at 4 °C for at least 2 h. Fixed cells were stained with 20 µg/mL propidium iodide (PI) containing 10 µg/mL RNase A for 30 min at room temperature in a light protected area. DNA content distribution was acquired using cytometry (FACSCalibur) and analyzed using FlowJo software (Treestar, Inc.).
Proliferation assay using Ki-67 staining
A total of 2 × 105 cells/mL were treated vehicle (Ø), GDP366 (2, 10, and 50 µM) or AD80 (0.4, 2, and 10 µM) for 24 h. The cells were then resuspended in 70 % ethanol and stored at -20 °C. Cells were stained with anti-Ki-67 according to the manufacturer’s instructions (Ki-67 FITC clone B56; BD Bioscience, San Jose, CA, USA) and the mean fluorescence intensity was measured using flow cytometry (FACSCalibur). The IgG isotype was used as a negative control. Ten thousand events were acquired for each sample.
Western blot analysis
Jurkat and NB4 cells were treated with vehicle (Ø), GDP366 (2 and 10 µM), or AD80 (0.4 and 2 µM) for 24 h and submitted for total protein extraction using a buffer containing 100 mM Tris (pH 7.6), 1 % Triton X-100, 150 mM NaCl, 2 mM phenylmethylsulfonyl fluoride (PMSF), 10 mM Na3VO4, 100 mM NaF, 10 mM Na4P2O7, and 4 mM ethylenediaminetetraacetic acid (EDTA). Equal amounts of protein (30 µg) from total extracts were subjected to SDS-PAGE and Western blot analysis with the indicated antibodies and a SuperSignalTM West Dura Extended Duration Substrate System (Thermo Fisher Scientific, San Jose, CA, USA) and a G:BOX Chemi XX6 gel doc system (Syngene, Cambridge, UK). Antibodies directed against phosphorylated Stathmin1S16 (sc-12,948-R), Stathmin 1 (OP18) (sc-55,531), PARP1 (sc-56,197), and α-tubulin (sc-5286) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies directed against γH2AX (#9718), phosphorylated S6 ribosomal protein (RP)S235/236 (#4858), S6RP (#2217), and Survivin (BIRC5; #2808) were obtained from Cell Signaling Technology (Danvers, MA, USA).
PCR array
Total RNA from Jurkat cells treated with vehicle (Ø), GDP366 (10 µM), or AD80 (2 µM) for 24 h was obtained using TRIzol reagent (Thermo Fisher Scientific) from two independent experiments. The cDNA was synthesized from 1 µg RNA using the RT2 First Strand Kit (Qiagen Sciences Inc., Germantown, MD, USA). PCR array analysis was performed using a Human Cytoskeleton Regulators RT2 Profiler PCR Array kit (#PAHS-088ZC; Qiagen Sciences Inc.), according to the manufacturer’s instructions. Amplification was performed using a QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific). The mRNA levels were normalized to those in untreated cells, and genes that presented a ≥ 1.5-fold change in expression in any direction were included in the heatmap prepared using multiple experiment viewer (MeV) 4.9.0 software. Networks for modulated genes were constructed using the GeneMANIA database (https://genemania.org/) and the main biological interactions and processes were indicated.
Statistical analyses
Statistical analyses were performed using GraphPad Instat 5 (GraphPad Software, Inc., San. Diego, CA, USA). For comparisons, ANOVA and the Bonferroni post-test, Kruskal-Wallis test and Dunn’s post-hoc test, or Student t-test were used. A p-value < 0.05 was considered statistically significant.
Results
Stathmin 1 is highly expressed in a large cohort of acute leukemia patients
Previous studies by our research group [7, 8] and others [20, 21] reported high STMN1 expression in AML and ALL patients. The high STMN1 expression may confer selectivity for compounds that inhibit the signaling pathway mediated by this protein against leukemia cells; therefore, the expression of STMN1 was investigated in a large cohort of patients with acute leukemia (n = 664). STMN1 mRNA was higher in samples from AML patients, independently of risk stratification, compared to healthy donor samples (all p < 0.001). Similarly, B-ALL and T-ALL showed aberrant STMN1 expression compared to healthy donor samples (all p < 0.0001, Fig. 1a). According to the molecular phenotype, AML patients with normal karyotype, other karyotype alteration, t(15,17), t(8,21), inv(16), and − 5/-7q had higher STMN1 levels compared to the healthy donor group (p < 0.05). AML patients with t(15,17) or t(8,21) showed greater STMN1 expression compared to the normal karyotype, other karyotype alteration, complex karyotype, or + 8 AML patients (p < 0.05). All B-ALL subtypes displayed high STMN1 expression (all p < 0.05) compared to healthy donor samples. B-ALL with t(12,21) patients present higher STMN1 levels compared to B-ALL patients with MLL rearrangements (p < 0.05, Supplementary Fig. 1).

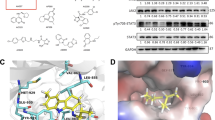
STMN1 is highly expressed in AML and ALL patient and cheminformatics analysis identifies GDP366 structural analogs. a STMN1 (200783_s_at and 217714_s_at) mRNA levels in samples from healthy donors (normal leukocytes, LN) and patients with acute myeloid leukemia (AML) (stratified into favorable, intermediate, and adverse groups according to European Leukemia Net [16]), B-acute lymphoblastic leukemia (ALL), and T-ALL. The “y” axis represents gene expression data obtained from the Amazonia! database 2008 (http://amazonia.transcriptome.eu), which were measured using Affymetrix HGU133 plus 2.0 arrays. Data sets were cross-referenced using tumor-specific identification numbers. The numbers of subjects for each group are indicated. ***p < 0.0001; Kruskal-Wallis test and Dunn’s post-hoc test. b The cheminformatics analysis based on the ligand-based similarity search to GDP366 identified three compounds: AD80 GSK2606414, and GW768505A. Chemical structure and Chemical Abstracts Service (CAS) numbers and ROCS setup for GDP366 are indicated. The superposition of GDP366 and AD80 are illustrated. c Dose- and time-response cytotoxicity analyzed using a methylthiazoletetrazolium (MTT) assay for ALL cell lines (Jurkat and Namalwa) and AML cell lines (NB4 and U937) treated with increased concentrations of GDP366, AD80, GSK2606414, or GW768505A (0.0032, 0.016, 0.08, 0.4, 2, 10, and 50 µM) for 72 h. IC50 values were calculated using nonlinear regression analysis. Values are expressed as the percentage of viable cells for each condition relative to vehicle-treated controls. IC50 values were calculated using nonlinear regression analysis
The cheminformatics analysis and cell‐based assays identify bioactive GDP366 structural analogs
Potential inhibitors of STMN1-mediated signaling were identified by cheminformatics analysis searching for compounds with structural similarity to the reference substance GDP366. One initial step was the identification of compounds similar to GDP366, based on its structural similarity using the similarity coefficient of 0.8 in the SciFinder® database (Chemical Abstract Services). The selected compounds from SciFinder® were analyzed together with the whole eMolecules database using the conformations calculated using the OMEGA classic setup. The lack of a three-dimensional structure for GDP366 complexed with a macromolecule led us to use the ligand-based drug design approach. Some models were obtained after the analysis of the similarity results from ROCS using one conformation of GDP366 as a reference to determine the shape (gray contour) and the features (shown as spheres) in Fig. 1b. GDP366 is similar to other known kinase inhibitors, so this information was then used to determine the weightings for the physicochemical properties (features) in the final model. In the actual model used, the maintenance of aromatic rings containing (green spheres indicating 3× and 5×), and a hydrogen bond donor (blue sphere, 5×) and an acceptor (red sphere, 5×) indicated in GDP366 (Fig. 1b) were overweighted in relation to the other features. The 100 compounds with the best ranking according to the combined Tanimoto index (shape and color) were visually analyzed to identify the key features and overall superposition to the reference substance (exemplified for AD80 in Fig. 1b). AD80 is a multikinase inhibitor with high similarity to GDP366, and this indirectly corroborates the initial hypothesis made for GDP366 as a kinase inhibitor [22]. Ultimately, in addition to GDP366, three other chemical structures were identified: AD80 (CAS number 1384071-99-1), GSK2606414 (337531-89-1), and GW768505A (CAS number 501693-25-0) (Fig. 1b).
The IC50 for each compound was determined using cell viability assays in acute leukemia cellular models: Jurkat (T-ALL), Namalwa (B-ALL), NB4 (AML), and U937 (AML). Among the tested compounds, AD80 was more potent and effective (IC50 ranged 1.1 to 6.3 µM) compared to GW768505A (IC50 ranged from 16 to > 50 µM), and GSK2606414 (IC50 ranged from 36 to > 50 µM). The IC50 for the reference compound, GDP366, ranged from 5.1 to > 50 µM. Based on these findings, we further investigated AD80 and GDP366 (Fig. 1c).
We also found that GDP366 and AD80 reduced cell viability in a dose- and time-dependent manner in all the leukemia cells evaluated, with AD80 more potent and effective than GDP366 (all p < 0.05; Fig. 2a-b).

GDP366 and AD80 reduce cell viability and induce apoptosis in a dose-dependent manner in acute leukemia cellular models. Dose- and time-response cytotoxicity analyzed using a methylthiazoletetrazolium (MTT) assay for Jurkat, Namalwa, NB4, and U937 cells treated with increased concentrations of (a) GDP366 (0.0032, 0.016, 0.08, 0.4, 2, 10, and 50 µM) or (b) AD80 (0.0032, 0.016, 0.08, 0.4, 2, 10, and 50 µM) for 24, 48, and 72 h. Values are expressed as the percentage of viable cells for each condition relative to vehicle-treated cells. Results are shown as mean ± SD of at least four independent experiments. The p values and cell lines are indicated in the graphs; * p < 0.05, ** p < 0.01, *** p < 0.0001; ANOVA and Bonferroni post-test. Apoptosis detected by flow cytometry in Jurkat, Namalwa, NB4, and U937 cells treated with (c) GDP366 (2, 10, and 50 µM) or (d) AD80 (0.4, 2, and 10 µM) for 24 h using an annexin V/7AAD staining method. Representative dot plots are shown for each condition. The upper and lower right quadrants (Q2 plus Q3) cumulatively contain the apoptotic population (annexin V + cells). Bar graphs representing the mean ± SD of at least four independent experiments quantifying apoptotic cell death. The p values and cell lines are indicated in the graphs; * p < 0.05, ** p < 0.01, *** p < 0.0001; ANOVA and Bonferroni post-test
GDP366 and AD80 induce apoptosis and reduce clonogenicity and cell cycle progression in acute leukemia cells
The cellular events involved in reduced cell viability triggered by GDP366 and AD80 were examined by investigating apoptosis, autonomous clonal growth, and cell cycle progression. GDP366 and AD80 caused time- and dose-dependent inductions of apoptosis (all p < 0.05, Fig. 2c-d) and reductions in autonomous clonal growth in Jurkat, Namalwa, NB4, and U937 cells (all p < 0.05, Fig. 3a-b), with Jurkat and NB4 cells more sensitive for both compounds. DNA content analysis corroborated the cytotoxic effects of GDP366 and AD80 at high concentrations, as observed by the accumulation of cells in the subG1 phase. Conversely, at lower concentrations of GDP366 (2 µM) and AD80 (0.4 µM), an increase was observed in cells in the G2/M phase, especially for the Jurkat, NB4, and U937 cells (Fig. 3c-f).

Autonomous clonal growth and cell cycle progression are inhibited by GDP366 and AD80 in acute leukemia cells. Colonies containing viable cells were detected by MTT assays after 10 days of culture of ALL or AML cells treated with (a) GDP366 (2, 10, and 50 µM) or (b) AD80 (0.4, 2, and 10 µM) for 10 days (long-term exposure). Colony images are shown for one experiment and the bar graphs show the mean ± SD of at least four independent experiments. * p < 0.05, ** p < 0.01, *** p < 0.0001; ANOVA and Bonferroni post-test. Cell cycle progression was determined in Jurkat, Namalwa, NB4, or U937 cells treated with (c-d) GDP366 (2, 10, and 50 µM) or (e-f) AD80 (0.4, 2, and 10 µM) for 24 h using a propidium iodide (PI) staining method. A representative histogram for each condition is shown. Bar graphs represent the mean ± SD of the percentage of cells in the subG1, G0/G1, S, and G2/M phases upon GDP366 or AD80 treatment of at least four independent experiments. The p values and cell lines are indicated in the graphs; * p < 0.05, ** p < 0.01, *** p < 0.001; ANOVA and Bonferroni post-test
GDP366 and AD80 inhibit the PI3K/STMN1 axis and modulate genes involved in cytoskeleton regulation
A more detailed analysis of the molecular scenario involved in the phenotypes observed after treatment with GDP366 and AD80 was conducted using Jurkat and NB4 cells. Flow cytometry analysis revealed that GDP366 and AD80 treatment resulted in a dose-dependent reduction of expression of Ki-67, a proliferation marker (all p < 0.05, Fig. 4a-b). In Jurkat and NB4 cells, GDP366 induced STMN1 phosphorylation at serine 16 (an inhibitory site), survivin expression, PARP1 cleavage (an apoptosis marker), and γH2AX (a DNA damage marker), while reducing phosphorylation of S6 ribosomal protein (RP) (a PI3K/AKT/mTOR effector) at serine 235/236 (Fig. 4c). AD80 also induced PARP1 cleavage and γH2AX, and reduced S6RP phosphorylation at serine 235/236, but it downregulated STMN1 and survivin expression (Fig. 4d).

Molecular characterization identifies the PI3K/STMN1 axis as a potential target of GDP366 and AD80 in Jurkat and NB4 cells. Ki-67 mean fluorescence intensity (M.F.I.) was determined by flow cytometry after incubation ALL (Jurkat) or AML (NB4) cellular models treated with (a) GDP366 (Ø, 2, 10, and 50 µM) or (b) AD80 (Ø, 0.4, 2, and 10 µM) for 24 h; histogram traces are illustrated. The bar graphs represent the Ki-67 M.F.I normalized to the respective vehicle-treated cells, and results are shown as mean ± SD of at least four independent experiments. The p values and cell lines are indicated in the graphs; ** p < 0.01, *** p < 0.0001; ANOVA and Bonferroni post-test. Western blot analysis for p-Stathmin1S16, Stathmin 1, Survivin, p-S6RPS235/236, S6RP, PARP1, and p-histone H2A.XS139 (γ-H2A.X) in total cell extracts from Jurkat and NB4 cells treated with increased concentrations of (c) GDP366 (vehicle, 2 and 10 µM) or (d) AD80 (vehicle, 0.4 and 2 µM) for 24 h. The membranes were re-probed with the antibody for the detection of the respective total proteins or α-tubulin and developed using a SuperSignal™ West Dura Extended Duration Substrate system and a G:BOX Chemi XX6 gel doc system. (e) Gene expression heatmap from qPCR array analysis of Jurkat cells treated with vehicle, GDP366 (10 µM), or AD80 (2 µM). The mRNA levels were normalized to those of vehicle-treated Jurkat cells and calculated as fold changes in expression; genes demonstrating ≥ 1.5-fold in either direction compared to vehicle-treated cells upon the treatment with GDP366 or AD80 are included in the heatmap. Two independent experiments for each condition were used for the analysis; green indicates repressed mRNA levels and red elevated mRNA levels. (f) Network analysis for genes modulated by GDP366 or AD80 constructed using the GeneMANIA database (https://genemania.org/). The upregulated and downregulated genes in the PCR array are illustrated as strikethrough circles, and the interacting genes included by the software modeling are indicated by strikethrough strands. The main interactions between genes are indicated by colored lines and the five main cellular processes are described in the Figure
A PCR array analysis was also conducted as an exploratory strategy for examining the potential effects of GDP366 and AD80 in cytoskeleton-related processes. A total of 18 of the 84 genes investigated were modulated after treatment, 5 were modulated by GPD366 (PHLDB2, CDC42EP2, FSCN2, ARHGAP6, and MAPT), 7 by AD80 (MAPK13, ARHGEF11, AURKB, PPP3CB, CDK5, CDC42EP3, and CTTN) and 6 by both drugs (AURKC, MAPRE2, BAIAP2, WASF1, CDC42BPA, and GSN) (Fig. 4e and Supplementary Table 1). Analysis using the GeneMANIA tool and the genes modulated in each treatment revealed involvement of the processes related to regulation and organization of the actin cytoskeleton for GDP366 and the processes related to the regulation of cytokinesis, organization of the actin cytoskeleton, and morphogenesis for AD80 (Fig. 4f).
Discussion
The potential pharmacological inhibitors for STMN1, an oncoprotein related to the malignant phenotype of ALL and AML [14], were investigated in the present study. Increased STMN1 expression has been confirmed in a large cohort of AML and ALL patients; however, the causes of this elevated expression in acute leukemia remains a topic of investigation. Previous studies have shown that STMN1 expression is a marker of proliferation in hematopoietic cells [7]. Two important leukemia-related oncogenes, PML-RARα and BCR-ABL1, were also able to induce STMN1 expression [10, 11]. Taken together, these findings suggest that the malignant transformation mediated by activation of signaling pathways that act under aberrant cell proliferation are capable of inducing STMN1 expression and, in turn, accelerating cell cycle progression and contributing to the leukemia phenotype.
Cheminformatics analysis was applied to identify compounds with structural similarity with GDP366, the only STMN1 inhibitor reported to date [15]. GDP366 has been described as capable of reducing STMN1 and BIRC5 mRNA levels, and both of these targets are of interest from point of view of antineoplastic therapy. In colon cancer models, GDP366 treatment induces mitotic catastrophe and senescence and reduces xenograft tumor growth [15]. In the present study, three compounds with structural similarities to GDP366 were selected for analysis in biological assays: AD80, GSK2606414, and GW768505A.
AD80 has been reported as a selective multikinase inhibitor for RET, RAF, SRC, and S6K [23]. In models of endocrine neoplasia, treatment with AD80 increases apoptosis [23]. In CCRF-CEM (T-ALL) and MOLT3 (AML) cells, both deficient for the PTEN protein and showing S6K1 hyperactivation, AD80 treatment reduces cell viability. AD80 treatment also reduces tumor formation and increases survival in animals with PTEN-/- lymphomas [24]. A recent report showed a decrease in G2/M-related genes following AD80 treatment of HUH7 cells [25]. These findings corroborate the antineoplastic effects observed in ALL and AML cellular models used in the present study.
GSK2606414 was first identified in a panel of 294 kinases as a potent inhibitor of KIT and PERK, and recent studies have demonstrated its ability to inhibit RIPK1, a kinase involved in TNF-α-mediated cell death [26]. KIT is a type III tyrosine kinase receptor expressed in several cell types, including hematopoietic progenitor cells, and is activated by stem cell factor (SCF) to trigger growth and development, cell survival, metabolism, and differentiation, by activating multiple signal transduction pathways [27]. PERK has been implicated in tumor development and resistance to chemotherapy, making it a target for anti-cancer treatments [26, 28]. Despite the relevant targets described for GSK2606414, its effects on cell viability in ALL and AML cellular models were less than desired for an antineoplastic drug. GW768505A has been reported as a potent inhibitor of the autophosphorylation of the TRKA, TRKB, and TRKC kinases in KM12 cells [29].
GDP366 and AD80 treatment decreased the levels of phosphorylated S6 ribosomal protein, an effector of the PI3K/AKT/mTOR signaling. A relationship between STMN1 expression and activation of this signaling cascade has been reported: PI3K inhibition by LY294002 or AKT inhibition by MK2206 reduces the expression of STMN1 [30, 31]. Similarly, the PI3K inhibitor, BKM120, induces STMN1 phosphorylation in ALL cellular models [32]. Our findings further reinforce the relationship between PI3K/AKT/mTOR and STMN1 in acute leukemia cells.
GDP366 and AD80 treatments appear to have different actions on STMN1: GDP366 increases the levels of STMN1 in its phosphorylated form (serine 16, an inhibitory site), while AD80 promotes a decrease in the expression of STMN1. From a functional point of view, both compounds would potentially inhibit the biological functions mediated by STMN1 [13].
An increase in the survivin expression, PARP1 cleavage, and γH2AX was observed after GDP366 treatment, whereas AD80 treatment decreased the expression of survivin and increased PARP1 cleavage and γH2AX expression. The survivin protein plays an important role in cell survival by inhibiting apoptosis, and its high expression has been associated with an aggressive cancer phenotype and poor clinical outcomes, while its expression is practically undetectable in normal tissues [33, 34]. In HCT116 cells, reduced survivin expression has been described following GDP366 treatment [15], but this response was not observed in the ALL and AML cell lines. This finding corroborates the lower potency and effectiveness of GDP366 in acute leukemia models.
The modulation of genes related to the regulation of the actin cytoskeleton, cytokinesis, and morphogenesis was highlighted after treatment with GDP366 and AD80 by PCR array and GeneMANIA analysis of processes related to the development and progression of cancer [35]. The CDK5 gene expression was reduced by AD80 treatment, whereas GSN gene expression was increased by both compounds in Jurkat cells. The CDK5 gene acts in the progression of the cell cycle and promotes proliferation [36], while high levels of the GSN gene have been associated with reduced proliferation, migration, and invasion in solid tumors [37, 38].
In summary, the therapeutic value of pharmacological inhibition of STMN1-mediated signaling has been investigated in AML and ALL cells. Our results indicated that GDP366 and AD80 reduce the activity of the PI3K/STMN1 axis. These compounds also show cytotoxic effects in acute leukemia cell models, with AD80 more potent and effective than GDP366 in the evaluated cellular and molecular events. Our findings further highlight STMN1-mediated signaling as a putative anticancer target for acute leukemia.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Dohner H, Weisdorf DJ, Bloomfield CD (2015) Acute myeloid leukemia. N Engl J Med 373:1136–1152
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW (2016) The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127:2391–2405
Terwilliger T, Abdul-Hay M (2017) Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J 7:e577
Malard F, Mohty M (2020) Acute lymphoblastic leukaemia. Lancet 395:1146–1162
Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ Jr, Laird PW, Baty JD, Fulton LL, Fulton R, Heath SE, Kalicki-Veizer J, Kandoth C, Klco JM, Koboldt DC, Kanchi KL, Kulkarni S, Lamprecht TL, Larson DE, Lin L, Lu C, McLellan MD, McMichael JF, Payton J, Schmidt H, Spencer DH, Tomasson MH, Wallis JW, Wartman LD, Watson MA, Welch J, Wendl MC, Ally A, Balasundaram M, Birol I, Butterfield Y, Chiu R, Chu A, Chuah E, Chun HJ, Corbett R, Dhalla N, Guin R, He A, Hirst C, Hirst M, Holt RA, Jones S, Karsan A, Lee D, Li HI, Marra MA, Mayo M, Moore RA, Mungall K, Parker J, Pleasance E, Plettner P, Schein J, Stoll D, Swanson L, Tam A, Thiessen N, Varhol R, Wye N, Zhao Y, Gabriel S, Getz G, Sougnez C, Zou L, Leiserson MD, Vandin F, Wu HT, Applebaum F, Baylin SB, Akbani R, Broom BM, Chen K, Motter TC, Nguyen K, Weinstein JN, Zhang N, Ferguson ML, Adams C, Black A, Bowen J, Gastier-Foster J, Grossman T, Lichtenberg T, Wise L, Davidsen T, Demchok JA, Shaw KR, Sheth M, Sofia HJ, Yang L, Downing JR, Eley G (2013) Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368:2059–2074
Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Bene MC, De Vos J, Hernandez JM, Hofmann WK, Mills KI, Gilkes A, Chiaretti S, Shurtleff SA, Kipps TJ, Rassenti LZ, Yeoh AE, Papenhausen PR, Liu WM, Williams PM, Foa R (2010) Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol 28:2529–2537
Machado-Neto JA, de Melo Campos P, Favaro P, Lazarini M, Lorand-Metze I, Costa FF, Olalla Saad ST, Traina F (2014) Stathmin 1 is involved in the highly proliferative phenotype of high-risk myelodysplastic syndromes and acute leukemia cells. Leuk Res 38:251–257
Machado-Neto JA, Rodrigues Alves APN, Fernandes JC, Coelho-Silva JL, Scopim-Ribeiro R, Fenerich BA, da Silva FB, Scheucher PS, Simoes BP, Rego EM, Traina F (2017) Paclitaxel induces Stathmin 1 phosphorylation, microtubule stability and apoptosis in acute lymphoblastic leukemia cells. Heliyon 3:e00405
Machado-Neto JA, de Melo Campos P, Favaro P, Lazarini M, da Silva Santos Duarte A, Lorand-Metze I, Costa FF, Saad ST, Traina F (2015) Stathmin 1 inhibition amplifies ruxolitinib-induced apoptosis in JAK2V617F cells. Oncotarget 6:29573–29584
Zada AA, Geletu MH, Pulikkan JA, Muller-Tidow C, Reddy VA, Christopeit M, Hiddemann WD, Behre HM, Tenen DG, Behre G (2006) Proteomic analysis of acute promyelocytic leukemia: PML-RARalpha leads to decreased phosphorylation of OP18 at serine 63. Proteomics 6:5705–5719
Unwin RD, Sternberg DW, Lu Y, Pierce A, Gilliland DG, Whetton AD (2005) Global effects of BCR/ABL and TEL/PDGFRbeta expression on the proteome and phosphoproteome: identification of the Rho pathway as a target of BCR/ABL. J Biol Chem 280:6316–6326
Jeha S, Luo XN, Beran M, Kantarjian H, Atweh GF (1996) Antisense RNA inhibition of phosphoprotein p18 expression abrogates the transformed phenotype of leukemic cells. Cancer Res 56:1445–1450
Belletti B, Baldassarre G (2011) Stathmin: a protein with many tasks. New biomarker and potential target in cancer. Expert Opin Ther Targets 15:1249–1266
Machado-Neto JA, Saad ST, Traina F (2014) Stathmin 1 in normal and malignant hematopoiesis. BMB Rep 47:660–665
Shi X, Wang D, Ding K, Lu Z, Jin Y, Zhang J, Pan J (2010) GDP366, a novel small molecule dual inhibitor of survivin and Op18, induces cell growth inhibition, cellular senescence and mitotic catastrophe in human cancer cells. Cancer Biol Ther 9:640–650
Dohner H, Estey EH, Amadori S, Appelbaum FR, Buchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA, Lo-Coco F, Naoe T, Niederwieser D, Ossenkoppele GJ, Sanz MA, Sierra J, Tallman MS, Lowenberg B, Bloomfield CD (2010) Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 115:453–474
Carrour TL, Assou S, Tondeur S, Lhermitte L, Lamb N, Reme T, Pantesco V, Hamamah S, Klein B, De Vos J (2010) Amazonia!: An online resource to google and visualize public human whole genome expression data. Open Biotechnol J 4:5–10
Hawkins PC, Skillman AG, Warren GL, Ellingson BA, Stahl MT (2010) Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J Chem Inf Model 50:572–584
Hawkins PC, Skillman AG, Nicholls A (2007) Comparison of shape-matching and docking as virtual screening tools. J Med Chem 50:74–82
Hanash SM, Strahler JR, Kuick R, Chu EH, Nichols D (1988) Identification of a polypeptide associated with the malignant phenotype in acute leukemia. J Biol Chem 263:12813–12815
Roos G, Brattsand G, Landberg G, Marklund U, Gullberg M (1993) Expression of oncoprotein 18 in human leukemias and lymphomas. Leukemia 7:1538–1546
Plenker D, Riedel M, Bragelmann J, Dammert MA, Chauhan R, Knowles PP, Lorenz C, Keul M, Buhrmann M, Pagel O, Tischler V, Scheel AH, Schutte D, Song Y, Stark J, Mrugalla F, Alber Y, Richters A, Engel J, Leenders F, Heuckmann JM, Wolf J, Diebold J, Pall G, Peifer M, Aerts M, Gevaert K, Zahedi RP, Buettner R, Shokat KM, McDonald NQ, Kast SM, Gautschi O, Thomas RK, Sos ML (2017) Drugging the catalytically inactive state of RET kinase in RET-rearranged tumors. Sci Transl Med 9:eaah6144
Dar AC, Das TK, Shokat KM, Cagan RL (2012) Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature 486:80–84
Liu H, Feng X, Ennis KN, Behrmann CA, Sarma P, Jiang TT, Kofuji S, Niu L, Stratton Y, Thomas HE, Yoon SO, Sasaki AT, Plas DR (2017) Pharmacologic targeting of S6K1 in PTEN-deficient neoplasia. Cell Rep 18:2088–2095
Yu JX, Craig AJ, Duffy ME, Villacorta-Martin C, Miguela V, Ruiz de Galarreta M, Scopton AP, Silber L, Maldonado AY, Rialdi A, Guccione E, Lujambio A, Villanueva A, Dar AC (2019) Phenotype-based screens with conformation-specific inhibitors reveal p38 gamma and delta as targets for HCC polypharmacology. Mol Cancer Ther 18:1506–1519
Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, Li WH, Heerding DA, Minthorn E, Mencken T, Atkins C, Liu Q, Rabindran S, Kumar R, Hong X, Goetz A, Stanley T, Taylor JD, Sigethy SD, Tomberlin GH, Hassell AM, Kahler KM, Shewchuk LM, Gampe RT (2012) Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem 55:7193–7207
Stankov K, Popovic S, Mikov M (2014) C-KIT signaling in cancer treatment. Curr Pharm Des 20:2849–2880
Mahameed M, Wilhelm T, Darawshi O, Obiedat A, Tommy WS, Chintha C, Schubert T, Samali A, Chevet E, Eriksson LA, Huber M, Tirosh B (2019) The unfolded protein response modulators GSK2606414 and KIRA6 are potent KIT inhibitors. Cell Death Dis 10:300
Elkins JM, Fedele V, Szklarz M, Abdul Azeez KR, Salah E, Mikolajczyk J, Romanov S, Sepetov N, Huang XP, Roth BL, Al Haj Zen A, Fourches D, Muratov E, Tropsha A, Morris J, Teicher BA, Kunkel M, Polley E, Lackey KE, Atkinson FL, Overington JP, Bamborough P, Muller S, Price DJ, Willson TM, Drewry DH, Knapp S, Zuercher WJ (2016) Comprehensive characterization of the Published Kinase Inhibitor Set. Nat Biotechnol 34:95–103
Jiang W, Huang S, Song L, Wang Z (2018) STMN1, a prognostic predictor of esophageal squamous cell carcinoma, is a marker of the activation of the PI3K pathway. Oncol Rep 39:834–842
Li M, Yang J, Zhou W, Ren Y, Wang X, Chen H, Zhang J, Chen J, Sun Y, Cui L, Liu X, Wang L, Wu C (2017) Activation of an AKT/FOXM1/STMN1 pathway drives resistance to tyrosine kinase inhibitors in lung cancer. Br J Cancer 117:974–983
Pereira JK, Machado-Neto JA, Lopes MR, Morini BC, Traina F, Costa FF, Saad ST, Favaro P (2015) Molecular effects of the phosphatidylinositol-3-kinase inhibitor NVP-BKM120 on T and B-cell acute lymphoblastic leukaemia. Eur J Cancer 51:2076–2085
Ambrosini G, Adida C, Altieri DC (1997) A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med 3:917–921
Altieri DC (2003) Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene 22:8581–8589
Ong MS, Deng S, Halim CE, Cai W, Tan TZ, Huang RY, Sethi G, Hooi SC, Kumar AP, Yap CT (2020) Cytoskeletal proteins in cancer and intracellular stress: a therapeutic perspective. Cancers (Basel) 12:238
Pozo K, Bibb JA (2016) The emerging role of Cdk5 in cancer. Trends Cancer 2:606–618
Li WX, Yang MX, Hong XQ, Dong TG, Yi T, Lin SL, Qin XY, Niu WX (2016) Overexpression of gelsolin reduces the proliferation and invasion of colon carcinoma cells. Mol Med Rep 14:3059–3065
Chen Z, Li K, Yin X, Li H, Li Y, Zhang Q, Wang H, Qiu Y (2019) Lower expression of gelsolin in colon cancer and its diagnostic value in colon cancer patients. J Cancer 10:1288–1296
Acknowledgements
The authors thank John De Vos and Tanguy Le Carrour for leading the initiative amazonia! (http://amazonia.transcriptome.eu), thereby providing a tool for analysis of genomic data.
Funding
This study was supported by grant #2019/23864-7, #2017/24993-0, #2018/19372-9, #2018/15904-6, and #2015/17177-6 from the São Paulo Research Foundation (FAPESP), and grant #402587/2016-2 form the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - Finance Code 001.
Author information
Authors and Affiliations
Contributions
J.A.E.G.C. designed, executed, and analyzed the experiments and prepared the manuscript. K.L. participated in experiments and analyzed, and prepared the manuscript. L.V.C.-L. provided inputs and participated in the interpretation of data, and edited the manuscript. A.L. designed and executed cheminformatics analyses, participated in the interpretation of data, and edited the manuscript. J.A.M.-N. supervised and participated in the overall design of the study, experiments, and analyzes. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Jorge Antonio Elias Godoy Carlos declares that he has no conflict of interest. Keli Lima declares that she has no conflict of interest. Leticia Veras Costa-Lotufo declares that she has no conflict of interest. Andrei Leitão declares that he has no conflict of interest. João Agostinho Machado-Neto declares that he has no conflict of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Code availability
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(PDF 369 kb)
Rights and permissions
About this article

Cite this article
Carlos, J.A.E.G., Lima, K., Costa-Lotufo, L.V. et al. AD80, a multikinase inhibitor, exhibits antineoplastic effects in acute leukemia cellular models targeting the PI3K/STMN1 axis. Invest New Drugs 39, 1139–1149 (2021). https://doi.org/10.1007/s10637-021-01066-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-021-01066-w