
Summary
R-etodolac is a novel pro-apoptotic agent with potential antitumor activity against B-cell chronic lymphocytic leukemia (B-CLL). This phase I clinical trial was conducted to determine the tolerability, safety, and maximum tolerated dose (MTD) of R-etodolac, administered orally twice a day (BID), in patients with B-CLL. Secondary objectives included evaluating clinical response, pharmacodynamic activity (reduction of lymphocytes), and pharmacokinetic (PK) profile. Forty-three patients were enrolled in the study. The most frequently reported adverse events were diarrhea, rash, pruritus, and headache. Increases in alanine aminotransferase (ALT) were also observed. Adverse events were generally mild and self-limiting, although in an apparent dose–response relationship, grade 2 and 3 gastrointestinal toxicities and grade 3 skin toxicities were reported with the highest dose regimens (1,800 and 2,400 mg BID). Hematologic toxicity was rare. The MTD was determined to be 1,200 mg BID. PK results indicated that oral absorption of R-etodolac was rapid (time to maximum concentration ranged from 2 to 4 h), and the half-life ranged from 5 to 7 h. The increase in maximum concentration, however, was not proportional to the increase in dose. R-etodolac significantly reduced absolute lymphocyte count (ALC) in B-CLL patients in a dose-dependent manner up to 1,800 mg BID and caused partial responses in 2 patients. Further study of R-etodolac as a possible new maintenance therapy or as a part of combination therapy of B-CLL appears warranted.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
B-cell chronic lymphocytic leukemia (B-CLL) is a slowly progressive disease, characterized by the accumulation of mature, malignant B lymphocytes in the bone marrow, blood, and lymphoid tissues. This incurable disease occurs primarily in elderly patients, with a median age at diagnosis of 65 years, as reviewed in Rozman and Montserrat [1] and Chiorazzi [2]. In advanced phases, B-CLL patients may experience severe morbidity such as transfusion-dependent anemia and thrombocytopenia, autoimmune complications, and immunosuppression leading to frequent infections [3, 4]. Median survival after diagnosis is between 1 and >10 years, according to risk stratification. Patients usually need many different and less toxic pharmacologic therapies over the complete disease course, and ultimately, most patients become refractory to conventional therapy.
The current standard of care includes treatment with alkylating agents, purine analogues, or both [5]. More recently, monoclonal antibody therapies have been entering mainstream treatment strategies. Allogeneic blood stem cell transplantation has also gained significant attention as a potential cure for B-CLL, although such a procedure is suitable only for a small proportion of patients due to associated high risk of complications. In general, current treatment regimens are not curative, and although they have been relatively successful at delaying disease progression [5– 9], a need remains for additional treatment options.
Nonsteroidal anti-inflammatory drugs (NSAIDs) are popular agents that are used worldwide because of their anti-inflammatory and pain relief effects. Recently, it was discovered that NSAIDs can exert potent antitumor activity by inhibiting cyclo-oxygenase 2 (COX-2) and through other COX-independent mechanisms [10–13]. Etodolac is an orally available, chiral NSAID that is currently marketed for the treatment of arthritis. Etodolac is unusual among most chiral NSAIDs in that the R- and S-enantiomers are not metabolically convertible. Also, the R-enantiomer is metabolized significantly more slowly than the S-enantiomer and thus can accumulate to up to 10-fold higher plasma levels [14, 15]. Only the S-enantiomer has COX-inhibitory activity [16], an activity common to many NSAIDs that is associated with serious, often dose-limiting, adverse effects, including gastric ulcers and renal dysfunction [17, 18].
It was serendipitously observed that etodolac lowered the malignant lymphocyte counts in a patient with B-CLL who was being treated for arthritis [19]. Further investigation revealed that this activity was unaffected by repeated or prolonged treatment; however, the antileukemic effect of 300 mg of etodolac twice daily (BID) was short-lived, with lymphocyte counts returning to pretreatment levels within 2 weeks of drug withdrawal. The same patient was treated with 14 different NSAIDs and 4 different COX-2 inhibitors. Lymphocyte counts were measured before and after treatment with each drug. Etodolac was unique in lowering lymphocyte counts, suggesting that the mechanism of etodolac’s antileukemic activity was not related to its anti-inflammatory or COX-two-inhibitory activities.
Additional in vitro studies have since shown that etodolac has tumor cell-specific antiproliferative activity on cultured tumor cells from patients with B-CLL and several other cancers, including lymphoma, myeloma, and prostate cancer [20– 25]. These and other preclinical studies have identified apoptosis as a primary mechanism of action for etodolac in B-CLL [25, 26]. Etodolac has been shown to induce apoptosis in different cancer cells through down-regulation of antiapoptotic proteins Bcl-2 and Mcl-1, activation of caspases, activation of PPARγ, suppression of cell adhesion molecules, and suppression of β-catenin and Wnt signaling [20, 22, 24, 26]. Both enantiomers of etodolac individually showed apoptotic activity against B-CLL cells that was comparable to that of the RS-etodolac mixture [25]. With these unique characteristics of antileukemic activity without COX-inhibitory activity, the R-enantiomer (R-etodolac) is an attractive candidate as a novel therapeutic agent for B-CLL.
The primary objectives of this phase I trial were to determine the tolerability, safety, and maximum tolerated dose (MTD) of orally administered R-etodolac in patients with B-CLL. Pharmacokinetics (PK), lymphocyte reduction pharmacodynamic activity, and clinical response were evaluated as secondary endpoints.
Design and methods
Patient selection
Male and female patients, aged 21 to 75 years, with clinically stable Binet stage B or C or progressive Binet stage A B-CLL were eligible for this study. B-cell CLL diagnosis was defined by standard clinical and immunophenotype criteria. After intensive discussion between the investigators, recruitment of Binet stage A patients was restricted to those individuals fulfilling progression criteria, because “nonprogressive” Binet stage A patients did not meet criteria for initiating treatment as generally accepted. Progressive disease was defined as: (1) worsening anemia or thrombocytopenia, defined as ≥25% decrease in ≤6 months; (2) splenomegaly >6 cm below the costal margin; (3) massive adenopathy >10 cm longest diameter; or (4) progressive lymphocytosis with >50% increase in 2 months or doubling time <6 months. Absolute lymphocyte count (ALC) ≥15 × 109/l and platelet count >75,000/μl were required for enrollment, and up to three prior antitumor treatment regimens were permitted. Other eligibility criteria included the following: Eastern Cooperative Oncology Group (ECOG) performance status of 0, 1, or 2; adequate liver function (aspartate aminotransferase [AST], alanine aminotransferase [ALT], alkaline phosphatase, γ-glutamyl transferase [GGT], lactate dehydrogenase [LDH], total bilirubin, and direct bilirubin no more than 1.5 times normal limits); and adequate renal function (blood urea nitrogen and serum creatinine within normal limits), including a calculated creatinine clearance of ≥40 ml/min.
Exclusion criteria included the following: active or progressive disease (for definition see above), Binet stage B or C, warranting cytoreductive therapy; evidence of central nervous system involvement; active autoimmune manifestation of the B-CLL; concurrent treatment with chemotherapy, corticosteroids, immunotherapy, aspirin, other NSAIDs, warfiarin, heparin, histamine H2 antagonists, proton-pump inhibitory drugs, or angiotensin-converting enzyme inhibitors; concurrent diagnosis of or previous treatment for other active malignancies within 5 years before start of the study; a history of excessive alcohol use or illicit drug use within 1 year before start of the study; use of any of the prohibited medications within specified washout times before start of the study or receipt of an investigational study drug within 30 days before study enrollment; active, clinically significant medical or psychiatric conditions; chronic immunosuppressive viral infections; any gastrointestinal (GI) bleeding, esophageal varices, gastric or duodenal ulcer, or bleeding dyscrasia within 2 years before start of the study; allergy to or GI intolerance of any NSAID; and recent GI surgery or medical condition that might affect absorption, metabolism, or distribution of R-etodolac. Pregnant or lactating patients were also excluded.
The protocol was approved by the Institutional Review Board/Ethics Committee for each investigational center. All patients provided signed informed consent, and the study was conducted in full accordance with good clinical practice (GCP) standards.
Treatment schema and trial design
R-etodolac was administered orally BID, at all dose levels, for 8 weeks. Gelatin-locking capsules containing 200 mg R-etodolac were used for the 600-, 800-, and 1,000-mg BID dosing regimens; and for doses exceeding 800 mg, 600 mg tablets were permitted to be substituted for the 200-mg capsules to aid patient compliance. Dose escalation included a stepwise approach across the 6 dosing regimens. Dosing assignments were sequential in the order in which they were received by a centralized Treatment Designation Center. The 600-mg BID regimen was selected as a conservative initial regimen based on more than 10 years of clinical experience with RS-etodolac (Lodine®) at doses up to 1,200 mg/day. The study protocol required enrollment to begin with the 600-mg BID regimen, and enrollment in the next-higher dosing regimen was contingent on 3 patients completing the first 4 weeks of treatment at the hitherto active dose without a hematologic or nonhematologic dose-limiting toxicity (DLT). A nonhematologic DLT, as defined by the National Cancer Institute (NCI) Cancer Therapy Evaluation Program’s Common Toxicity Criteria (CTC), was any grade 3 (or greater) toxicity or grade 2 toxicity that did not resolve to grade 1 within 1 week despite medical intervention. A hematologic DLT, as defined by the modified CTC criteria for hematologic malignancies, was defined as a grade 3 (or greater) toxicity or grade 2 toxicity that did not resolve to grade 1 within 1 week despite medical intervention. Adverse events classified as not related to study drug administration were not assigned a CTC grade.
If a DLT was observed in 1 patient during the first 4 weeks of treatment with any dosing regimen, dose escalation to the next cohort was prohibited until a minimum of 6 patients completed 4 weeks of treatment without a second occurrence of a DLT. Further dose escalation ceased if 2 patients experienced a DLT anytime during the 8-week treatment period. Per study protocol, the MTD was defined as the highest dose level that had no more than 1 reported DLT at completion of the 8-week treatment period.
Baseline and follow-up assessment procedures
The study was performed in an outpatient setting. Patients were screened prior to the first study visit, not to exceed 60 days from screening to first dosing. Screening consisted of ECOG performance assessment; physical examination including evaluation of lymph nodes and spleen; 12-lead ECG; vital sign measurements including blood samples for hematology, serum chemistry, serology and stool sample for occult blood test, and urine sample; drug screening; and pregnancy test if appropriate. These assessments were repeated at study visit 1. Patients were examined immediately before start of therapy and then at weekly intervals until end of study. Adverse events were assessed and graded at each visit. Tests conducted at weekly visits included complete blood counts, serum chemistries, urinalysis, stool analysis, and vital signs. Every other week, patients were given a complete physical examination, including spleen and lymph node assessment, and checked with a 12-lead electrocardiogram.
Response evaluation and pharmacodynamic activity
Clinical response was evaluated by the reduction in tumor burden using the Revised NCI-Working Group Response (WGR) criteria [27]. Patients who met the complete response (CR) criteria at the 8-week assessment were to have a bone marrow biopsy to confirm CR as specified by standard hospital procedure. Patients were evaluated at weekly intervals during the posttreatment follow-up period. If, at the end of the 4-week posttreatment follow-up period, patients preliminarily met the criteria for either CR or partial response (PR), they were required to continue an additional 4-week posttreatment assessment (two additional study visits 2 weeks apart).
Pharmacodynamic activity was determined by the percent change from baseline in the ALC and by flow cytometry for changes in B- and T-cell counts from baseline. The ALCs for patients treated with R-etodolac were recorded weekly over the course of treatment. Flow cytometry measurements were conducted at weeks 0 (baseline), 5, and 8.
Pharmacokinetic sampling and methods
Blood samples (each sample 7 ml volume) for PK analysis were obtained at week 8 before the first daily dose and at 0.5, 1, 2, 4, 8, and 12 h after the last dose. R-etodolac in plasma was analyzed using a validated method developed at MDS Pharma Services (Montreal, Quebec, Canada). The analytical range was 0.2 to 200.0 μg/ml. To verify whether there was in vivo isomeric conversion of R-etodolac to S-etodolac, plasma concentrations of S-etodolac were also assayed using a validated method developed at MDS Pharma Services. For each study participant who completed 8 weeks of treatment, the following PK parameters of R-etodolac in plasma were calculated using noncompartmental methods: area under the curve to the last quantifiable concentration (AUC0 − t ), maximum concentration (C max), time to maximum concentration (T max), and terminal elimination half-life (t 1/2).
Study endpoints
The primary objectives of this study were to determine the tolerability, safety, and MTD of R-etodolac when given in a multidose regimen to patients with clinically stable B-CLL. Secondary objectives included the following: (1) to evaluate clinical response as determined by the reduction in tumor burden using the revised NCI-WGR criteria; (2) to assess the pharmacodynamic activity (i.e., the percent change in lymphocyte count from baseline) of R-etodolac in patients with documented B-CLL who had received 8 weeks of R-etodolac therapy; and (3) to determine the steady-state PK profile of various R-etodolac dosing regimens in patients with B-CLL.
Statistical methods
All statistical tests were performed against a one-sided alternative hypothesis, with statistical significance defined as P < 0.05. All analyses and tabulations were performed using SAS® Version 8.2 on a PC platform. Demographic and background information was summarized by treatment group for all study participants and presented as means, ranges, and percentages where appropriate. Any finding indicative of significant differences in baseline characteristics between groups that might potentially affect the endpoint variables was accounted for by subsequent analysis of covariance (ANCOVA).
Sample-size calculations indicated that six patients would be adequate to assess the initial pharmacodynamic activity of R-etodolac when given in a multidose treatment regimen. For pharmacodynamic analyses, the percent change from baseline in lymphocyte count was calculated for each patient in each group, and descriptive statistical analysis was performed on each group. Statistical comparisons were used to compare the response in each of the four dose groups. Percent changes from baseline for each visit were compared by ANCOVA, with effects for treatment group and baseline lymphocyte count. Percentages of patients with 25 and 50% lymphocyte reduction were summarized and compared between treatment groups using the Fisher exact test. Reductions in lymphocyte count at nadir and days on treatment to nadir were summarized and compared between treatment groups for all patients and for responders (all patients with ≥25% reduction in lymphocyte count from baseline); analysis of variance (ANOVA) was used to detect differences in the means.
Parametric (normal-theory) general linear model procedures were used to analyze PK data. An ANOVA was performed on the PK parameters log-transformed (if appropriate) AUC and C max. Dose proportionality was evaluated using a regression approach on log-transformed parameters.
Results
Forty-three patients were enrolled in the following R-etodolac cohorts: 600 mg BID (n = 7), 800 mg BID (n = 6), 1,000 mg BID (n = 6), 1,200 mg BID (n = 8), 1,800 mg BID (n = 8), and 2,400 mg BID (n = 8). Patient demographics are shown in Table 1. At enrollment, 25.6% of patients overall were Binet Stage A-progressive, 58.1% were Binet B, and 16.3% were Binet C. The majority had an ECOG status of 0 (83.7%); all others had an ECOG status of 1. The majority of patients (60.5%) had received no prior CLL therapy, 27.9% had received 1 prior treatment, 9.3% had received 2 prior treatments, and 2.3% had received 3 prior treatments. Baseline characteristics were similar across treatment groups.
Thirty-eight (88%) patients completed 4 weeks of treatment, and 23 (54%) completed 8 weeks of treatment and 4 weeks of follow-up. Eleven patients (26%) discontinued the study because of adverse events (AEs). Data from all patients were included in the pharmacodynamic analyses, and data from the 23 patients who completed the 8-week treatment study were included in PK analyses.
Safety
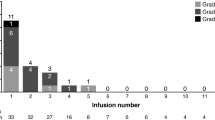
The most frequently reported AEs for all patients were gastrointestinal (GI) events, including loose stools or diarrhea (16 and 37% of patients, respectively), followed by abdominal pain not otherwise specified (NOS) (16%), nausea (14%), positive test for occult blood in stool (14%), and flatulence (12%) (Table 2). Frequency and severity of GI AEs increased with higher R-etodolac doses: only 16 of 47 (34%) GI AEs were reported in the first 4 dose cohorts, while 31 of 47 (65%) reported AEs were documented in the 2 highest (1,800- and 2,400-mg BID) cohorts. In particular, the incidence of diarrhea was highest at 1,800 mg BID (88%) and abdominal pain (NOS) at 2,400 mg BID (50%). Treatment-related GI events were generally mild and self-limiting in the 600- to 1,200-mg BID cohorts. Most GI AEs were CTC grade 1/2, and 1 case of diarrhea in the 2,400-mg BID cohort was reported as CTC grade 3.
Skin disorders were the second most frequently documented AE complex. Erythema, pruritus, and rash were reported in 14, 19, and 23% of patients, respectively. Skin disorders were in general mild and self-limiting in the 600- to 1,200-mg BID cohorts. One case each of grade 3 rash and pruritus was reported in the 1,800 and 2,400-mg BID cohorts, respectively (Table 3).
Headache was seen in 26% of all patients. Although this AE was found at all dose levels, headache was thought to be dose dependent because 7 of 13 events reported were seen in the 2 highest-dose (1,800 and 2,400 mg BID) cohorts. Notably, increased ALT levels were reported in 11 of 43 (26%) patients; however, a correlation with R-etodolac dosage was not identified (Table 2). Other occasionally occurring AEs were fever (12%), fatigue (14%), nasopharyngitis (12%), pain in the limb (7%), bilirubinuria (7%), and hypouricemia (7%). Lymph node pain was a remarkable but manageable AE seen in 12% of all patients, but only in the higher dose levels (1,000–2,400 mg BID). It was suspected that this AE reflects pharmacodynamic activity of R-etodolac.
Hematologic AEs were rare (Tables 2 and 3). One case of CTC grade 4 thrombocytopenia, most likely not related to R-etodolac treatment, was observed in a patient receiving the 800-mg BID regimen. Other observed grade 1 or grade 2 hematologic adverse events included anemia (at 600 mg and 800 mg BID), thrombocytopenia (at 2,400 mg BID) and aggravated thrombocytopenia (at 800 mg BID), and splenomegaly (at 1,800 mg BID). No signs of clinically significant T-cell suppression were seen. All hematologic AEs observed showed no obvious correlation to the R-etodolac dose administered.
Dose limiting toxicity
During the course of this R-etodolac study, 12 patients experienced 15 DLTs that were related to the investigational drug.
Eleven of these DLTs experienced by 9 patients met the per-protocol DLT grade criteria and are described as follows. In the 600-mg BID group, 1 patient had deep vein thrombosis (DVT) and pneumonia, and 1 patient had pruritic rash; both patients were treated and the events resolved. In the 1,200-mg BID group, 1 patient had increased ALT, and 1 patient had lymphadenopathy (lymph node pain) and fatigue. Lymphadenopathy was treated, and in both patients study medication was stopped and all events resolved. In the 1,800-mg BID group, two patients had events that met DLT grade criteria (grade 3): one patient had rash that resolved with treatment, and one patient had DVT that was treated and resolved. In the 2,400-mg BID group, one patient had pain that resolved after treatment and after study medication was stopped, one patient had diarrhea that resolved with treatment, and one patient had pruritus that resolved after study medication was stopped.
Additionally three patients experienced four events that did not meet the per-protocol DLT criteria but were nonetheless identified as DLTs by local investigators’ judgment: rash and limb pain (1,800 mg BID), erythema (1,800 mg BID), and gout (2,400 mg BID), all of which resolved either with treatment or after study medication was stopped.
Occurrence of three DLTs in the lowest treatment cohort (600 mg BID) was intensively discussed by the study investigators and sponsor because, per strict protocol, this would have constituted criteria to abort the study. Finally it was decided to continue the study because the relationship to R-etodolac was estimated to be weak, and DLTs did not reflect prior experiences with R-etodolac. In fact, DLTs were then not found in the next two treatment cohorts (800 and 1,000 mg BID) again. In the 1,200-mg BID cohort, three DLTs were reported, and four DLTs were reported in each of the 1,800 and 2,400-mg BID cohorts.
Reasons for study discontinuation
Eleven patients (26%) discontinued the study prematurely because of AEs, which included pruritic rash (600 mg BID); decreased weight, macular rash, oral candidiasis, vomiting, pneumonia, and DVT (600 mg BID); abnormal liver function tests (1,000 mg BID); hepatotoxicity (1,000 mg BID); increased ALT (1,200 mg BID); back pain, limb pain, and DVT (1,200 mg BID); abdominal pain (1,800 mg BID); arthralgia, rash, limb pain, and peripheral swelling (1,800 mg BID); erythema and pruritus (1,800 mg BID); pain and hypersensitivity (2,400 mg BID); and erythema and pruritus (2,400 mg BID). No clear correlation between discontinuations from the study and the R-etodolac dose was found. Some of these AEs were classified as SAEs (see next section).
Serious adverse events
Six patients experienced 7 serious adverse events (SAEs). One patient treated with the 600-mg BID regimen experienced both DVT of the leg and pneumonia. Other SAEs were Pneumocystis carinii pneumonia at 800 mg BID, hepatotoxicity at 1,000 mg BID, painful lymphadenopathy (that resolved with discontinuation of study drug) and DVT of the left leg at 1,200 mg BID, and pneumonia at 1,800 mg BID. All but the pneumonia at 1,800 mg BID were considered DLTs. The P. carinii pneumonia at 800 mg BID and the DVT at 1,200 mg BID were regarded as unrelated to R-etodolac treatment. Notably, no SAEs were observed among patients treated with the highest dose of 2,400 mg BID.
Maximum tolerated dose
Based strictly on protocol, the MTD could be defined as 1,000 mg BID, which is 1 dose level immediately below the first cohort (1,200 mg BID) with more than 1 DLT. This issue was discussed by the study investigators and sponsor, and it was found that two DLTs reported at 1,200 mg BID, increase of ALT and fatigue, were most likely non-dose-dependent AEs and consequently should not be used to define the MTD. In contrast, the 1,800- and 2,400-mg BID dose levels were associated with increasing numbers of DLTs and were found to be toxic. As a result, the 1,200-mg BID dose was defined as the MTD.
Pharmacokinetics of R-etodolac
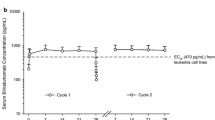
Blood samples for a PK assessment were provided during a 12-h visit at week 8 by the 23 patients who remained in the study. Mean PK profiles of R-etodolac following treatment at week 8 are depicted in Fig. 1. Mean PK plasma parameters are presented in Table 4. The AUC0 − t and C max of R-etodolac increased with dose (600–2,400 mg BID); however, these increases were not proportional to dose. The T max for all dose regimens was 2 to 4 h, and the mean t 1/2 of R-etodolac ranged from 5 to 7 h (data not available for the 600-mg BID regimen). All plasma S-etodolac concentrations were below the limit of quantification, with the exception of trace amounts observed at the 0.5- and 2-h plasma samples from 1 patient in the 1,200-mg BID cohort.

Mean pharmacokinetic profiles for R-etodolac twice-daily dosing regimens following 8 weeks of treatment
R-etodolac-mediated reduction of absolute lymphocyte counts
Statistically significant trends in decreased ALCs with increasing doses of R-etodolac were observed in terms of the number of patients achieving at least 50% reduction in ALC and the percentage reduction at nadir for all patients (Table 5). The magnitude of the mean decrease in ALC from baseline was greatest in the 1,800-mg BID cohort at weeks 2, 3, 7, and 8 (−28.1 to −46.0%) and in the 1,000-mg BID cohort at weeks 9, 10, and 12 (−36.0 to −42.6%); sample sizes in these groups were two to eight patients at each time point.
Overall, 26% of patients had at least a 50% reduction in ALC, and 58% of patients had at least a 25% reduction in ALC. The proportions of patients experiencing at least a 50 or 25% reduction in ALC at some point during treatment appeared to be dose dependent up to 1,800 mg BID (63 and 88%, respectively) and dropped with the 2,400-mg BID regimen (38 and 63%, respectively). Similarly, the percent reduction of ALC at nadir was dose dependent, both overall and specifically for responders, up to 1,800 mg BID (Fig. 2).

Mean percent reduction in absolute lymphocyte count (ALC) at nadir (BID, twice daily)
Clinical response
According to NCI criteria, two patients (5%) had a PR at the end of treatment: one patient in the 1,800-mg BID cohort and one patient in the 2,400-mg BID cohort. Neither of these responses, however, was maintained during the posttreatment follow-up period. Additionally, 32 patients (74%) had stable disease, and nine patients (21%) showed disease progression at treatment completion.
Discussion
This phase I study was conducted to determine the tolerability, safety, and MTD of R-etodolac in clinically stable B-CLL patients. Results suggest that R-etodolac has a generally acceptable safety profile with rare hematologic toxicities. Most AEs were mild to moderate and self-limiting (grade 1/2). The severity and frequency of AEs markedly increased above 1,200 mg BID, which was determined to be the MTD.
In this study, 11 patients discontinued treatment because of AEs, the most common of which were diarrhea, rash, pruritus, and headache. However, study discontinuation due to AEs was found through all dose levels and does not accurately reflect the safety profile of R-etodolac. The tolerability of R-etodolac can be more accurately estimated by the total number and severity of AEs recorded during the complete study than by the number of study discontinuations.
A large number of different AEs were recorded, many of which occurred at low frequency, and some had questionable relation to the study drug. Two symptom complexes clearly showed dose dependency: (1) GI events (diarrhea, abdominal pain, nausea, flatulence, and positive test for occult blood in stool); and (2) severe skin disorders (erythema, rash, and pruritus). Headache also appeared to be somewhat more common at the higher dose levels. These dose-dependent AEs guided determination of the MTD of R-etodolac in B-CLL patients (1,200 mg BID). Overall, this study showed that R-etodolac has an acceptable safety profile at doses up to 1,200 mg BID.
The occurrence of three incidences of DVT (one at 600 mg BID, one at 1,200 mg BID, one at 1,800 mg BID) was striking. DVTs lead to hospitalization and can cause serious harm. It could not be clearly concluded whether R-etodolac caused the DVTs or if they occurred coincidentally. However no preexisting coagulation abnormalities were reported for these patients. The possibility that COX-2 inhibition through the S-enantiomer of etodolac could be related to the observed DVTs was ruled out because the concentration of S-etodolac was below detection levels. Furthermore, although COX-2 inhibition has been associated with an increased risk of cardiovascular atherothrombotic events, which resulted in market withdrawal of NSAIDs such as rofecoxib and celecoxib [28], DVTs were not reported in large safety analyses of rofecoxib and celecoxib. Prospective monitoring of patients for DVTs in future investigations may shed more light on the relationship of this AE to R-etodolac. Evaluations of the clinical activity and the PK profile of R-etodolac in patients with B-CLL were secondary objectives of this study. The two transient PRs (5%) observed in this study occurred in the 1,800- and 2,400-mg BID cohorts. The 1,800-mg BID regimen also correlated with the greatest pharmacodynamic efficacy (25–50% reduction in ALC) of R-etodolac in the B-CLL patients studied. The antileukemic activity of R-etodolac did not increase with the subsequent higher dose regimen (2,400 mg BID). By comparison, the therapeutic dose of RS-etodolac, which is limited by AEs related to COX inhibition, is typically 600 to 1,000 mg daily. These findings show that R-etodolac can safely be administered at much higher doses than RS-etodolac. Notably, despite significant reductions in ALC in response to R-etodolac treatment, significant effects on lymph node status and spleen size were not observed, suggesting that R-etodolac may have a compartment-specific effect on the circulating B-CLL pool. Histological examination of lymph nodes during R-etodolac therapy may be of interest in future studies of R-etodolac in B-CLL patients.
Increased rates of infections were not an obvious problem during the study, since frequency and severity of the few cases of infections as recorded does not exceed the rate seen in other CLL populations for our feeling. However a single Pneumocystis carinii pneumonia is reported, which should be reason to keep an eye on opportunistic infections and impaired T cell function in future studies covering larger patient numbers.
The PK data analysis revealed a nonlinear relationship between dosing and AUC0 − t and C max parameters. The AUC0 − t and C max of R-etodolac following BID oral administration seemed to increase in a less-than-proportional manner after 8 weeks of treatment. These nonlinear PK data may partly explain the observation that increases in dose did not elicit proportional increases in pharmacodynamic activity. This finding differs from previously published PK results describing a linear increase in AUC with the RS-etodolac mixture in healthy volunteers [15]. The mean t 1/2 of R-etodolac ranged from 5 to 7 h after 8 weeks of treatment, similar to the previously observed 6- to 8-h elimination t 1/2 with each of the etodolac enantiomers [15]. Plasma concentrations of S-etodolac were not detectable, confirming that R-etodolac does not undergo racemic conversion to S-etodolac in patients with B-CLL.
Current treatment options for B-CLL offer patients hope of sustained response or stable disease; however, no treatment regimen currently offers a cure for this disease. In a retrospective comparison study, the historically used front-line treatment chlorambucil achieved ∼4% CR, whereas frontline fludarabine achieved 20% CR. The improved response rate with front-line fludarabine, however, failed to translate into a significant benefit in median overall survival over chlorambucil (66 vs 56 months), and fludarabine was associated with increased risk for infection and neutropenia [6]. Other studies have reported up to 63% CR with front-line fludarabine and prednisone, and 13 to 37% CR in previously treated B-CLL patients [7, 8]. Alemtuzumab (CD52 monoclonal antibody) induces an overall response rate of 33 to 50% in advanced-phase B-CLL patients [29], and recent clinical data show that the addition of alemtuzumab or rituximab monoclonal antibody therapies to alkylators and purine analogues in second-line or subsequent salvage therapy may provide additive or synergistic effects [30, 31].
An important finding of this study is that, unlike more aggressive current therapies, R-etodolac treatment up to 2,400 mg BID does not lead to hematologic toxicity. In addition, antileukemic activity of R-etodolac has been demonstrated in this study by significant reductions in ALC. Together with previous reports that etodolac activity is maintained even after multiple treatments [19], the results of this study may provide a rationale for further investigation of the efficacy of R-etodolac as maintenance therapy or in combination therapy strategies.
Other compounds in a family of R-etodolac analogues have shown antitumor activity, as well as synergy with standard cytotoxics, against B-CLL cells in preclinical studies [32, 33]. SDX-308 and SDX-309 in particular have greater potency than R-etodolac. Further clinical investigation of R-etodolac and these analogues in the treatment of B-CLL may be warranted.
References
Rozman C, Montserrat E (1995) Chronic lymphocytic leukemia. N Engl J Med 333:1052–1057
Chiorazzi N, Rai KR, Ferrarini M (2005) Chronic lymphocytic leukemia. N Engl J Med 352:804–815
Robertson TI (1990) Complications and causes of death in B cell chronic lymphocytic leukaemia: a long term study of 105 patients. Aust N Z J Med 20:44–50
Anaissie EJ, Kontoyiannis DP, O’Brien S, Kantarjian H, Robertson L, Lerner S, Keating MJ (1998) Infections in patients with chronic lymphocytic leukemia treated with fludarabine. Ann Intern Med 129:559–566
Zent CS, Kay NE (2004) Update on monoclonal antibody therapy in chronic lymphocytic leukemia. Clin Adv Hematol Oncol 2:107–113
Rai KR, Peterson BL, Appelbaum FR Kolitz J, Elias L, Shepherd L, Hines J, Threatte GA, Larson RA, Cheson BD, Schiffer CA (2000) Fludarabine compared with chlorambucil as primary therapy for chronic lymphocytic leukemia. N Engl J Med 343:1750–1757
O’Brien S, Kantarjian H, Beran M, Smith T, Koller C, Estey E, Robertson LE, Lerner S, Keating M (1993) Results of fludarabine and prednisone therapy in 264 patients with chronic lymphocytic leukemia with multivariate analysis-derived prognostic model for response to treatment. Blood 82:1695–1700
Wierda W, O’Brien S, Faderl S, Ferrajoli A, Wang X, Do KA, Garcia-Manero G, Thomas D, Cortes J, Ravandi-Kashani F, Giles F, Lerner S, Kantarjian H, Keating M (2006) A retrospective comparison of three sequential groups of patients with Recurrent/Refractory chronic lymphocytic leukemia treated with fludarabine-based regimens. Cancer 106:337–345
Keating MJ, Chiorazzi N, Messmer B, Damle RN, Allen SL, Rai KR, Ferrarini M, Kipps TJ (2003) Biology and treatment of chronic lymphocytic leukemia. Hematology (Am Soc Hematol Educ Program) 153–175
Bellosillo B, Pique M, Barragan M, Castano E, Villamor N, Colomer D, Montserrat E, Pons G, Gil J (1998) Aspirin and salicylate induce apoptosis and activation of caspases in B-cell chronic lymphocytic leukemia cells. Blood 92:1406–1414
Klampfer L, Cammenga J, Wisniewski HG, Nimer SD (1999) Sodium salicylate activates caspases and induces apoptosis of myeloid leukemia cell lines. Blood 93:2386–2394
Shiff SJ, Rigas B (1999) The role of cyclooxygenase inhibition in the antineoplastic effects of nonsteroidal antiinflammatory drugs (NSAIDs). J Exp Med 190:445–450
Pereg D, Lishner M (2005) Non-steroidal anti-inflammatory drugs for the prevention and treatment of cancer. J Intern Med 258:115–123
Brocks DR, Jamali F (1990) The pharmacokinetics of etodolac enantiomers in the rat. Lack of pharmacokinetic interaction between enantiomers. Drug Metab Dispos 18:471–475
Brocks DR, Jamali F (1994) Etodolac clinical pharmacokinetics. Clin Pharmacokinet 26:259–274
Demerson CA, Humber LG, Abraham NA, Schilling G, Martel RR, Pace-Asciak C (1983) Resolution of etodolac and antiinflammatory and prostaglandin synthetase inhibiting properties of the enantiomers. J Med Chem 26:1778–1780
Allison MC, Howatson AG, Torrance CJ, Lee FD, Russell RI (1992) Gastrointestinal damage associated with the use of nonsteroidal antiinflammatory drugs. N Engl J Med 327:749–754
Ahmad SR, Kortepeter C, Brinker A, Chen M, Beitz J (2002) Renal failure associated with the use of celecoxib and rofecoxib. Drug Saf 25:537–544
Nardella FA, LeFevre JA (2002) Enhanced clearance of leukemic lymphocytes in B-cell chronic lymphocytic leukemia with etodolac. Blood 99:2625–2626
Hedvat M, Jain A, Carson DA, Leoni LM, Huang G, Holden S, Lu D, Corr M, Fox W, Agus DB (2004) Inhibition of HER-kinase activation prevents ERK-mediated degradation of PPARgamma. Cancer Cell 5:565–574
Kolluri SK, Corr M, James SY, Bernasconi M, Lu D, Liu W, Cottam HB, Leoni LM, Carson DA, Zhang XK (2005) The R-enantiomer of the nonsteroidal antiinflammatory drug etodolac binds retinoid X receptor and induces tumor-selective apoptosis. Proc Natl Acad Sci U S A 102:2525–2530
Kobayashi M, Nakamura S, Shibata K, Sahara N, Shigeno K, Shinjo K, Naito K, Ohnishi K (2005) Etodolac inhibits EBER expression and induces Bcl-2-regulated apoptosis in Burkitt’s lymphoma cells. Eur J Haematol 75:212–220
Yasui H, Hideshima T, Hamasaki M, Roccaro AM, Shiraishi N, Kumar S, Tassone P, Ishitsuka K, Raje N, Tai YT, Podar K, Chauhan D, Leoni LM, Kanekal S, Elliott G, Munshi NC, Anderson KC (2005) SDX-101, the R-enantiomer of etodolac, induces cytotoxicity, overcomes drug resistance, and enhances the activity of dexamethasone in multiple myeloma. Blood 106:706–712
Nakamura S, Kobayashi M, Shibata K, Sahara N, Shigeno K, Shinjo K, Naito K, Hayashi H, Ohnishi K (2006) Etodolac induces apoptosis and inhibits cell adhesion to bone marrow stromal cells in human myeloma cells. Leuk Res 30:123–135
Adachi S, Leoni LM, Carson DA, Nakahata T (2004) Apoptosis induced by molecular targeting therapy in hematological malignancies. Acta Haematol 111:107–123
Lu D, Zhao Y, Tawatao R, Cottam HB, Sen M, Leoni LM, Kipps TJ, Corr M, Carson DA (2004) Activation of the Wnt signaling pathway in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 101:3118–3123
Cheson BD, Bennett JM, Grever M, Kay N, Keating MJ, O’Brien S, Rai KR (1996) National Cancer Institute-sponsored Working Group guidelines for chronic lymphocytic leukemia: revised guidelines for diagnosis and treatment. Blood 87:4990–4997
Drazen JM (2005) COX-2 inhibitors—a lesson in unexpected problems. N Eng J Med 352:1131–1132
Robak T (2006) New agents in chronic lymphocytic leukemia. Curr Treat Options Oncol 7:200–212
Elter T, Borchmann P, Schulz H, Reiser M, Trelle S, Schnell R, Jensen M, Staib P, Schinkothe T, Stutzer H, Rech J, Gramatzki M, Aulitzky W, Hasan I, Josting A, Hallek M, Engert A (2005) Fludarabine in combination with alemtuzumab is effective and feasible in patients with relapsed or refractory B-cell chronic lymphocytic leukemia: results of a phase II trial. J Clin Oncol 23:7024–7031
Wierda WG, Kipps TJ, Keating MJ (2005) Novel immune-based treatment strategies for chronic lymphocytic leukemia. J Clin Oncol 23:6325–6332
Lindhagen E, Rickardson L, Elliott G, Leoni L, Nygren P, Larsson R, Åleskog A (2007) Pharmacological profiling of novel non-COX-inhibiting indole-pyran analogues of etodolac reveals high solid tumour activity of SDX-308 in vitro. Invest New Drugs 25:297–303
Lindhagen E, Nissle S, Leoni L, Elliott G, Chao Q, Larsson R, Åleskog A (2006) R-etodolac (SDX-101) and the related indole–pyran analogues SDX-308 and SDX-309 potentiate the antileukemic activity of standard cytotoxic agents in primary chronic lymphocytic leukaemia cells. Cancer Chemother Pharmacol 60:545–553
Acknowledgements
We thank Jennifer Wright Oliver, MD, for assistance with data interpretation and editorial guidance. We thank Margret Platz, Achim Rothe, and Sven Trelle for indispensable help in patient management and data collection. We also acknowledge the literature research and editorial contributions of Bridget O’Keeffe, Ph.D., and Mona Lee, Ph.D., of Helix Medical Communications LLC in the development of this manuscript. Dr. Österborg received research support from Salmedix, Inc.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jensen, M., Engert, A., Weissinger, F. et al. Phase I study of a novel pro-apoptotic drug R-etodolac in patients with B-cell chronic lymphocytic leukemia. Invest New Drugs 26, 139–149 (2008). https://doi.org/10.1007/s10637-007-9106-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-007-9106-z