
Abstract
Background
Patients with irritable bowel syndrome (IBS) frequently have meal-related symptoms and can recognize specific trigger foods. Lactose intolerance is a well-established carbohydrate malabsorption syndrome that causes symptoms similar to IBS such as bloating, abdominal pain, and diarrhea. However, the prevalence of sucrase-isomaltase deficiency (SID) in this population is poorly defined. SID is a condition in which sucrase-isomaltase, an enzyme produced by brush border of small intestine to metabolize sucrose, is deficient. Just like lactase deficiency, SID causes symptoms of maldigestion syndromes including abdominal pain, bloating, gas, and diarrhea. In this study, we aim to determine the prevalence of SID in patients with presumed IBS-D/M and characterize its clinical presentation.
Methods
Patients with a presumed diagnosis of IBS-D/M based on symptoms of abdominal pain, diarrhea, and/or bloating who underwent esophagogastroduodenoscopy with duodenal biopsies and testing for disaccharidase deficiency were included. Patients with a history of inflammatory bowel disease, gastrointestinal malignancy, or celiac disease were excluded. Odds ratio was calculated for abdominal pain, diarrhea, and bloating in patients with versus without SID.
Results
A total of 31 patients with clinical suspicion for IBS-D/M were included with a median age of 46 years (IQR 30.5–60) and with 61% females. SID was present in 35% of patients. Among patients with SID, 63.6% had diarrhea, 45.4% had abdominal pain, and 36.4% had bloating. Patients with SID were less likely than controls to have abdominal pain (OR 0.16, 95% CI 0.03–0.81, p = 0.04) although no difference in diarrhea or bloating was found. Only two patients with SID underwent sucrose breath testing of which only one had a positive result. However, this patient also had a positive glucose breath test and may have had small intestinal bacterial overgrowth as a confounder.
Conclusion
SID was found in 35% of patients with presumed IBS-D/M and should be considered in the differential diagnosis of patients presenting with abdominal pain, diarrhea, or bloating. Further studies should better characterize the clinical features of SID and investigate the effects of dietary modification in this group of patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Irritable bowel syndrome (IBS) affects up to 11% of people throughout the world, with 30% of patients seeking medical attention for symptoms related to IBS [1]. There is significant morbidity associated with IBS, with loss of quality of life and missing workdays. IBS is a chronic functional gastrointestinal disorder of the gut–brain axis. With the Rome IV criteria, clinicians are able to diagnose IBS and its subtypes, including IBS-C (constipation predominant), IBS-D (diarrhea predominant), and IBS-M (mixed bowel habits) by clinical history. The pathophysiology of IBS remains poorly understood despite an increased recognition over the last few decades. Proposed etiologies include altered gut microbiota, visceral hypersensitivity, altered gastrointestinal motility, bacterial overgrowth, post-infectious changes, carbohydrate malabsorption, food sensitivity, and intestinal inflammation.
Patients with IBS frequently have meal-related symptoms and can recognize specific trigger foods. Lactose intolerance is a well-established carbohydrate malabsorption syndrome that causes similar symptoms to IBS such as bloating, abdominal pain, and diarrhea, and lactose avoidance is oftentimes a recommendation made in patients with IBS. Of note, prevalence of lactose intolerance among patients with IBS was noted to be 38%. A diet low in fermentable oligosaccharides, disaccharides, monosaccharides, and polyols (FODMAP) is also often recommended and improves symptoms of bloating and pain.
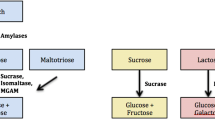
For absorption to occur, starches need to be hydrolyzed to monosaccharides as they are complex molecules of polysaccharides and oligosaccharides with long linear (α1–4) and branched (α1–6) chains of glucose molecules. Digestion of starch starts with salivary and pancreatic α-amylase followed by the α-glucosidases, such as maltase-glucoamylase and sucrase-isomaltase in the small intestine. Alpha-amylase is the primary enzyme cleaving large starches into medium chains, called α-limit dextrins. These α-limit dextrins are digested by α-glucosidase complexes such as sucrase-isomaltase and maltase-glucoamylase. These two complexes produce four maltase activities, all producing free glucose for mucosal absorption. Glucoamylase mainly cleaves medium linear chains (approximately 12–20 glucose molecules), whereas sucrase, maltase, and isomaltase cleave short linear chains and isomaltase (assayed with palatinase) cleaves virtually all α1–6 branched linkages [2].
Disaccharidase enzyme deficiency may induce symptoms of bloating, pain, and diarrhea. Sucrase-isomaltase (SI) is an intestinal brush border enzyme complex. The sucrase subunit hydrolyzes sucrose to fructose and glucose, while the isomaltase subunit hydrolyzes the α1–6 bond of α-limit dextrins to two glucose molecules [3]. Congenital sucrase-isomaltase deficiency (CSID) is a well-defined pediatric syndrome caused by mutations in the SI gene and inherited in an autosomal recessive fashion with more than 37 pathogenic mutations described [4]. Heterozygotes can be symptomatic [4], and heterozygosity can be diagnosed with breath test or disaccharide assay. It is also important to note that normal genetic test does not rule out a genetic CSID diagnosis since not all mutations have been identified. Patients with SID cannot hydrolyze sucrose which is allowed to reach the colon where bacterial fermentation leads to increased gas production and gastrointestinal symptoms. Symptoms of SID can range from mild to severe chronic watery acidic diarrhea, bloating, and abdominal pain. Recently, adult heterozygous carriers have been described who develop symptoms of CSID [5, 6], including diarrhea, abdominal pain, and bloating [7]. Typical diagnosis for SID includes chronic watery diarrhea with acidic pH, and small intestinal biopsy with disaccharidase level showing sucrase activity of less than 10% of control specimen (< 25 μ) [8]. Severity of symptoms is associated with residual sucrase-isomaltase enzyme activity, amount of sugar and starch consumption, and the extent of buffering by other foods in pediatric population [7]. However, in adult population, association of enzyme level and severity of symptoms are not well described.
The incidence of sucrase-isomaltase deficiency (SID) in the adult population remains ill-defined. Emerging evidence has correlated genetic polymorphisms in the SI complex with IBS-D/M in adult populations [9]. Garcia-Etxebarria et al. [10] recently reported increase in prevalence of SI genetic variants in IBS patients. Due to the significant overlap in symptoms of disaccharidase deficiency and IBS such as gas, bloating, abdominal pain, and diarrhea, it is important to consider this entity in individuals presenting with IBS-like symptoms.
The gold standard for diagnosing SID is duodenal biopsies for disaccharidase deficiency testing [11, 12]. Alternative modalities include sucrase breath testing and genetic testing for SI gene mutations. The American College of Gastroenterology Task Force recommends considering lactose breath testing in patients with presumed IBS, but there is no consensus regarding sucrase deficiency testing given the paucity of data in the adult population [13].
Methods
Consecutive patients undergoing esophagogastroduodenoscopy (EGD) for evaluation of chronic diarrhea and/or abdominal pain who were seen at the University of Miami between July 2016 and August 2017 were included (University of Miami Miller School of Medicine IRB20160876, Table 1). All patients were over the age of 18 years and had a presumed diagnosis of IBS-D/M based on Rome III criteria. At the time of the recruitment, patients with IBS-M had diarrhea-predominant phase. Patients with lactose intolerance on a strict lactose-free diet were also included. We excluded patients with a history of inflammatory bowel disease, gastrointestinal malignancy, or celiac disease, as well as patients with concurrent antibiotic use, including rifaximin use for treatment of IBS-D.
Patients underwent EGD with routine duodenal biopsies for testing for disaccharidase deficiency. Duodenal tissue samples were collected and homogenized. Substrate was added to the homogenate and incubated for 60 min at 37 °C (15 min for maltase); then, reactions were terminated by incubating at 100 °C for 5 min [14]. The liberated glucose was measured by reaction with 1.0 mL glucose oxidase reagent. After 20 min at room temperature, the reaction mixture was read at 505 nm with a reference reading at 655 nm [14]. The liberated glucose concentration was then determined using a glucose standard. The final enzyme activity was calculated by subtracting a blank sample in which the homogenates were incubated at 100 °C to inactivate the enzymes before the addition of substrate [14]. Duplicates were performed for each sample, including two assays and two blanks and standards. Regardless of the enzymes tested, depending on the substrate added in the assay, the free glucose generated in the assay indicated the activity of the enzymes. Our patients were tested for lactase, sucrase, palatinases, and maltases. Sucrase level of < 25 μ/g is considered to meet the criteria for sucrase deficiency (Table 2, Normal disaccharidase values).
Histology can be used as a tool to approximate a likely etiology of genetic sucrase deficiency. Normal lactase levels can also be used, but a low lactase should not rule out CSID due to high frequency of adult-onset hypolactasia which varies by race or ethnicity. An abnormal histology with abnormal sucrase has historically been interpreted as a secondary (non-genetic) deficiency, but this theory has not been confirmed with genetic testing.
To compare the accuracy of three different methods to diagnose SID, we performed genetic testing and sucrose breath testing on patients who were diagnosed with SID by duodenal biopsy on EGD. Genetic testing was performed by sequencing the SI gene from samples obtained from buccal swab. Genetic testing was done at LabCorp, using Snapshot chip array that will identify certain known SNPs in the SI gene. The presence of any one of the 37 known pathogenic mutations (Table 3) in the SI gene associated with SID regardless of genotype could confirm the genetic diagnosis. Even heterozygotes can be symptomatic [4]. SID heterozygotes should be confirmed with a function assay (breath test or disaccharidase assay). However, a normal genetic test does not rule out a genetic CSID diagnosis since not all mutations have been characterized.
Our definition was based on the North American Consensus for definition of small bowel bacterial overgrowth syndrome based on glucose breath testing. This consensus defines an abnormal hydrogen breath test as a rise of breath hydrogen of 20 ppm or more above baseline after consuming 50 g of sucrose or 75 g of glucose within 3 h, or a rise of methane of 12 ppm or both hydrogen and methane of > 15 ppm [15].
Statistical analysis was performed using R version 3.4.2 (R Foundation for Statistical Computing, Vienna, Austria). Proportions were compared using Pearson’s Chi-squared test or Fisher’s exact tests where appropriate.
Results
A total of 31 patients were enrolled, age range 18–82 years of age, mean age 46 years (females = 19). All patients had clinical suspicion for IBS-D/M based on meeting the Rome III criteria and exclusion of other potential etiologies. Celiac disease was ruled out in all patients by serologic testing (n = 8) and/or duodenal biopsies (n = 28).
Among patients who tested positive for SID (n = 11), 63.6% had diarrhea (n = 7), 45.4% had abdominal pain (n = 5), and 36.3% reported bloating (n = 4). When comparing patients with SID versus those with normal sucrase activity, those with SID had less abdominal pain with an odds ratio (OR) of 0.16 (95% CI 0.03–0.81, p = 0.0377). We found no differences for diarrhea (OR 2.54, 95% CI 0.57–12.00, p = 0.2734) or bloating (OR 0.39, 95% CI 0.08–1.74, p = 0.2734) in patients with or without SID (Table 4).
However, diarrhea has been commonly reported in children with SID.
Only two patients with SID underwent SBT, one of which had a positive sucrose breath test. However, this same only positive case also had a positive glucose breath test, suggesting that this patient could have had small intestinal bacterial overgrowth as a confounder for the positive SBT. Another patient with a positive SBT had normal enzymatic activity on duodenal biopsies. Given the small sample size, no firm conclusions about the accuracy of SBT in SID could be made.
Only two patients with IBS-M were included of which only one had SID by duodenal biopsy disaccharidase. However, given the small sample size, no firm conclusions could be made regarding the utility of SID testing in IBS-M.
None of the genetic testing among 11 patients was abnormal or heterozygous. Once they have met the criteria for SID with levels < 25 μ, there was no correlation between severity of symptoms and sucrase level.
Based on disaccharidase testing (see Table 5), we found SID in 11 patients (35%) undergoing EGD for presumed IBS-D/M. Interestingly, all of these patients had concomitant lactase deficiency by duodenal biopsies. In the group of patients without SID, 12 patients (60%) were identified who had lactase deficiency. Of interest, symptoms of diarrhea, abdominal pain, and bloating were similar between patients with and without lactase or sucrase deficiency (Fig. 1).

Flow diagram—patients with SID diagnosed with duodenal biopsy, subsequent test results including celiac panel (biopsy/serology), sucrose breath test, and genetic testing
Discussion
Our results demonstrated that about one-third of patients with a presumed diagnosis of IBS-D/M had SID diagnosed by disaccharidase testing on duodenal biopsies. Sucrose maldigestion may contribute to symptoms in patients with presumed IBS and often coexists with lactose intolerance. Our study suggests that abdominal pain is seen less commonly in IBS patients with SID compared to those without SID; however, abdominal pain is still present in 45.4% of patients with SID and does not exclude this entity. We do not believe that all SID patients have less abdominal pain than the non-SID population, and it is perplexing why our patients with SID had less bloating although all patients were found to be lactose intolerant, and therefore, perhaps the degree of lactose deficiency may have been the deciding factor for this discrepancy in results. In patients with IBS-D/M or lactose intolerance with refractory symptoms, SID should be considered in the differential diagnosis. Interestingly, the finding of lactase deficiency on duodenal biopsies did not correlate with symptoms of abdominal pain, diarrhea, or bloating, although our sample size was small (n = 11). Diarrhea and bloating did not distinguish between IBS patients with and without SID, although this study may have been underpowered to find a statistically significant difference.
Two distinct populations have been found to have SID. Patients with primary (hereditary) SID are heterozygous for SI mutations and are not discovered until adult age. It has been estimated that 2–9% of Americans of European descent may be affected [16, 17], suggesting that hereditary SID is greatly underdiagnosed in the adult population. Unfortunately, our study did not have many subjects that underwent genetic testing for us to make any conclusions. Conversely, secondary or acquired SID is more common than the genetic form and is secondary to duodenal villous atrophy and/or inflammation. Potential causes of secondary/acquired SID include celiac disease, immunodeficiency [18], malnutrition, SIBO, and infections. This form of SID is reversible with resolution of the underlying etiology. Genetic testing was done at LapCorp, using Snapshot chip array that will only identify certain known SNPs in the SI gene. We did not use lactase persistency gene testing for our genetic testing since abnormal lactase does not always translate to a secondary disaccharidase deficiency in adult patients due to high frequency of adult-onset hypolactasia. However, this would be a future direction for further research.
SID has been reported in several pediatric studies. For example, Nichols et al. performed a retrospective study involving 27,875 duodenal biopsies in a pediatric population with a mean age of 11 years over a 6-year period. They reported an overall prevalence of SID in 9.3% of patients and the majority were thought to be acquired SID. Unfortunately, no clinical or endoscopic correlation was reported [19]. In another retrospective study, 963 duodenal biopsies with disaccharidase testing were performed among 5368 patients in another pediatric population. SID was diagnosed in 7.6%, and these patients presented with symptoms of abdominal pain (85%), diarrhea (53%), constipation (26%), nausea (29%), and flatulence (9%) [20]. This is in contrast to our adult population where abdominal pain was present in 45.4%. This may be due to differences in clinical presentation of SID between adults and children, different underlying etiologies for secondary SID, and the possibility that children may describe bloating as pain.
Daileda et al. performed a systemic review evaluating the histopathological correlation of disaccharidase deficiency in pediatric populations. They included 29 studies which reported duodenal inflammatory changes in 6–24% and villous atrophy in 8.7–100%. Of note, four studies reported no histopathological findings [21,22,23,24]. The wide variation in histological findings is due to the heterogeneity of the included patients [25]. The overall prevalence of sucrase deficiency in this systematic review was 9.0%, similar to that reported by Nichols et al. The higher prevalence of SID in our study likely reflects a referral bias in the setting of a tertiary hospital with a motility clinic. It is likely that in patients with histological changes, the presence of SID may be acquired in the context of a nonspecific loss of several disaccharidases. However, in patients without histopathological changes, the loss of disaccharidases may represent a more subtle injury to the brush border. Importantly, the lack of histopathological changes does not rule out a disaccharidase deficiency. In clinical practice, this subgroup of patients may be misclassified as having IBS in the absence of disaccharidase testing.
Another common disease to be evaluated with histopathology in duodenum is celiac disease. Approximately 10% of duodenal biopsies of patients with celiac disease meet criteria for Marsh I or II, which by definition, having intact villi with inflammation with or without crypt hyperplasia [26]. It has also been described that patients with celiac disease with normal villi have a higher prevalence of disaccharidase deficiencies [26]. Whether persistent symptoms in patients with celiac disease may be due to concurrent SI is currently an unexplored area of research. Only 8 of the 31 patients had serology done for ruling out celiac disease. The other 28/31 patients had negative duodenal biopsies performed, while 6 of those also had serologic testing that was negative.
In our study, about one-third of patients had lactase deficiency, a prevalence similar to prior studies [19, 25]. Interestingly, the prevalence of SID in our patient cohort was higher than expected (35% compared to approximately 10% in prior studies). This may be due to a selection and referral bias as we included patients with IBS undergoing EGD in a tertiary referral motility center.
Interestingly, two of our patients had combined lactase, sucrase, maltase, and palatinase deficiency (pan-disaccharidase deficiency). We suspect that these patients had acquired SID in the setting of a nonspecific injury to the mucosa. Our histological data showed nonspecific changes without clinical significance, and this remains to be our limitation of the study due to small sample size (Fig. 2). None of these patients had SI gene mutations.

Nonspecific mucosal injury noted in duodenal biopsy of a patient with pan-disaccharidase deficiency patient (high power field)
The finding of coexisting lactase deficiency in patients with SID was not surprising given the high prevalence of lactase deficiency in patients with IBS-D. Lactase deficiency is the most common disaccharidase deficiency followed by pan-disaccharidase deficiency [19]. The coexistence of lactase deficiency in all of our SID patients and the absence of palatinase (isomaltase) deficiency (Table 5) argue for a nonspecific mucosal brush border injury in a subgroup of patients with IBS leading to sucrose and/or lactose intolerance due to nonspecific chronic inflammation. The extent to which patients who are heterozygous carriers of SI gene mutations remains largely unknown.
In our small pilot study, we showed that SID is found in a subset of patients with IBS-D and IBS-M (when diarrheal predominant at the time of the encounter) and may be considered if an upper endoscopy is being performed. Diarrheal symptoms did not correlate with the level of sucrase in our patients. However, the clinical significance of this finding is yet to be determined. In patients with IBS-D, it is unknown to what extent sucrase deficiency is a pathogenic mechanism as opposed to an epiphenomenon in the setting of a nonspecific mucosal injury. None of our patients were heterozygote/homozygote for sucrase-isomaltase deficiency; therefore at this time, we are unable to correlate any genetic results with sucrase mucosal activity. Only one patient with sucrase-isomaltase deficiency had histological finding with blunted villi with sucrase level of 19.8. This particular patient did not have celiac disease with negative serology and was not heterozygous for the SI gene (Fig. 3). The prevalence and clinical significance of heterozygous SI gene mutations in patients with presumed IBS-D also remain to be determined. Future studies should aim to better characterize the prevalence and role of SID in patients with IBS-D. The possibility of heterozygous SI gene mutations predisposing to acquired SID should be explored as should the role of medications or substances which cause intestinal damage such as non-steroidal medications or alcohol. Finally, studies investigating the effects of dietary modification and/or digestive enzyme supplementation in this subgroup of patients are warranted.

Patients with SID with focal mild villous blunting/broadening due to focal increased lymphoplasmacytic infiltrate of lamina propria (high power)
Abbreviations
- CSID:
-
Congenital sucrase-isomaltase deficiency
- EGD:
-
Esophagogastroduodenoscopy
- FODMAP:
-
Fermentable oligosaccharides, disaccharides, monosaccharides, and polyols
- IBS:
-
Irritable bowel syndrome
- IBS C:
-
Irritable bowel syndrome—constipation predominant
- IBS D:
-
Irritable bowel syndrome—diarrhea predominant
- IBS M:
-
Irritable bowel syndrome—mixed bowel habits
- SBT:
-
Sucrase breath testing
- SI:
-
Sucrase-isomaltase
- SID:
-
Sucrase-isomaltase deficiency
References
Card T, Canavan C, West J. The epidemiology of irritable bowel syndrome. Clin Epidemiol.. 2014;6:71. https://doi.org/10.2147/CLEP.S40245.
Hurtado CW, Waasdorp CF. Carbohydrate Digestion and Absorption NASPGHAN Physiology Series. https://www.naspghan.org/files/documents/pdfs/training/curriculum-resources/physiology-series/Carbohydrate_digestion_NASPGHAN.pdf. Accessed November 19, 2018.
Xu H, Collins JF, Ghishan FK. Molecular physiology of gastrointestinal function during development. In: Said Hamid M, ed. Physiology of the Gastrointestinal Tract. Amsterdam: Elsevier; 2012:415–449. https://doi.org/10.1016/b978-0-12-382026-6.00014-2.
Uhrich S, Wu Z, Huang JY, Scott CR. Four mutations in the SI gene are responsible for the majority of clinical symptoms of CSID. J Pediatr Gastroenterol Nutr.. 2012;55:34–35. https://doi.org/10.1097/01.mpg.0000421408.65257.b5.
Naim HY, Heine M, Zimmer K-P. Congenital sucrase-isomaltase deficiency: Heterogeneity of inheritance, trafficking, and function of an intestinal enzyme complex. J Pediatr Gastroenterol Nutr.. 2012;55:S13–20. https://doi.org/10.1097/01.mpg.0000421402.57633.4b.
Chumpitazi BP, Robayo-Torres CC, Opekun AR, Nichols BL, Naim HY. Congenital sucrase-isomaltase deficiency: summary of an evaluation in one family. J Pediatr Gastroenterol Nutr.. 2012;55:S36. https://doi.org/10.1097/01.mpg.0000421409.65257.fc.
Cohen SA. The clinical consequences of sucrase-isomaltase deficiency. Mol Cell Pediatr.. 2016;3:5. https://doi.org/10.1186/s40348-015-0028-0.
Treem WR, McAdams L, Stanford L, Kastoff G, Justinich C, Hyams J. Sacrosidase therapy for congenital sucrase-isomaltase deficiency. J Pediatr Gastroenterol Nutr.. 1999;28:137–142. https://doi.org/10.1097/00005176-199902000-00008.
Henström M, Diekmann L, Bonfiglio F, et al. Functional variants in the sucrase-isomaltase gene associate with increased risk of irritable bowel syndrome. Gut. 2018;. https://doi.org/10.1136/gutjnl-2016-312456.
Garcia-Etxebarria K, Zheng T, Bonfiglio F, et al. Increased Prevalence of rare sucrase-isomaltase (SI) pathogenic variants in irritable bowel syndrome patients. Clin Gastroenterol Hepatol.. 2018;. https://doi.org/10.1016/j.cgh.2018.01.047.
Treem WR. Congenital sucrase-isomaltase deficiency. J Pediatr Gastroenterol Nutr.. 1995;21:1–14.
Treem WR. Clinical aspects and treatment of congenital sucrase-isomaltase deficiency. J Pediatr Gastroenterol Nutr.. 2012;55:S7–13. https://doi.org/10.1097/01.mpg.0000421401.57633.90.
Brandt LJ, Chey WD, Foxx-Orenstein AE, et al. An evidence-based position statement on the management of irritable bowel syndrome. Am J Gastroenterol.. 2008;104:S1–S35. https://doi.org/10.1038/ajg.2008.122.
He Z, Bolling L, Tonb D, Nadal T, Mehta DI. An automated method for the determination of intestinal disaccharidase and glucoamylase activities. J Autom Methods Manag Chem.. 2006;2006:1–4. https://doi.org/10.1155/JAMMC/2006/93947.
Rezaie A, Buresi M, Lembo A, et al. Hydrogen and methane-based breath testing in gastrointestinal disorders: The North American Consensus. Am J Gastroenterol.. 2017;112:775–784. https://doi.org/10.1038/ajg.2017.46.
DeJonge AM, Norris KS, Hernandez K, Elser H, Opekun AR. Mo1971 sucrase-isomaltase genetic variant carriers can be symptomatic. Gastroenterology.. 2014;146:S-705. https://doi.org/10.1016/s0016-5085(14)62560-9.
Puntis JWL, Zamvar V. Congenital sucrase–isomaltase deficiency: Diagnostic challenges and response to enzyme replacement therapy. Arch Dis Child.. 2015;100:869–871. https://doi.org/10.1136/archdischild-2015-308388.
Taylor C, Hodgson K, Sharpstone D, et al. The prevalence and severity of intestinal disaccharidase deficiency in human immunodeficiency virus-infected subjects. Scand J Gastroenterol.. 2000;35:599–606.
Nichols BL, Adams B, Roach CM, Ma C-X, Baker SS. Frequency of sucrase deficiency in mucosal biopsies. J Pediatr Gastroenterol Nutr.. 2012;55:S28–S30. https://doi.org/10.1097/01.mpg.0000421405.42386.64.
Cohen SA, Oloyede H. Variable use of disaccharidase assays when evaluating abdominal pain. Gastroenterol Hepatol (N Y).. 2018;14:26–32.
Shulman RJ, Langston C, Lifschitz CH. Histologic findings are not correlated with disaccharidase activities in infants with protracted diarrhea. J Pediatr Gastroenterol Nutr.. 1991;12:70–75.
Welsh JD, Poley JR, Hensley J, Bhatia M. Intestinal disaccharidase and alkaline phosphatase activity in giardiasis. J Pediatr Gastroenterol Nutr.. 1984;3:37–40.
Horváth K, Horn G, Bodánszky H, Tóth K, Váradi S. Disaccharidases in coeliac disease. Acta Paediatr Hung.. 1983;24:131–136.
Harrison M, Walker-Smith JA. Reinvestigation of lactose intolerant children: lack of correlation between continuing lactose intolerance and small intestinal morphology, disaccharidase activity, and lactose tolerance tests. Gut.. 1977;18:48–52.
Daileda T, Baek P, Sutter ME, Thakkar K. Disaccharidase activity in children undergoing esophagogastroduodenoscopy: A systematic review. World J Gastrointest Pharmacol Ther.. 2016;7:283–293. https://doi.org/10.4292/wjgpt.v7.i2.283.
Mones RL, Yankah A, Duelfer D, Bustami R, Mercer G. Disaccharidase deficiency in pediatric patients with celiac disease and intact villi. Scand J Gastroenterol.. 2011;46:1429–1434. https://doi.org/10.3109/00365521.2011.619276.
Funding
Dr. Moshiree has disclosed she received grant funding from QOL Medical, this study was unfunded and investigator initiated.
Author information
Authors and Affiliations
Contributions
SK was involved in writing of manuscript, editing, design of study, and graphs/tables. FC performed statistical data analysis and proofreading manuscript. JG helped with study design, editing, and patient recruitment. MG contributed to pathology review and provided slides. BM performed study design and writing, manuscript editing, patient recruitment, and proofreading.
Corresponding author
Ethics declarations
Ethical statement
I testify on behalf of all the co-authors that our article submitted to Digestive Diseases and Sciences above has not previously been published in whole or in part elsewhere and that it is not being considered for publication in another journal. All others have been personally and actively involved in the substantive work leading to the manuscript and will hold themselves jointly and individually responsible for its content.
Conflict of Interest
None.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kim, S.B., Calmet, F.H., Garrido, J. et al. Sucrase-Isomaltase Deficiency as a Potential Masquerader in Irritable Bowel Syndrome. Dig Dis Sci 65, 534–540 (2020). https://doi.org/10.1007/s10620-019-05780-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-019-05780-7