
Abstract
Hepatocellular carcinoma (HCC) has an increasing incidence and dismal prognosis, with few systemic treatments approved, including several small molecule tyrosine kinase inhibitors. The application of immune checkpoint inhibitors (ICIs) to HCC has resulted in durable activity, and further evaluation is ongoing. In this review, we discuss the immunologic principles and the mechanism of action of the ICIs and present the relevant clinical data. Furthermore, we provide an overview of the current and emerging immunotherapeutic approaches for HCC, such as combination treatments, vaccines, and cellular therapies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.

Charalampos S. Floudas

Gagandeep Brar

Tim F. Greten
Key Messages
-
ICIs achieve durable responses in HCC, with safety even in HCC of viral etiology.
-
The ICI-induced responses seem to plateau at 10–20%. There is an unmet need for identification of predictive biomarkers.
-
Strategies under study aiming to improve the efficacy of ICIs include combinations with TKIs, vaccines, locoregional therapies in liver-limited disease, and dual checkpoint inhibition.
-
ICIs are also currently being tested in the adjuvant setting in HCC.
-
An emerging and highly promising immunotherapy modality is the CAR T cell treatment, currently targeting predominantly the protein GPC3, which is expressed specifically in HCC.
Introduction
Hepatocellular carcinoma (HCC), the most common primary liver cancer, is also one of the most common malignancies worldwide, ranking as the sixth most commonly diagnosed cancer in 2018, with 841,000 new cases annually [1]; and with 782,000 deaths annually worldwide, it is also a leading cause of cancer-associated mortality. In the United States (USA), the estimate for 2018 is for 42,220 new cases of liver cancer and 30,200 liver cancer-related deaths [2]. While anti-hepatitis B virus (HBV) vaccination has resulted in the decline of HCC incidence in traditional high-incidence regions such as Southeast Asia and Sub-Saharan Africa, in the USA and in Europe the incidence and mortality have doubled over the previous decades, a trend expected to continue [3].
Most commonly HCC will develop in the presence of chronic liver damage and cirrhosis, as the chronic fibroinflammation leads to continuous remodeling, transformation of hepatocytes, and an immunosuppressive microenvironment promoting liver cancer development [4]. Significant causes are chronic viral hepatitis, alcohol abuse, and nonalcoholic fatty liver disease [5], the latter being a major risk factor for HCC development in the USA [6] and of particular importance given its increasing incidence [7].
In early stage disease, there is potential for cure with surgical resection and transplantation, and 5-year survival rate is greater than 70% [2]; however, despite surveillance programs, more than 20% of the patients are diagnosed with advanced disease [8]. Locoregional therapies such as transhepatic arterial chemoembolization or ablation are applicable to liver-limited disease, while for patients with extrahepatic disease or those who fail locoregional therapy systemic therapy is used [9].
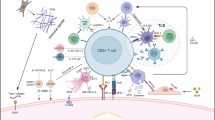
Systemic treatments for HCC constitute mostly of multi-targeted tyrosine kinase inhibitors (TKIs), all blocking the vascular endothelial growth factor receptor (VEGFR) plus various other tyrosine kinases that vary for each molecule. Standard first-line treatment approved for inoperable or metastatic HCC is sorafenib [10], while lenvatinib was approved based on a multicenter, randomized, open-label, non-inferiority trial comparing it to sorafenib [11]. For patients that fail first-line treatment, regorafenib is approved and another one, cabozantinib, is undergoing review by the Food and Drug Administration (FDA) for approval [12]. The survival benefit from the TKIs over the best supportive care is limited though, indicating the critical need for development of novel treatment approaches for advanced HCC. Following the advancements of immunotherapy in solid tumors over the last few years, as shown by the results of targeting the cytotoxic T lymphocyte-associated protein 4 (CTLA-4) or the programmed cell death-1/programmed cell death ligand 1 (PD-1/PD-L1) axis in melanoma, lung, bladder, and kidney [13,14,15], the anti-PD-1 immune checkpoint inhibitor (ICI) nivolumab was approved for advanced HCC, based on the CheckMate 040 trial [16]. In this review, we discuss the available clinical data on immune-based approaches to the treatment of HCC (Fig. 1) and we describe future perspectives.

Immune-based approaches to the treatment of HCC. APC antigen-presenting cells, CTLA4 cytotoxic T lymphocyte-associated protein 4, GPC3 glypican 3, ICI immune checkpoint inhibitor, LAG-3 lymphocyte activation gene 3 protein, PD-1 programmed cell death-1, PD-L1 programmed cell death ligand 1, RT radiation therapy, TCR T-cell receptor, TGF-βR1 transforming growth factor beta receptor 1, TIM-3 T cell immunoglobulin and mucin-domain-containing molecule 3, TKI tyrosine kinase inhibitor, VEGF vascular endothelial growth factor
Immunotherapy in HCC
The antigenically enriched portal vein blood provides a constant immunologic stimulus to the liver, which is prevented from overactivation through liver-intrinsic tolerogenic mechanisms [17]. The persistent inflammation of the chronically diseased liver leads to an immunosuppressed microenvironment and facilitates the development of HCC [18]. Additionally, the escape from immunosurveillance allows the tumor to progress, and depends on multiple mechanisms, including the upregulation of immune checkpoints and of immune inhibitory factors such as arginase-1 and galectin-9 [19]. Immune checkpoint molecules are expressed on T cells and in normal physiologic processes they prevent overactivation of T-lymphocytes by interacting with their respective ligands on antigen-presenting cells (APCs) and other cells, and include CTLA-4 and CD80/86, PD-1 and PD-L1, killer-cell immunoglobulin-like receptor (KIR) and major histocompatibility complex I and II (MHC I/II), lymphocyte activation gene 3 protein (LAG-3) and MHC I/II, and T cell immunoglobulin and mucin-domain-containing molecule 3 (TIM-3) with galectin 9 (GAL9) [20]. In HCC PD-L1, overexpression has been correlated with tumor aggressiveness, evidenced by increased postoperative recurrences [21, 22]. Similarly, the recruitment of regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) to the HCC microenvironment exerts suppressive effects on the antitumor immunity [23]: both Tregs and MDSCs have been found to be increased in HCC and are correlated with worse outcome [24,25,26,27].
Cytokines
Initial immunotherapy efforts in HCC-involved cytokines, specifically interferon alpha-2b (IFN-α-2b), which in patients with advanced disease, resulted in poor response rates without a survival benefit, with an unfavorable adverse event profile [28]. Subsequently, intratumoral delivery of dendritic cells engineered to secrete interleukin-12 (IL-12) or an IL-12-encoding adenovirus was tried, but did not result in encouraging tumor control [29, 30]. More promising results were obtained in a phase-II trial of the transforming growth factor beta receptor 1 (TGF-βR1) kinase inhibitor galunisertib (LY2157299), with a median overall survival (OS) of 93.1 weeks in patients with high (> 200 ng/mL) serum alpha-fetoprotein (AFP) who responded with > 20% reduction in AFP while on treatment versus 29.6 weeks in non-responders [31] and 16.8 months (73 weeks) in patients with low serum AFP [32]. Trials of galunisertib in HCC are ongoing (NCT02240433, NCT02906397, NCT02178358, NCT02423343, NCT01246986).
Immune Checkpoint Inhibitors
A class of agents that in the recent years has been at the epicenter of immunotherapy approaches in HCC is the monoclonal antibodies (mAbs) against the immune checkpoint inhibitors CTLA4, PD-1, and PD-L1. CTLA-4 is expressed on activated T cells and participates in CD4 + T cell activation and the immune response priming phase, but also in decreasing T cell activation upon antigen presentation and mediating the Treg suppressive activity [33]. PD-1 is expressed by activated CD8 + and CD4 + T cells, B cells, natural killer cells, Tregs, MDSCs, monocytes, and dendritic cells, and participates in the immune response effector phase. Its ligands are PD-L1 and PD-L2, both expressed in hematopoietic cells, with PD-L1 being additionally expressed in APCs, MDSCs, and in different types of parenchymal cells [20]. PD-1 and ligand binding result in inhibition of CD8 + and CD4 + T cell activation, a mechanism used for immune evasion by cancer cells that express PD-L1 and PD-L2, utilizing T cell exhaustion, a process of reduced T cell receptor (TCR) signaling and T cell proliferation [34].
The results of clinical trials of ICIs in HCC patients are summarized in Table 1. The first clinical trial of an ICI in advanced HCC was a trial of tremelimumab, a fully human IgG2 mAb antagonist of CTLA-4 [35]. In this phase-II multicenter trial, patients with advanced HCC and chronic hepatitis C virus (HCV) infection were treated with a dosing regimen that is currently considered suboptimal (15 mg/kg every 90 days), and were evaluated for safety and tumor response. Of the 17 evaluable patients, there were three partial responses (17.6%) and an additional 10 patients (58.8%) were found to have stable disease, while the time to progression was 6.48 months and the median OS reached 8.2 months. The agent was tolerated quite well, with no patient requiring systemic glucocorticoids and no treatment-related deaths, while it was shown to have a significant antiviral effect.
Tremelimumab was further tested in combination with incomplete tumor ablation, by percutaneous radiofrequency (RFA) or transarterial chemoembolization (TACE), hypothesizing that the tumor necrosis can induce antigenic stimulation and systemic immune response, potentiated by the immune checkpoint inhibition [36]. The dosing regimen used in this trial involved optimal interval of administrations, every 4 weeks, at two dose levels, of 3.5 and 10 mg/kg. The RFA or TACE were performed 5 weeks after the first infusion of tremelimumab. In this phase-I/II trial, there were 19 evaluable patients, with five patients (26%) achieving a partial response and 12 patients (63%) having stable disease, with time to progression 7.4 months and median overall survival of 12.3 months, with the treatment being well tolerated. An important observation in both these studies was the absence of HCV viremia worsening. Given the small sample sizes of these two trials, it has not been possible to ascertain whether the ablation enhances the antitumor effect.
Given the encouraging results of the anti-CTLA-4 ICI tremelimumab, the trial of anti-PD-1/PD-L1 ICI was a logical consequence. Early-phase trial results have shown that monotherapy with anti-PD-1/PD-L1 is efficacious and tolerable, with a favorable safety profile, while responses are durable, and phase-III trials are expected to provide more data on survival (Table 2).
Nivolumab, a fully human IgG4 anti-PD-1 mAb, was tested in patients with advanced HCC and Child-Pugh A cirrhosis who progressed on or were intolerant to sorafenib, in a dose escalation cohort and in an expansion cohort, the latter at a dose of 3 mg/kg every 2 weeks in the CheckMate 040 trial [16]. The objective response rate in the expansion cohort was 20% (95% CI 15–26%) with three complete responses and 39 partial responses, with response duration ranging from 3.2 to 38.2 + months, and responses observed in HCC of all etiologies (viral and non-viral), regardless of prior treatment with sorafenib. Nivolumab was well tolerated, with frequencies of grade 3/4 treatment-related AEs and treatment-related serious AEs at 20% and 7%, respectively, while immune-related hepatitis was rare and there were no treatment-related deaths. Furthermore, there was no viral reactivation or viremia worsening of viremia in patients with HBV or HCV. Based on these results, it received accelerated USFDA approval for patients with advanced HCC who have received sorafenib. The results of trial NCT02576509, an open-label phase-III multicenter randomized study of nivolumab 240 mg every 2 weeks versus sorafenib 400 mg twice daily in patients with HCC are expected in 2018, with the goal to confirm the clinical benefit of nivolumab.
Pembrolizumab is a humanized IgG4 anti-PD-1 mAb, which was tested in a phase-II trial, KEYNOTE-224, in patients with advanced HCC who had progressed on sorafenib [37]. Among 104 patients that were treated, the overall response rate was 17%, with one complete response and 16 partial responses. The treatment was safe, without viral reactivation (where applicable), and the effect was observed in HCC of all etiologies (viral and non-viral). A phase-III study of pembrolizumab in advanced HCC, comparing against placebo (NCT03062358, KEYNOTE-240), has completed accrual and results are pending.
Durvalumab is a human IgG1κ mAb to PD-L1, which was tested in a phase-I/II trial in advanced HCC patients [38] with an overall response rate of 10.3% (95% CI 2.9–24.2%) in 39 evaluable patients. Similar to the results from the anti-PD-1 mAbs nivolumab and pembrolizumab, durvalumab had also a favorable safety profile, with grade 3/4 treatment-related AEs reported in 20% of patients and no death occurring due to treatment-related AEs.
Other anti-PD-1/PD-L1 mAbs, such as avelumab, atezolizumab, PDR001, SHR-121, LY3300054, REGN2810, and BGB-A317, are currently under evaluation in clinical trials either alone or in combinations. Table 2 lists the clinical trials of anti-CTLA-4 and anti-PD-1/PD-L1 agents which are actively accruing patients.
Since the minority of HCC patients who respond to ICIs is coupled with the potential for immune-mediated adverse events, the identification of predictive biomarkers for response and toxicity is very important. PD-L1 expression in the tumor was not found to be correlated with response in the nivolumab trial [16], while the mutational burden in HCC is moderate [39], and consequently unlikely to be of predictive value as it is in other malignancies [40]. Host factors such as the gut microbiome have recently been shown to influence the response to ICIs in melanoma [41] and other epithelial tumors [42]. In the case of HCC, it was recently shown that the gut microbiome regulates liver tumor immunosurveillance by primary-to-secondary bile acids conversion [43].
The results of ICI treatment in metastatic HCC have generated interest in the administration of ICIs as adjuvant treatment, with an ongoing phase-III study in the USA of adjuvant nivolumab versus placebo after complete resection or complete response following ablation (NCT03383458). Furthermore, one phase-II trial in the USA is ongoing, exploring nivolumab versus nivolumab plus ipilimumab in the neoadjuvant setting (NCT03222076), while a phase-II study in Japan aims to study recurrence of HCC with pembrolizumab administration before and after curative surgery or ablation (NCT03337841). The same combination of neoadjuvant and adjuvant administration, though with nivolumab, will be utilized by a phase-II study in France in the context of curative-intent electroporation (NCT03630640).
Vaccines
Efforts to utilize the antitumor immune response in cancer treatment have also included vaccination with specific antigens. The rationale for cancer vaccines is that by presenting tumor-specific mutation-derived neoantigens or tumor-associated antigens, immune recognition of these antigens and consequently immune activity against them will increase [44]. An example of a tumor-associated antigen is alpha-fetoprotein (AFP), which is expressed by HCC cells but typically not by normal adult tissues. AFP has been the first tumor-associated antigen utilized for vaccine-based trials in HCC, but this approach was unsuccessful: Early studies with AFP peptides or AFP-pulsed dendritic cells led to a T cell response, but no clinical benefit [45, 46]. A more recent phase-I clinical trial of an AFP-derived peptide vaccine conducted in 15 HCC patients led to T cell response and additionally to clinical response: There was one complete response and suppressed tumor growth in eight patients, while no serious adverse events were reported [47].
Another tumor antigen that has been evaluated in clinical trials is the liver-cancer-specific antigen glypican-3 (GPC3), which is expressed in nearly all hepatocellular cancers. GPC3 is a glycosylphosphatidylinositol (GPI)-anchored cell surface protein that contains a core protein and two heparan sulfate (HS) chains. It functions as a co-receptor for Wnt3a/b-catenin and promotes cell proliferation [48, 49]. It is also participating in other pathways, including the transforming growth factor beta 2 (TGF-β2) and (extracellular signal-regulated kinase) ERK pathways, and promotes epithelial-mesenchymal transition [50,51,52].
In early studies, a GPC3 peptide vaccine was safe and effective in inducing infiltration of the tumor by CD8 + T cells. The clinical benefit was limited though, as there was a single partial response out of the 33 treated patients, and the median time to progression was 3.4 months [53]. The same group used a pre-clinical model to show that the combination of the anti-GPC3 peptide vaccine with anti-PD-1 mAb increased the immune response and antitumor effects of the vaccine [54]. A trial has also been conducted in the adjuvant setting, where it has demonstrated improvement in the recurrence rate at 1 year after surgery compared to surgery alone, but not after 2 years [55]. GPC3 has been targeted also directly, with a humanized IgG1 anti-GPC3 antibody [56], in a phase-I trial in which it was well tolerated and displayed preliminary evidence of clinical benefit [57].
Other vaccine trials included vaccines of dendritic cells pulsed with hepatoblastoma or with autologous tumor lysates, which failed to demonstrate a significant clinical benefit, while a cell-free plasmid DNA vaccine platform followed by AFP-expressing replication-deficient adenovirus was tested in the adjuvant setting had unclear clinical benefit [18]. A telomerase peptide vaccine, GV1001, was tested in patients with advanced HCC in a phase-II trial in combination with low dose cyclophosphamide and granulocyte–macrophage colony-stimulating factor (GM-CSF) as immune sensitizers [58]. While the vaccination elicited an immune response, it failed to show any radiologically detectable tumor responses.
Another immunotherapeutic vaccine is the targeted oncolytic poxvirus product pexastimogene devacirepvec (JX-594, PexaVec). PexaVec is derived from a vaccinia virus vaccine strain engineered to replicate (and therefore lyse) preferentially in cancer cells by the deletion of the thymidine kinase gene, and it also expresses human GM-CSF to enhance recruitment of APCs. Its administration has been found to be safe, with promising results as the intrahepatic disease control rate has been reported at 46% [59, 60]. Currently, there are two ongoing clinical trials of PexaVec, a phase-III clinical trial evaluating PexaVec followed by sorafenib versus sorafenib alone in patients with advanced HCC (NCT02562755) as well as a phase-I/II trial of nivolumab with PexaVec (NCT03071094).
Combinations of Immunotherapeutic Agents
While anti-CTLA-4 and anti-PD-1/PDL-1 monotherapy have been efficacious, the percentage of patients that will achieve a durable response is low, and consequently, there has been great interest in approaches that would improve on that. Since the immune checkpoints CTLA-4 and PD-1/PD-function at different phases of the effector T cell activity, we can hypothesize that by combining them we may increase T cell activation and tumor killing, though maybe with increased toxicity. This is in agreement with the positive results from the combination of nivolumab with ipilimumab in melanoma [61] and has prompted trials following the same approach in HCC, where the combinations of nivolumab with ipilimumab and durvalumab with tremelimumab are currently being tested in phase-I/II trials (Table 2). Preliminary safety and efficacy data have been reported for the combination of durvalumab with tremelimumab [62], indicating good safety and tolerability, with a response rate of 15% (6 out of 40 patients), and the trial is ongoing as phase-II expansion (NCT02519348). There is also an ongoing randomized, open-label, multicenter phase-III study of durvalumab with or without tremelimumab versus sorafenib in advanced HCC (NCT03298451), planning to enroll about 1200 patients and explore two dose schedules of durvalumab and tremelimumab.
Novel checkpoint inhibitors are combined with PD-1/PD-L1 in early-phase basket clinical trials that include patients with HCC (Table 2), such as the anti-TIM-3 antibody LY3321367 with the anti-PD-L1 antibody LY3300054 (NCT03099109), the anti-LAG-3 antibody REGN3767 with or without the anti-PD1 antibody REGN2810 (NCT03005782).
Other teams are attempting to improve the efficacy of ICIs by combining them with other classes of therapeutic agents, such as TKIs, oncolytic viruses and ablative therapies (Table 2). The effects of TKIs on immune pathways and the tumor microenvironment result in differential effects on the immune response to the tumor [63]. Currently, several early-phase studies are ongoing exploring the safety and tolerability of ICIs when combined with a TKI, including nivolumab with sorafenib (NCT03439891), pembrolizumab with sorafenib (NCT03211416), PDR001 with sorafenib (NCT02988440), nivolumab with lenvatinib (NCT03418922), pembrolizumab with lenvatinib (NCT03006926), pembrolizumab with regorafenib (NCT03347292), avelumab with regorafenib (NCT0347595), nivolumab with cabozantinib (NCT03299946), SHR-1210 with apatinib (NCT02942329) and avelumab with axitinib (NCT03289533).
A further strategy combines targeting the PD-L1 and vascular endothelial growth factor (VEGF), as it has been found that VEGF inhibition enhances antigen presentation and intratumoral T-cell infiltration [64]. In a phase-I/II study, the combination of atezolizumab and bevacizumab resulted in partial responses in 62% of patients with treatment-naïve advanced HCC [65]. There is also an ongoing phase-III randomized study of atezolizumab and bevacizumab versus sorafenib in patients with advanced HCC looking at ORR and OS (NCT03434379).
Ongoing early-phase trials are also evaluating the combination of ICI with the DNA methyltransferase (DNMT) inhibitor guadecitabine, (NCT03257761), the anti-OX40 mAb INCAGN01949 (NCT03241173), the anti-phosphatidylserine mAb bavituximab (NCT03519997), the heat shock protein (Hsp90) inhibitor XL888 (NCT03095781), the MET inhibitor INC280 (NCT02795429), the fibroblast growth factor receptor 4 (FGFR4) inhibitor FGF401 (NCT02325739), and the anti-transforming growth factor beta (TGFbeta) mAb NIS793 (NCT02947165).
Combination with Locoregional Therapies
Locoregional treatments are recommended as the primary form of treatment in liver-limited unresectable HCC, but they are also utilized in advanced HCC in combination with ICIs. The rationale for this originates from the proved assumption that tumor destruction, e.g., by RFA or TACE, promotes immunogenic cell death, alters the local immune microenvironment, and through the systemic release of antigens changes the peripheral immune response as well; this peripheral immune response can be enhanced by the administration of an ICI [36, 66]. Consequently, studies of combination of ICIs with liver-directed treatments (ablation, radiation, embolization, chemoembolization, or radioembolization) are underway to evaluate the safety, tolerability, and efficacy of ICI combinations with ablative therapies (Table 2), including trials of dual anti-ICI (anti-CTLA-4 and anti-PD-L1) with ablation/TACE (NCT02821754) or radiation (NCT03482102).
Cellular Therapies
Lastly, a novel immunotherapeutic modality in HCC is adoptive cell transfer (ACT), which is promising, highly personalized, but also highly challenging. ACT is a cellular treatment consisting as a general principle in expanding host immune cells and administering them back to the patient [67], and it uses the natural antigen recognition and elimination potential of the T cells. T cells recognize the diseased cells via the interaction of the T cell receptor (TCR) with the major histocompatibility complex class I (MHC-I molecules) present on the diseased cell. The use of autologous tumor-infiltrating lymphocytes (TIL) for ACT has resulted in complete, durable tumor regressions in melanoma patients [68] and in a patient with cholangiocarcinoma [69]. The use of tumor-specific cytotoxic T cells (CTL) for ACT in advanced HCC had better results than the ACT with lymphokine-activated killer cells when compared in an early trial [70]. More recently, cytokine-induced killer (CIK) cells, which are peripheral blood mononuclear cell-derived and ex vivo expanded, were used for ACT in HCC in combination with RFA or TACE, displaying an improvement in OS over RFA or TACE alone; however, treatment allocation was not randomized [71]. A subsequent phase-II randomized trial compared ACT using CIKs with standard treatment in treatment-naïve HCC patients and reported prolonged OS and progression free survival (PFS) [72]. Additionally, a multicenter, randomized, open-label phase-III trial of CIK ACT in HCC after resection or ablation (RFA or percutaneous ethanol injection) with curative intent, reported increases in recurrence-free survival and in overall survival [73].
Better targeting of the transferred cells to the tumor cells has been achieved with TCR engineering to produce TCR-recognizing tumor-specific antigens. MHC-restricted tumor antigens were identified when using TILs in melanoma [74], and their identification allowed the generation of TCR-engineered tumor reactive T cells with TIL-derived TCR genes in cases where not enough TILs could be isolated from the tumor [75, 76].
In HCCs of viral etiology, tumor-expressed viral antigens can be utilized for TCR selection. This was tested with targeting HBV-infected tumor hepatocytes with HBV-targeted TCR cells and was found to reconstitute antiviral T cell activity directed against the infected HCC cells [77]. An ongoing first-in-human phase-I trial is evaluating the safety and efficacy of AFP-targeted in patients with advanced HCC (NCT03132792).
The applicability of TCR therapy is limited to a subset of patients due to MHC restriction, a limitation absent from the chimeric antigen receptor (CAR) T cells. The chimeric receptor consists of an antibody single-chain variable fragment connected to the T cell receptor and costimulatory receptor signaling domains, creating a transmembrane complex able to recognize and bind cell surface antigens directly, without the requirement of antigen processing and MHC presentation (and therefore restriction) [78]. The antigen-binding part of the CAR can be targeted against tumor antigens, and in the case of HCC, the selected tumor-specific antigen is GPC3. Pre-clinical models of anti-GPC3 CAR T cells have led to eradication of HCC high GPC3-expressing xenografts and have halted the growth of low GPC3-expressing xenografts [49]. A phase-I clinical trial of anti-GPC3 CAR T cells, with or without lymphodepletion (fludarabine and cyclophosphamide) in relapsed or refractory GPC3 positive HCC, resulted in progressive disease for all non-lymphodepleted patients (n = 5), while in the lymphodepleted group (n = 8), of the six evaluable patients there were one patient with partial response (duration 385 days) and three with stable disease (duration 384 and 563 days, third patient died at 108 days). The authors report only one serious AE, a grade-1 fever, without any dose limiting toxicities [79]. The currently ongoing, actively accruing early-phase clinical trials of CAR T cells in HCC, all in China, either only HCC or basket trials are five of anti-GPC3 CAR T cells (NCT03146234, NCT03198546, NCT02715362, NCT03130712, NCT02959151) one which with intratumoral administration (NCT03130712) and one with transarterial administration (NCT02715362), one of anti-AFP CAR T cell trial (NCT03349255), one of anti-mucin 1, cell surface associated (MUC1) CAR T cells (NCT03198546), one of anti-epithelial cell adhesion molecule (EpCAM) CAR T cells (NCT03013712), and one of anti-DR5 and anti-C-met for HCC (NCT03638206). One GPC3 CAR T cell trial (NCT02723942) is reported as completed, and one terminated for undisclosed reasons (NCT02395250), while two GPC3 CAR T cell trials in China (NCT03084380, NCT03302403) and one in the USA (NCT02905188) are registered but not yet accruing patients. Furthermore, a trial of c-Met/PD-L1 CAR T cells in HCC is also registered but not yet accruing (NCT03672305). Consequently, the global landscape of registered CAR T cell trials for HCC is presented in Fig. 2a, whereas Fig. 2b presents the distribution of CAR T cell trials for HCC in China. Of note, in 2017 there were 121 CAR T cell trials reported and/or registered at ClinicalTrials.gov from China [80].

a The global landscape of registered CAR T cell trials for HCC. b The distribution of CAR T cell trials for HCC in China
Conclusion
As tumor immunology has found its way from the bench to the bedside, it has resulted in significant breakthroughs in cancer immunotherapy, and HCC immunotherapy in particular. In HCC, immune checkpoint inhibitors lead to durable disease control in a subset of patients and currently many studies are evaluating different combination strategies as well as emerging cellular therapies to improve outcomes.
References
Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians; 2018.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30.
Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90.
Karin M. Nuclear factor-κB in cancer development and progression. Nature. 2006;441:431–436.
Llovet JM, Zucman-Rossi J, Pikarsky E, et al. Hepatocellular carcinoma. Nat Rev Dis Prim. 2016;2:16018.
Makarova-Rusher OV, Altekruse SF, McNeel TS, et al. Population attributable fractions of risk factors for hepatocellular carcinoma in the United States: US HCC-attributable risk factors. Cancer. 2016;122:1757–1765.
Masuoka HC, Chalasani N. Nonalcoholic fatty liver disease: an emerging threat to obese and diabetic individuals. Ann N Y Acad Sci. 2013;1281:106–122.
Bruix J, Sherman M. Management of hepatocellular carcinoma: an update. Hepatology. 2011;53:1020–1022.
Benson AB, D’Angelica MI, Abbott DE, et al. NCCN guidelines insights: hepatobiliary cancers, version 1.2017. J Natl Compr Canc Netw. 2017;15:563–573.
Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390.
Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391:1163–1173.
Abou-Alfa GK, Meyer T, Cheng A-L, et al. Cabozantinib (C) versus placebo (P) in patients (pts) with advanced hepatocellular carcinoma (HCC) who have received prior sorafenib: results from the randomized phase III CELESTIAL trial. J Clin Oncol. 2018;36:207.
Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373:1803–1813.
Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–461.
Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375–384.
El-Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389:2492–2502.
Knolle PA, Thimme R. Hepatic immune regulation and its involvement in viral hepatitis infection. Gastroenterology. 2014;146:1193–1207.
Makarova-Rusher OV, Medina-Echeverz J, Duffy AG, et al. The yin and yang of evasion and immune activation in HCC. J Hepatol. 2015;62:1420–1429.
Prieto J, Melero I, Sangro B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2015;12:681–700.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264.
Gao Q, Wang X-Y, Qiu S-J, et al. Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin Cancer Res. 2009;15:971–979.
Calderaro J, Rousseau B, Amaddeo G, et al. Programmed death ligand 1 expression in hepatocellular carcinoma: relationship with clinical and pathological features. Hepatology. 2016;64:2038–2046.
Schmidt N, Thimme R. Role of immunity in pathogenesis and treatment of hepatocellular carcinoma. Dig Dis. 2016;34:429–437.
Ormandy LA, Hillemann T, Wedemeyer H, et al. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Can Res. 2005;65:2457–2464.
Arihara F, Mizukoshi E, Kitahara M, et al. Increase in CD14 + HLA-DR −/low myeloid-derived suppressor cells in hepatocellular carcinoma patients and its impact on prognosis. Cancer Immunol Immunother. 2013;62:1421–1430.
Greten TF, Wang XW, Korangy F. Current concepts of immune based treatments for patients with HCC: from basic science to novel treatment approaches. Gut. 2015;64:842–848.
Hoechst B, Ormandy LA, Ballmaier M, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4 + CD25 + Foxp3 + T cells. Gastroenterology. 2008;135:234–243.
Llovet JM, Sala M, Castells L, et al. Randomized controlled trial of interferon treatment for advanced hepatocellular carcinoma. Hepatology. 2000;31:54–58.
Sangro B, Mazzolini G, Ruiz J, et al. Phase I trial of intratumoral injection of an adenovirus encoding interleukin-12 for advanced digestive tumors. J Clin Oncol. 2004;22:1389–1397.
Mazzolini G, Alfaro C, Sangro B, et al. Intratumoral injection of dendritic cells engineered to secrete interleukin-12 by recombinant adenovirus in patients with metastatic gastrointestinal carcinomas. J Clin Oncol. 2005;23:999–1010.
Faivre SJ, Santoro A, Kelley RK, et al. A phase 2 study of a novel transforming growth factor-beta (TGF-β1) receptor I kinase inhibitor, LY2157299 monohydrate (LY), in patients with advanced hepatocellular carcinoma (HCC). J Clin Oncol. 2014;32:LBA173.
Faivre SJ, Santoro A, Gane E, et al. A phase 2 study of galunisertib, a novel transforming growth factor-beta (TGF-β) receptor I kinase inhibitor, in patients with advanced hepatocellular carcinoma (HCC) and low serum alpha fetoprotein (AFP). J Clin Oncol. 2016;34:4070.
Wing K, Onishi Y, Prieto-Martin P, et al. CTLA-4 control over Foxp3 + regulatory T cell function. Science. 2008;322:271–275.
Barber DL, Wherry EJ, Masopust D, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687.
Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59:81–88.
Duffy AG, Ulahannan SV, Makorova-Rusher O, et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J Hepatol. 2017;66:545–551.
Zhu AX, Finn RS, Edeline J, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol. 2018;. https://doi.org/10.1016/s1470-2045(18)30351-6.
Wainberg ZA, Segal NH, Jaeger D, et al. Safety and clinical activity of durvalumab monotherapy in patients with hepatocellular carcinoma (HCC). J Clin Oncol. 2017;35:4071.
Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome landscapes. Science. 2013;339:1546–1558.
Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–2199.
Gopalakrishnan V, Spencer CN, Nezi L, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2017;359:97–103.
Routy B, Chatelier EL, Derosa L, et al. Gut microbiome influences efficacy of PD-1—based immunotherapy against epithelial tumors. Science. 2018;359:91–97.
Ma C, Han M, Heinrich B, et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018;360:eaan5931.
Sahin U, Türeci Ö. Personalized vaccines for cancer immunotherapy. Science. 2018;359:1355–1360.
Butterfield LH, Ribas A, Meng WS, et al. T-cell responses to HLA-A*0201 immunodominant peptides derived from alpha-fetoprotein in patients with hepatocellular cancer. Clin Cancer Res. 2003;9:5902–5908.
Butterfield LH, Economou JS, Gamblin T, et al. Alpha fetoprotein DNA prime and adenovirus boost immunization of two hepatocellular cancer patients. J Transl Med. 2014;12:86.
Nakagawa H, Mizukoshi E, Kobayashi E, et al. Association between high-avidity T-cell receptors, induced by α-fetoprotein-derived peptides, and anti-tumor effects in patients with hepatocellular carcinoma. Gastroenterology. 2017;152:e10.
Feng M, Gao W, Wang R, et al. Therapeutically targeting glypican-3 via a conformation-specific single-domain antibody in hepatocellular carcinoma. Proc Natl Acad Sci. 2013;110:E1083–E1091.
Gao H, Li K, Tu H, et al. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin Cancer Res. 2014;20:6418–6428.
Acloque H, Adams MS, Fishwick K, et al. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Investig. 2009;119:1438–1449.
Sun CK, Chua M-S, He J, et al. Suppression of glypican 3 inhibits growth of hepatocellular carcinoma cells through up-regulation of TGF-β2. Neoplasia. 2011;13:735–747.
Wu Y, Liu H, Weng H, et al. Glypican-3 promotes epithelial-mesenchymal transition of hepatocellular carcinoma cells through ERK signaling pathway. Int J Oncol. 2015;46:1275–1285.
Sawada Y, Yoshikawa T, Nobuoka D, et al. Phase I trial of a glypican-3-derived peptide vaccine for advanced hepatocellular carcinoma: immunologic evidence and potential for improving overall survival. Clin Cancer Res. 2012;18:3686–3696.
Sawada Y, Yoshikawa T, Shimomura M, et al. Programmed death-1 blockade enhances the antitumor effects of peptide vaccine-induced peptide-specific cytotoxic T lymphocytes. Int J Oncol. 2015;46:28–36.
Sawada Y, Yoshikawa T, Ofuji K, et al. Phase II study of the GPC3-derived peptide vaccine as an adjuvant therapy for hepatocellular carcinoma patients. OncoImmunology. 2016;5:e1129483.
Nakano K, Ishiguro T, Konishi H, et al. Generation of a humanized anti-glypican 3 antibody by CDR grafting and stability optimization. Anticancer Drugs. 2010;21:907.
Zhu AX, Gold PJ, El-Khoueiry AB, et al. First-in-man phase I study of GC33, a novel recombinant humanized antibody against Glypican-3, in patients with advanced hepatocellular carcinoma. Clin Cancer Res. 2013;19:920–928.
Greten TF, Forner A, Korangy F, et al. A phase II open label trial evaluating safety and efficacy of a telomerase peptide vaccination in patients with advanced hepatocellular carcinoma. BMC Cancer. 2010;10:209.
Park B-H, Hwang T, Liu T-C, et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. 2008;9:533–542.
Heo J, Reid T, Ruo L, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19:329–336.
Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372:2006–2017.
Kelley RK, Abou-Alfa GK, Bendell JC, et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): phase I safety and efficacy analyses. J Clin Oncol. 2017;35:4073.
Harding JJ, Dika IE, Abou-Alfa GK. Immunotherapy in hepatocellular carcinoma: primed to make a difference? Cancer. 2016;122:367–377.
Wallin JJ, Bendell JC, Funke R, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun. 2016;7:12624.
Stein S, Pishvaian MJ, Lee MS, et al. Safety and clinical activity of 1L atezolizumab + bevacizumab in a phase Ib study in hepatocellular carcinoma (HCC). J Clin Oncol. 2018;36:4074.
Greten TF, Sangro B. Targets for immunotherapy of liver cancer. J Hepatol. 2018;68:157–166.
Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62–68.
Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–4557.
Tran E, Turcotte S, Gros A, et al. Cancer immunotherapy based on mutation-specific CD4 + T cells in a patient with epithelial cancer. Science. 2014;344:641–645.
Haruta I, Yamauchi K, Aruga A, et al. Analytical study of the clinical response to two distinct adoptive immunotherapies for advanced hepatocellular carcinoma: comparison between LAK cell and CTL therapy. J Immunother Emphas Tumor Immunol. 1996;19:218–223.
Huang Z-M, Li W, Li S, et al. Cytokine-induced killer cells in combination with transcatheter arterial chemoembolization and radiofrequency ablation for hepatocellular carcinoma patients. J Immunother. 2013;36:287–293.
Yu X, Zhao H, Liu L, et al. A randomized phase II study of autologous cytokine-induced killer cells in treatment of hepatocelluar carcinoma. J Clin Immunol. 2014;34:194–203.
Lee JH, Lee J-H, Lim Y-S, et al. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology. 2015;148:e6.
Kershaw MH, Westwood JA, Darcy PK. Gene-engineered T cells for cancer therapy. Nat Rev Cancer. 2013;13:525–541.
Clay TM, Custer MC, Sachs J, et al. Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J Immunol. 1999;163:507–513.
Morgan RA, Dudley ME, Yu YYL, et al. High efficiency TCR gene transfer into primary human lymphocytes affords avid recognition of melanoma tumor antigen glycoprotein 100 and does not alter the recognition of autologous melanoma antigens. J Immunol. 2003;171:3287–3295.
Gehring AJ, Xue S-A, Ho ZZ, et al. Engineering virus-specific T cells that target HBV infected hepatocytes and hepatocellular carcinoma cell lines. J Hepatol. 2011;55:103–110.
Hinrichs CS, Rosenberg SA. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev. 2014;257:56–71.
Zhai B, Shi D, Gao H, et al. A phase I study of anti-GPC3 chimeric antigen receptor modified T cells (GPC3 CAR-T) in Chinese patients with refractory or relapsed GPC3 + hepatocellular carcinoma (r/r GPC3 + HCC). J Clin Oncol. 2017;35:3049.
Liu B, Song Y, Liu D. Clinical trials of CAR-T cells in China. J Hematol Oncol. 2017;10:166.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Floudas, C.S., Brar, G. & Greten, T.F. Immunotherapy: Current Status and Future Perspectives. Dig Dis Sci 64, 1030–1040 (2019). https://doi.org/10.1007/s10620-019-05516-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-019-05516-7