
Abstract
Background and Aims
Peutz–Jeghers syndrome (PJS) is an autosomal-dominant genetic disease caused by mutations in the tumor suppressor gene, STK11, which is characterized by gastrointestinal hamartomas, melanin spots on the lips and the extremities, and an increased risk of developing both gastrointestinal and extraintestinal malignancies.
Methods and Results
We treated a PJS patient without a positive family history, who possessed typical clinical manifestations including polyp canceration. In order to explore the genotype of this patient, blood samples were collected from all the available family members. The whole coding region and the flanking regions of the STK11 gene were amplified by polymerase chain reaction and analyzed by Sanger sequencing. Molecular analysis of the STK11 gene here revealed a 23-nucleotide deletion (c.426–448delCGTGCCGGAGAAGCGTTTCCCAG) in exon 3, resulting in a change of 13 codons and a truncating protein (p.S142SfsX13). This mutation was not found in normal individuals in this family including her parents or in 100 control individuals. Protein structure prediction indicated a dramatic loss of the kinase domain and complete loss of the C-terminal regulatory domain.
Conclusions
The results presented here enlarge the spectrum of STK11 mutation both disease-causing and malignancy-causing.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Peutz–Jeghers syndrome (PJS, OMIM 175200) is a autosomal-dominant disorder characterized by gastrointestinal (GI) hamartomatous polyps, mucocutaneous pigmentation, and an increased risk for the development of gastrointestinal and various extra-GI malignancies [1, 2]. PJS is a rare condition with an incidence of 1/50,000–1/200,000 [3], but its incidence in China is not been reported. The elevated cancer risk in PJS patients has been observed in several cohort studies, and it is estimated to be 9–18 times higher than the general population [4, 5]. The main clinical symptoms of PJS include abdominal pain, rectal blood loss, anemia, small bowel obstruction, and intussusception leading to a high laparotomy rate [6].
The clinical condition connecting mucocutaneous pigmentation and multiple gastrointestinal polyps together was first described in 1921 by Peutz, and Peutz reported this association in a family [7]. Jeghers reported a series of cases and established the syndrome as an autosomal-dominant disease in 1940s [8]. In 1997, serine-threonine kinase 11 (STK11/LKB1, OMIM 602216) on chromosome 19p13.3 was cloned by Jenne et al. [9] and Hemminki et al. [10], respectively, which has been considered to be the pathogenic gene of PJS. The gene is divided into nine coding exons that encode a 433-amino-acid protein. STK11 has a wide mutation spectrum in PJS, and a considerable uncharacterized genetic heterogeneity remains in this syndrome. Up to now, STK11 is the only confirmed pathogenic gene of PJS, and Buchet-Poyau et al. [11] has excluded several candidate genes as a second PJS locus in the 19q13.3–q13.4 region and other regions.
Most mutations are frameshift or nonsense changes, which result in an abnormal truncated protein and kinase activity impaired, and they can be detected in PJS patients despite of family histories [12]. However, the genotype–phenotype correlation is still poorly understood in PJS. Here, we report a novel STK11 mutation in a PJS patient without a family history, which is associated with very likely cancer risk.
Materials and Methods
Patient and Sample Collection
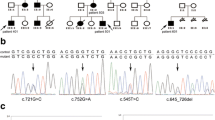
The index patient was a 26-year-old female from East China. The characteristic pigmentation on his lips and fingertips appeared shortly after birth. Clinical diagnosis of PJS was determined at the age of 6 after her partial ileal resection because of intestinal obstruction, and multiple hamartomatous polyps was confirmed in her postoperative pathological specimens [13]. Before regular endoscopy surveillance was available, the patient received open surgeries twice at the age of 12 and 23. The patient came to our department for double-balloon enteroscopy (DBE) at the age of 24. But the enteroscope could not pass through due to serious intestinal adhesion, so she had to receive polypectomy by another open surgery. The diameters of the resected polyps were as large as 4 cm. Unfortunately, adenocarcinoma infiltration within hamartomas was confirmed in surgical specimens (Fig. 1a). After that the patient has regular endoscopy surveillance, and her health is well maintained. Though the index patient possessed typical manifestations of PJS, her parents, brother and sister had no PJS-related complaints (Fig. 1b).

Genogram, pathology and electropherogram of the index patient. a Representative hematoxylin–eosin-stained tissue slices of the GI polyp resection specimens showed carcinoma infiltration. Up, ×100 magnification; low, ×400 magnification. b The genogram showed an isolated PJS patient. Roman numerals indicate generations, and Arabic numbers indicate individuals. Squares males, circles females. Affected individuals are denoted by solid symbols and unaffected individuals are denoted by open symbols. The index patient is indicated by an arrow. c The structure of STK11 gene. This novel mutation is within exon 3. d Sanger sequencing with the help of T vector assay revealed a heterozygous frameshift mutation, c.426–448delCGTGCCGGAGAAGCGTTTCCCAG. The codons in red cause amino acid residues changed in the mutant haplotype. Red box, 23 nucleotides deleted in the mutant haplotype; black bar the site where the deletion starts; blue bar the site where the deletion ends; red arrow the new junction site in mutant haplotype
This study (AFGHEC2016-145) was approved by the Medical Ethics Committee, Airforce General Hospital of PLA. After obtaining informed consent, blood samples were collected from all available family members (relatives I:1, I:2, II:1, and II:3) including the patient.
Genomic DNA Isolation and Mutation Analysis
Genomic DNA of peripheral blood leukocytes was extracted routinely by Isolation Kit (DP318, Tiangen Biotech, Beijing, China) according to the manufacturer’s instructions. All nine coding exons and flanking introns of the STK11 gene were amplified by the use of primers listed in Table 1. Polymerase chain reactions (PCR) of STK11 exons were performed in a 50-μl reaction which contained 0.4 μM of each primer, 50 ng genomic DNA, and 25 μl 2× premix Ex Taq DNA polymerase (RR030A, Takara Bio Inc., Dalian, China). The PCRs were performed under the following conditions: denaturation at 95 °C for 4 min, followed by 35 thermal cycles, each composed of 95 °C for 30 s, 58 °C for 30 s, and 72 °C for 45 s.
All available family members underwent STK11 germline mutation testing to confirm cosegregation of the mutation with the disease. For frameshift mutation, T vector assay (CV15, Aidlab Inc., Beijing, China) was used to identify each haplotype by constructing monoclonal cells. In order to rule out polymorphisms and to confirm the pathogenic effects of variations, 100 chromosomes from 50 unrelated healthy individuals were also screened for the presence of the mutation.
The PCR products were gel- and column-purified and directly sequenced. The purified PCR fragments were then sequenced using BigDye Terminator (Applied Biosystems, Foster City, CA, USA) on an ABI Prism 3100 genetic analyzer (Applied Biosystems, Foster City, CA, USA) by Majorbio Co. Ltd.(Shanghai, China). The results were used to performance sequence alignment according to STK11 gene sequence (NP_000446.1 and NM_000455.4 in GRCh38.p7). In addition, samples from 50 unrelated ethnicity-matched healthy controls were sequenced to exclude the mutations as non-disease associated variations in the Chinese population.
Structure Predictions of the Mutant Protein and Analysis of Evolutionary Conservation of Amino Acid Residues
The homology modeling programs, Swiss-Model (http://swissmodel.expasy.org), was used to develop an appropriate model to mimic the effects of the mutated region [14]. Evolutionary conservation of amino acid residues altered was analyzed by comparing across different species (https://www.ncbi.nlm.nih.gov/protein/).
Results
Direct sequencing analysis of the DNA from the proband revealed a heterozygous germline frameshift mutation in exon 3. Further, we performed T vector assay and identified the exact haplotype, which is a 23-nucleotide deletion (c.426–448delCGTGCCGGAGAAGCGTTTCCCAG) (Fig. 1c, d). To our knowledge, this mutation has not been reported in literatures. Then we checked public databases such as dbSNP (database of single nucleotide polymorphisms), OMIM (Online Mendelian Inheritance in Man), ClinVar and HGMD (Human Gene Mutation Database), and we confirm that this mutation has not been discovered or recorded.
Through sequence alignment, we find this mutation results in a translational frameshift (p.S142SfsX13), and the premature stop codon appears in codon 154, which causes largely partial loss of kinase domain and C-terminal regulatory domain (Fig. 2a). Evolutionary conservation analysis of amino acid residues revealed that these impaired amino acid residues were most evolutionary conserved, indicating the mutation was likely pathological (Fig. 2b). The predicted 3D structure of the mutant protein is dramatically impaired compared with the wide type (Fig. 2c).

Mutant protein structure and evolutionary conservation of amino acid residues. a Schematics of the secondary structure of functional domains of the STK11 protein. NLS nuclear localization signal, NRD or CRD, N- or C-terminal regulatory domain. b Evolutionary conservation of amino acid residues altered by c.426–448delCGTGCCGGAGAAGCGTTTCCCAG (p.S142SfsX13) across different species. c The mutant protein (p.S142SfsX13) was predicted to result in loss of part of the kinase domain and complete C-terminal regulatory domain by Swiss-Model online software compared to the wild type
The mutation was only found in the patient, but not in her parents, which means it is a de-novo mutation. Moreover, this genetic variation was not detected in her healthy relatives and 50 unrelated ethnicity-matched healthy individuals. Since it clearly co-segregates with the disease phenotype in the family, and the mutation is predicted to affect the key structure of STK11 protein, we conclude that this mutation is disease-specific and not a polymorphic variant of the STK11 gene.
Discussion
PJS is mainly caused by heterozygous germline mutation in STK11, and in this study we identified a novel de-novo mutation of STK11, c.426–448delCGTGCCGGAGAAGCGTTTCCCAG, in a Chinese PJS patient without a family history. This mutation possesses cancer risk, which also broadens the spectrum of cancer-related STK11 mutation.
The STK11 protein is mainly comprised of three major domains: an N-terminal regulatory domain, a catalytic kinase domain, and a C-terminal regulatory domain [15]. Among 396 reported STK11 mutations detected in patients with PJS or other disorders (HGMD Professional 2016.2) [16], most variants are located in the region of catalytic kinase domain (amino acids 49–309) and result in the absence of kinase activity and disrupting the formation of a complex to maintain kinase activation [17]. Several autophosphorylation sites within this area have been described [18]. Sequence alignment shows the mutation detected here causes translational frameshift (p.S142SfsX13), and the premature stop codon (codon 154) leads to large-scale protein truncation including most kinase domain. It is reasonable to conclude the pathogenicity of this mutation. Moreover, the C-terminal domain of STK11 protein is important for binding STRADα/β [19, 20], so lack of this part could also lessen the AMPKα activity, affect cell polarity, and impair downstream signaling [19].
The exact function of STK11 needs further investigating, and it is currently thought to play a role in cell signaling and apoptosis [21]. The elimination of STK11 kinase activity and the impaired interaction between STK11 and several proteins (P53, Cdc37, Hsp90, and PTEN) are probably responsible for PJS phenotype, which are mainly associated with a loss of growth suppression function [22]. STK11 induces cell cycle arrest and apoptosis on the dependence of P53. Overexpression of STK11 could elevate the transcriptional activity of P53, while its depletion inhibited P53 functions, which has been observed in patients of PJS-associated GI carcinomas [23, 24]. Transgenic mice experiments have proven a greater life span reduction and tumor incidence increasing in STK11 and P53 both knockout than single gene knockout [25]. In PJS, a mutation in key domain of STK11 gene may induce the function impaired of STK11 protein and the disturbance in its interaction with the partners, and eventually cause PJS phenotype, including the cancerogenesis.
Generally, identification of a STK11 gene mutation in an index patient offers the possibility of a predictive diagnosis in PJS pedigrees, but sometimes a patient without a positive family history is found clinically. Genetic testing often discovers a STK11 germline mutation carried only by the proband but not the parents, which is called a de-novo mutation. In these cases, clinicians should take de-novo mutation into consideration and be aware of the possible cancer risk, so that an appropriate surveillance recommendation can be suggested. The genotype–phenotype correlation in PJS is indeed existing but largely obscure, and PJS-associated cancer should be considered as a complex disease, with gene–gene and gene-environment interactions. So surveillance is as important as diagnosis for clinical management in PJS.
To treat PJS, DBE is the key method to use. When DBE is absent, PJS patients often suffer from recurrent intestinal obstruction and come to the surgical emergency [26]. With the widely use of DBE, PJS patients can be managed intensively to avoid emergency condition and potential cancer risk. People with positive family histories should receive first upper GI endoscopy and colonoscopy at age 8 years, and regular follow-up is necessary [27]. DBE is the most effective method to detect and remove small bowel polyps nonoperatively [28,29,30,31]. In our clinical center, 131 PJS patients who had abdominal surgery for intestinal obstruction before received DBE intervention, and 113 of them (86.3%) avoid a second open surgery, indicating proper follow-up ensures the patients free of intestinal obstruction and malignancy [32].
In conclusion, we identified a novel heterozygous mutation (c.426–448delCGTGCCGGAGAAGCGTTTCCCAG, p.S142SfsX13) in the STK11 gene causing PJS in a Chinese female without a PJS family history, and this mutation possesses high cancer risk. This study also expanded the mutation spectrum of PJS, which formed the fundament of genetic counseling.
Abbreviations
- AMPKα:
-
AMP-activated catalytic subunit alpha
- DBE:
-
Double-balloon enteroscopy
- dbSNP:
-
Database of single nucleotide polymorphisms
- GI:
-
Gastrointestinal
- HGMD:
-
Human Gene Mutation Database
- OMIM:
-
Online Mendelian Inheritance in Man
- PCR:
-
Polymerase chain reaction
- PJS:
-
Peutz–Jeghers syndrome
- STK11 :
-
Serine/threonine kinase 11
- STRADα/β:
-
STE20-related kinase adaptor alpha/beta
References
Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz–Jeghers syndrome. Gastroenterology. 2000;119:1447–1453.
Hearle N, Schumacher V, Menko FH, et al. Frequency and spectrum of cancers in the Peutz–Jeghers syndrome. Clin Cancer Res. 2006;12:3209–3215.
Giardiello FM, Trimbath JD. Peutz–Jeghers syndrome and management recommendations. Clin Gastroenterol Hepatol. 2006;4:408–415.
van Lier MG, Wagner A, Mathus-Vliegen EM, Kuipers EJ, Steyerberg EW, van Leerdam ME. High cancer risk in Peutz–Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol. 2010;105:1258–1264. (author reply 1265).
van Lier MG, Westerman AM, Wagner A, et al. High cancer risk and increased mortality in patients with Peutz–Jeghers syndrome. Gut. 2011;60:141–147.
Gao Y, Zhang FM, Huang S, et al. A De Novo mutation of STK11 gene in a Chinese patient with Peutz–Jeghers syndrome. Dig Dis Sci. 2010;55:1032–1036.
Peutz JL. Very remarkable case of familial polyposis of the mucous membrane of the intestinal tract and nasopharynx accompanied by peculiar pigmentation of the skin and mucous membrane. Ned Tijdschr Geneeskd. 1921;10:134–146.
Jeghers H, Mc KV, Katz KH. Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits: a syndrome of diagnostic significance. N Engl J Med. 1949;241:993. (illust; passim).
Jenne DE, Reimann H, Nezu J, et al. Peutz–Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998;18:38–43.
Hemminki A, Tomlinson I, Markie D, et al. Localization of a susceptibility locus for Peutz–Jeghers syndrome to 19p using comparative genomic hybridization and targeted linkage analysis. Nat Genet. 1997;15:87–90.
Buchet-Poyau K, Mehenni H, Radhakrishna U, Antonarakis SE. Search for the second Peutz–Jeghers syndrome locus: exclusion of the STK13, PRKCG, KLK10, and PSCD2 genes on chromosome 19 and the STK11IP gene on chromosome 2. Cytogenet Genome Res. 2002;97:171–178.
Wang ZJ, Churchman M, Avizienyte E, et al. Germline mutations of the LKB1 (STK11) gene in Peutz–Jeghers patients. J Med Genet. 1999;36:365–368.
Giardiello FM, Welsh SB, Hamilton SR, et al. Increased risk of cancer in the Peutz–Jeghers syndrome. N Engl J Med. 1987;316:1511–1514.
Biasini M, Bienert S, Waterhouse A, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–W258.
Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241:42–52.
Stenson PD, Mort M, Ball EV, et al. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136:665–677.
Boudeau J, Baas AF, Deak M, et al. MO25alpha/beta interact with STRADalpha/beta enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J. 2003;22:5102–5114.
Korsse SE, Peppelenbosch MP, van Veelen W. Targeting LKB1 signaling in cancer. Biochem Biophys Acta. 2013;1835:194–210.
Forcet C, Etienne-Manneville S, Gaude H, et al. Functional analysis of Peutz–Jeghers mutations reveals that the LKB1 C-terminal region exerts a crucial role in regulating both the AMPK pathway and the cell polarity. Hum Mol Genet. 2005;14:1283–1292.
Baas AF, Kuipers J, van der Wel NN, et al. Complete polarization of single intestinal epithelial cells upon activation of LKB1 by STRAD. Cell. 2004;116:457–466.
Shaw RJ, Kosmatka M, Bardeesy N, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA. 2004;101:3329–3335.
Mehenni H, Gehrig C, Nezu J, et al. Loss of LKB1 kinase activity in Peutz–Jeghers syndrome, and evidence for allelic and locus heterogeneity. Am J Hum Genet. 1998;63:1641–1650.
Liu L, Du X, Nie J. A novel de novo mutation in LKB1 gene in a Chinese Peutz Jeghers syndrome patient significantly diminished p53 activity. Clin Res Hepatol Gastroenterol. 2011;35:221–226.
Korsse SE, Biermann K, Offerhaus GJ, et al. Identification of molecular alterations in gastrointestinal carcinomas and dysplastic hamartomas in Peutz–Jeghers syndrome. Carcinogenesis. 2013;34:1611–1619.
Wei C, Amos CI, Stephens LC, et al. Mutation of Lkb1 and p53 genes exert a cooperative effect on tumorigenesis. Can Res. 2005;65:11297–11303.
van Lier MG, Mathus-Vliegen EM, Wagner A, van Leerdam ME, Kuipers EJ. High cumulative risk of intussusception in patients with Peutz–Jeghers syndrome: time to update surveillance guidelines? Am J Gastroenterol. 2011;106:940–945.
Beggs AD, Latchford AR, Vasen HF, et al. Peutz–Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010;59:975–986.
Ohmiya N, Taguchi A, Shirai K, et al. Endoscopic resection of Peutz–Jeghers polyps throughout the small intestine at double-balloon enteroscopy without laparotomy. Gastrointest Endosc. 2005;61:140–147.
May A, Nachbar L, Ell C. Double-balloon enteroscopy (push-and-pull enteroscopy) of the small bowel: feasibility and diagnostic and therapeutic yield in patients with suspected small bowel disease. Gastrointest Endosc. 2005;62:62–70.
Burke CA, Santisi J, Church J, Levinthal G. The utility of capsule endoscopy small bowel surveillance in patients with polyposis. Am J Gastroenterol. 2005;100:1498–1502.
Parsi MA, Burke CA. Utility of capsule endoscopy in Peutz–Jeghers syndrome. Gastrointest Endosc Clin N Am. 2004;14:159–167.
Zhang ZC, Li BR, Li X, et al. Location, growth and clinical outcome of polyps of patients with Peutz–Jeghers syndrome. Chin J Dig. 2016;36:593.
Acknowledgments
This work was supported by Application Research of Capital Clinical Character (Z151100004015215), Annual Project of Air Force General Hospital (KZ2015026 and KZ2016021) and National Natural Science Foundation of China (81500490). The Authors thank the subjects for their participation.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
None.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhao, ZY., Jiang, YL., Li, BR. et al. A 23-Nucleotide Deletion in STK11 Gene Causes Peutz–Jeghers Syndrome and Malignancy in a Chinese Patient Without a Positive Family History. Dig Dis Sci 62, 3014–3020 (2017). https://doi.org/10.1007/s10620-017-4741-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-017-4741-5