
Abstract
Background
The cystic fibrosis transmembrane conductance regulator (CFTR) gene, responsible for the development of cystic fibrosis, is known as a pancreatitis susceptibility gene. Direct DNA sequencing of PCR-amplified CFTR gene segments is a first-line method to detect unknown mutations, but it is a tedious and labor-intensive endeavor given the large size of the gene (27 exons, 1,480 amino acids). Next-generation sequencing (NGS) is becoming standardized, reducing the cost of DNA sequencing, and enabling the generation of millions of reads per run. We here report a comprehensive analysis of CFTR variants in Japanese patients with chronic pancreatitis using NGS coupling with target capture.
Methods
Exon sequences of the CFTR gene from 193 patients with chronic pancreatitis (121 idiopathic, 46 alcoholic, 17 hereditary, and nine familial) were captured by HaloPlex target enrichment technology, followed by NGS.
Results
The sequencing data covered 91.6 % of the coding regions of the CFTR gene by ≥20 reads with a mean read depth of 449. We could identify 12 non-synonymous variants including three novel ones [c.A1231G (p.K411E), c.1753G>T (p.E585X) and c.2869delC (p.L957fs)] and seven synonymous variants including three novel ones in the exonic regions. The frequencies of the c.4056G>C (p.Q1352H) and the c.3468G>T (p.L1156F) variants were higher in patients with chronic pancreatitis than those in controls.
Conclusions
Target sequence capture combined with NGS is an effective method for the analysis of pancreatitis susceptibility genes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Chronic pancreatitis (CP) is a progressive inflammatory disease that eventually leads to impairment of the exocrine and endocrine functions of the organ [1, 2]. Since the identification of mutations in the cationic trypsinogen (protease, serine, 1; PRSS1) gene as a cause of hereditary pancreatitis in 1996 [3], several pancreatitis susceptibility genes have been identified [3–6]. Gain-of-function mutations in the PRSS1 gene as well as loss-of-function variants in the serine protease inhibitor Kazal type 1 (SPINK1) gene and the trypsin-degrading enzyme chymotrypsin C (CTRC) increase the risk of CP [3–5]. In 2013, carboxypeptidase A1 (CPA1) gene was identified as a novel pancreatitis susceptibility gene [6]. These studies have been replicated in the Japanese population [7–10].
The cystic fibrosis transmembrane conductance regulator (CFTR) gene is another pancreatitis susceptibility gene [11, 12]. Acute recurrent pancreatitis and CP have been accepted as CFTR-related disorders (CFTR-RDs), a clinical entity associated with CFTR dysfunction that does not fulfill the diagnostic criteria for cystic fibrosis (CF; MIM# 219700) [13]. The CFTR gene, responsible for the development of CF, encodes for a cyclic adenosine monophosphate-dependent chloride channel that is located in the apical membrane of secretory and absorptive epithelial cells of the pancreas, intestine, liver, airways, vas deferens, and sweat glands [14]. In general, the clinical manifestations of CF arise from ductal and glandular obstruction because of an inability to hydrate macromolecules within the ductal lumens [15]. Until now, more than 1,900 variants have been identified in the Cystic Fibrosis Mutation Database (http://www.genet.sickkids.on.ca/cftr). The human CFTR gene spans 250 kb and contains 27 exons that encode for a protein with a total length of 1,480 amino acids [14]. Direct DNA sequencing of polymerase chain reaction (PCR)-amplified CFTR gene segments is a first-line method to detect unknown CFTR mutations [16], but this is a tedious and labor-intensive endeavor given the large size of the gene.
A new approach that uses massive parallel sequencing called next-generation sequencing (NGS) is becoming standardized, and the cost is rapidly dropping [17]. Using the ultimate platforms, such systems are able to perform billions of sequencing reactions with a read length of 150–250 nucleotides. For most research groups, whole-genome sequencing of many samples remains a costly endeavor, and targeted capture of selected regions of interest followed by sequencing provides a more efficient and cost-effective option. This strategy has allowed identification of causal variants in several Mendelian disorders, variants associated with complex diseases, and recurrently mutated cancer genes [18–20]. The HaloPlex target enrichment technology is a selective circularization-based method that is a further development of the principle of selector probes [21]. In the HaloPlex technology, genomic DNA is fragmented by restriction enzyme digestion and circularized by hybridization to probes whose ends are complementary to the target fragments. Compared to hybrid capture methods, the HaloPlex system requires smaller amounts of starting DNA, has higher specificity (fraction of sequence reads in the region of interest), and provides more uniform genome coverage [22]. Using the bench-top Illumina MiSeq platform, comprehensive analysis of many samples can be easily done. We here report the comprehensive analysis of CFTR variants in Japanese patients with CP.
Materials and Methods
Subjects
One hundred and ninety-three patients with CP (121 idiopathic, 46 alcoholic, 17 hereditary, and nine familial) were enrolled in this study. Because we initially aimed to identify novel pancreatitis-associated genes using the HaloPlex system, majority of the patients recruited were nonalcoholic. To extend our findings, we additionally recruited patients with alcoholic CP who had developed CP at relatively younger ages (mean: 44.1 years old). The diagnosis of CP was based on at least two separate episodes of abdominal pain and radiological findings of pancreatic calcifications by computed tomography, endoscopic ultrasonography, and/or morphological findings such as pancreatic ductal irregularities and dilatations revealed by endoscopic retrograde cholangiopancreatography (ERCP) or by magnetic resonance imaging [23]. Hereditary pancreatitis was diagnosed when one first-degree relative or two or more second-degree relatives had recurrent acute pancreatitis or CP without any apparent predisposing factor [24]. Patients with CP in whom the criteria for hereditary pancreatitis were not met but who had at least two affected family members were classified as having familial pancreatitis. Idiopathic CP was diagnosed in the absence of a positive family history or possible predisposing factors such as alcohol abuse, trauma, medication, and anatomical abnormalities. Patients who consumed alcohol over 80 g/day (for men) or 60 g/day (for women) for more than 2 years were classified as alcoholic CP. All subjects were Japanese. This study was performed with the informed consent of the patients in accordance with the principles of the declaration of Helsinki. This study was approved by the Ethics Committee of Tohoku University School of Medicine (article#: 2013-1-498).
Peripheral Blood Collection and DNA Preparation
After written informed consent was obtained, 5–10 mL of peripheral blood was collected in disposable vacuum tubes for genetic testing. Genomic DNA was extracted from peripheral blood leukocytes using the Wizard genomic DNA purification kit (Promega, Madison, WI, USA).
Targeted Next-Generation Sequencing
We used the online design tool SureDesign to generate a customized HaloPlex target enrichment system (Agilent Technologies, Santa Clara, CA, USA) targeting the regions including CFTR exons and 50 base pairs (bp) of flanking introns. The expected coverage of the CFTR coding region based on the amplicon design was 99.6 %. The HaloPlex target enrichment system relies on a tailored cocktail of restriction enzymes and customized probes to capture genomic regions of interest, which are subsequently amplified by PCR. Sequencing libraries were prepared according to the manufacturer’s instructions. Briefly, genomic DNA was digested with restriction enzymes, followed by hybridization to the biotinylated HaloPlex probe library in the presence of the indexing primer cassette. Hybridization results in the circularization of genomic DNA fragments and incorporation of indices and Illumina sequencing motifs. Hybridized probes were captured with streptavidin-coated magnetic beads. Subsequently, libraries were amplified by PCR to produce a sequencing-ready, target-enriched sample.
All libraries of target-enriched DNA were analyzed on a 2200 TapeStation (Agilent Technologies) to verify successful enrichment. All samples were sequenced on the Illumina MiSeq platform (Illumina Japan, Tokyo, Japan) with paired-end 151-bp reads according to the manufacturer’s instruction.
Bioinformatic Analysis of Sequencing Data
The reads were trimmed with the utility program Trim Galore (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) to remove possible adapter sequences, based on the Illumina TruSeq adapter index sequences. If either read from a pair was shorter than 20 bp after trimming, that pair was removed from the analysis. The remaining quality reads were mapped to the GRCh37 primary assembly of the human genome (http://ensembl.org/) using the Burrows–Wheeler Alignment tool 0.6.1 (http://bio-bwa.sourceforge.net/). Further sequence data processing, assessment of coverage rates, variant calling, and filtration were performed with the Genome Analysis Toolkit, version 1.6 software (Broad Institute, Cambridge, MA, USA; http://www.broadinstitute.org/gatk/). SNPs and insertions/deletions (indels) were annotated using the ANNOVAR (http://www.openbioinformatics.org/annovar/; BIOBASE, Wolfenbüttel, Germany).
Sanger Sequencing
Sanger sequencing was performed to analyze the DNA sequences which included any nucleotide variant identified by NGS. Exons and adjacent intronic regions of the CFTR gene containing the nucleotide variants were amplified by PCR using the primer sets (Table 1). The cycle conditions were as follows: preheating at 95 °C for 5 min, followed by 40 cycles of 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s; and then a final extension at 72 °C for 5 min. PCR products were cleaned up using the Illustra ExoProStar S (GE Healthcare Life Sciences; Little Chalfont, UK). The PCR products were sequenced using an ABI Prism BigDye Terminator Cycle Sequencing Kit, version 3.1 on ABI3730xl DNA Analyzer (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s instructions. The results were compared with the reference sequence derived from the GenBank (http://www.ncbi.nlm.nih.gov/genbank, reference sequence NM_000492) to identify nucleotide substitutions. The A of the ATG start codon was used as nucleotide +1. The mutations are described according to the nomenclature recommended by the Human Genome Variation Society (http://www.hgvs.org/mutnomen).
In addition, all exons and adjacent intronic regions of the PRSS1, SPINK1, CTRC, and CPA1 genes were amplified by PCR and directly sequenced as previously reported [5, 6, 8, 25].
In Silico Prediction
SIFT (Sorting Intolerant From Tolerant; http://sift.jcvi.org/) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) were used to predict whether an amino acid substitution would affect the structure and function of a protein. SIFT uses sequence homology, whereas PolyPhen-2 offers predictions based on conservation, protein folding, and crystal structure [26, 27]. The SIFT scores range from zero to one, with zero predicted to be the most deleterious mutation and one the least deleterious. The PolyPhen-2 scoring predicts three outcomes for mutations: “benign” (most likely lacking any phenotypic effect), “possibly damaging” (may affect protein structure or function), and “probably damaging” (high degree of confidence that protein structure function will be affected).
Statistical Analysis
The variant frequencies in the Japanese population were obtained from the Human Genetic Variation Database (www.genome.med.kyoto-u.ac.jp/SnpDB/). The significance of the differences in variant frequencies between patients and controls was tested by two-tailed Fisher’s exact test. A P value <0.05 was considered significant. All statistical analyses were performed using the SPSS version 17.0 statistical analysis software (SPSS Inc., Chicago, IL, USA).
Results
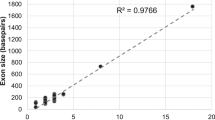
Approximately 10 kb of the coding regions and the adjacent noncoding regions of the CFTR gene were analyzed in this study. On average, 98.8, 97.0, and 95.1 % of the coding regions of the CFTR gene were covered by at least one, five, and 10 sequence reads, respectively. The sequencing data covered 91.6 % of the coding regions of the CFTR gene by ≥20 reads with a mean read depth of 449 and a median depth of 412 (Fig. 1). These results indicate a high-resolution capability for the identification of variants, such as mutations.

Graph of the mean depth, median depth, and sequencing coverage for all the 27 exons in the CFTR gene. By MiSeq NGS, a high-quality sequence was obtained for 27 exons and the flanking sequences from the CFTR gene, including a mean depth of ×516 and a median depth of ×442. On average, 90.3 % of the coding region was successfully covered by ≥20 reads
In our cohort of 193 CP patients, we identified 12 non-synonymous and seven synonymous variants in the exons of the CFTR gene by targeted NGS (Tables 2, 3, 4, 5). The presence of these variants was confirmed by Sanger sequencing. Based on the presence in dbSNP137, Exome Variant Server (NHLBI GO Exome Sequencing Project, Seattle, WA, USA; URL: http://evs.gs.washington.edu/EVS/), and the Human Genetic Variation Database, three non-synonymous variants [c.1231A>G (p.K411E), c.1753G>T (p.E585X) and c.2869delC (p.L957fs)] and three synonymous variants (c.372C>T, c.3975A>G and c.4254G>A) were novel. The frameshift variant c.2869delC (p.L957fs) leads to a stop codon afterward at amino acid 967, to premature termination of translation and a heavily truncated protein missing more than one-third of its amino acids. This variant was found in a 22-year-old female with idiopathic CP. She was admitted due to a pancreatitis attack and diagnosed as having CP. She had suffered from back and abdominal pain since 20 years old. The value of the n-benzoyl-l-tyrosyl-p-aminobenzoic acid test was 52 % (normal: >70 %), suggesting pancreatic exocrine dysfunction. The patient also had the c.4056G>C (p.Q1352H) variant in a heterozygous form (Table 6). The nonsense variant c.1753G>T (p.E585X) was found in a patient with alcoholic CP. He was diagnosed as having alcoholic CP at 28 years old. The c.1231A>G (p.411E) variant was found in a 19-year-old male with idiopathic CP. He had suffered from pancreatitis attacks since 12 years old. ERCP showed multiple stones in the main pancreatic duct. He underwent extracorporeal shock wave lithotripsy for the treatment of pancreatic stones. The patient also had the c.3468G>T (p.L1156F) variant in a heterozygous form. None of these three patients had known pancreatitis susceptibility mutations in the PRSS1, SPINK1, CTRC, or CPA1 genes (Table 6). All of the patients carrying the novel synonymous variants were idiopathic CP (Table 4).
The frequency of the c.4056G>C (p.Q1352H) variant was higher in all patients with CP than that in controls (P = 0.009; Table 3). Stratification based on the etiologies showed that the association was significant in patients with nonalcoholic CP (combination of cases with idiopathic, hereditary, and familial CP) (P = 0.037). The frequency of the c.3468G>T (p.L1156F) variant was also higher in patients with CP than that in controls (P = 0.04). There were no significant difference for any other non-synonymous or synonymous variants detected in the exons between all patients with CP and controls (Tables 3, 5). The frequency of the c.2562T>G variant was different between the controls and the patients with idiopathic or hereditary/familial CP.
The 5T and, more rarely, 3T splicing variants of the intron 9 acceptor splice site [c.1210-12T(5_9)] are considered to be variants associated with CFTR-RD [16]. The 5T or 3T allele is a polymorphic variant with variable penetrance, causing less efficient exon 10 splicing and a lower CFTR transcript level [28]. The splicing efficiency of exon 10 is further affected by the length of the adjacent TG repeat [c.1210-34TG(9_13)]. The distribution of the c.1210-34TG(9_13) and c.1210-12T(5_9) variants is shown in Table 7. In our cohort, nine patients with CP had the 5T allele, all in a heterozygous form. Four patients (two alcoholic, one idiopathic, one hereditary) had the 5T-TG13. No patient had the haplotype TG10-7T-M470, which was reported to increase the risk of idiopathic CP [28].
It has been increasingly recognized that compound and trans-heterozygosity in the pancreatitis susceptibility genes are an overt risk factor for idiopathic CP [29–32]. Among the 193 patients with CP enrolled in this study, 29 patients had pancreatitis-associated mutations in the PRSS1, SPINK1, CTRC, and CPA1 genes. Among these, nine patients had the non-synonymous CFTR variants, which are probably damaging based on the SIFT and/or the PolyPhen-2 prediction (Table 8).
Discussion
In this study, we performed comprehensive analysis of the variants in the CFTR gene by targeted NGS. To our knowledge, this is the first study to analyze pancreatitis susceptibility genes by targeted NGS. Comprehensive analysis by targeted NGS enabled us to identify novel and rare variants in the CFTR gene. The c.1753G>T (p.E585X) variant is a nonsense variant, and the c.2869delC (p.L957fs) variant leads to a stop codon afterward at amino acid 967. These variants result in a heavily truncated protein missing nearly two-thirds (p.E585X) or more than one-third (p.L957fs) of its amino acids. Because we did not perform functional assays, we do not have direct evidence that these two variants cause loss of the CFTR expression and/or function. However, a general acknowledgment has been agreed that mutations of this type, called class I mutations, are associated with complete loss or near complete loss of the CFTR function (<3 % of wild-type CFTR function) [33, 34]. The pathogenic potential of another novel variant, c.1231A>G (p.K411E), is currently unknown, but the in silico analyses suggest that this variant is deleterious. Importantly, the clinical phenotype of this patient might be complicated by the presence of another variant, p.L1156F. Noone et al. [30] reported that pancreatitis risk was increased approximately 40-fold by having two CFTR mutations. This is also the case with the c.374T>C (p.I125T) variant. Two of the three patients carrying this variant had other non-synonymous variants (p.I556V and p.R1453W). This p.I125T variant was originally reported in Chinese patients with idiopathic bronchiectasis and considered to be associated with CFTR-RD [35].
There are considerable regional and ethnic variations in the spectrum of the CFTR mutations [15]. Approximately 70 % of individuals with CF in the Caucasian population are homozygous for the F508del mutation, and almost 90 % of the patients have at least one F508del allele [36]. This mutation is extremely rare in the Japanese population, accounting for the rare presentation of classical CF in this region (approximately 1/350,000 live births) [37]. It is not surprising that the CF-causing mutations are frequently found in Caucasians, but very rarely in East Asia. Audrézet et al. [29] reported from France that at least 20 % of the patients with idiopathic CP carried one of the most common CFTR mutations. Fujiki et al. [38] reported from Japan that none of the 20 common CF-causing mutations was found in 65 Japanese patients with CP (51 alcoholic and 14 idiopathic). Wang et al. [39] reported comprehensive screening of pancreatitis susceptibility genes including CFTR in 75 pediatric patients with idiopathic CP from China. They identified a novel 8-bp deletion in exon 4, but not the common CF-causing mutations. In this study, we found no common severe CF-causing mutations, in agreement with these previous studies from East Asia.
We found a significant association between the p.Q1352H variant and CP. This finding confirms the previous reports from Japan and Korea showing that this variant was over-presented in patients with CP compared to controls [38, 40]. Fujiki et al. [38] from Japan reported that the frequency of this variant was higher in patients with CP (8/65, 12.3 %) than in controls (6/162, 3.7 %). Lee et al. [40] reported from Korea that 14.3 % (4/28) of the patients with CP had this variant, whereas only 0.9 % (1/117) of the controls did. Glutamine at 1,352 is located in the second nucleotide-binding fold of CFTR, and its change to histidine (p.Q1352H) causes reductions in both the protein expression and channel activity of CFTR [40]. Similarly, we found that the p.L1156F variant was overexpressed in patients with CP. A functional study reported reduced Cl−/HCO3 − permeability in the presence of the p.L1156F variant [41].
Gene–gene interactions of known pancreatitis susceptibility genes, especially between the CFTR and SPINK1 genes, have been increasingly recognized. Indeed, seven out of 25 patients carrying the SPINK1 variant(s) had the CFTR p.Q1352H and/or p.L1156F variants. One patient was trans-heterozygous for the CTRC p.R29Q and CFTR p.I125T/p.L1156F variants. Noone et al. [30] reported that pancreatitis risk was increased approximately 40-fold by having two CFTR mutations, 20-fold by having the SPINK1 p.N34S variant, and 900-fold by having both. Trans-heterozygosity of the SPINK1 p.N34S with the CFTR p.R75Q was reported to increase CP risk [31]. 6.5 % of the patients with idiopathic or hereditary CP carried variants in at least two pancreatitis susceptibility genes [32]. Whether the coinheritance of variants/mutations in pancreatitis susceptibility gene is a bona fide example of digenic inheritance or interaction between a disease-causing gene and a genetic modifier is unclear in most cases [42].
We used targeted sequence capture and high-throughput NGS to detect variants in the CFTR gene. Due to the large size (27 exons, 1,480 amino acids), traditional technologies, such as PCR and capillary sequencing, are time- and cost-consuming. A major advantage of the HaloPlex-targeted enrichment system is the convenient workflow, integrating both capture and library preparation. The protocol allows one person to prepare a set of finished libraries within two working days and requires no larger specialized instruments. Sequence capture eliminates the necessity of setting up hundreds of PCR, instead allowing for parallel enrichment of target regions in a single experiment. A weakness of this method is that the detection of larger copy number variations would require different methods. We have designed the HaloPlex platform for more than 70 genes, including the known pancreatitis susceptibility genes such as CFTR, PRSS1, SPINK1, CTRC, and CPA1. This system has allowed us to perform rapid screening of the known susceptibility genes simultaneously and gives an overview of potentially pathogenic variants in patients with pancreatitis. In addition, our HaloPlex platform includes candidates of novel pancreatitis susceptibility genes such as pancreatic digestive enzymes, those highly expressed in the pancreas and those related to autophagy and endoplasmic reticulum stress. This system might contribute to the identification of novel pancreatitis susceptibility genes in the future.
Abbreviations
- bp:
-
Base pair
- CF:
-
Cystic fibrosis
- CFTR:
-
Cystic fibrosis transmembrane conductance regulator
- CP:
-
Chronic pancreatitis
- ERCP:
-
Endoscopic retrograde cholangiopancreatography
- NGS:
-
Next-generation sequencing
- PCR:
-
Polymerase chain reaction
- RD:
-
Related disorder
- SIFT:
-
Sorting Intolerant From Tolerant
References
Steer ML, Waxman I, Freedman S. Chronic pancreatitis. N Engl J Med. 1995;332:1482–1490.
Witt H, Apte MV, Keim V, Wilson JS. Chronic pancreatitis: challenges and advances in pathogenesis, genetics, diagnosis, and therapy. Gastroenterology. 2007;132:1557–1573.
Whitcomb DC, Gorry MC, Preston RA, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet. 1996;14:141–145.
Witt H, Luck W, Hennies HC, et al. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet. 2000;25:213–216.
Rosendahl J, Witt H, Szmola R, et al. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat Genet. 2008;40:78–82.
Witt H, Beer S, Rosendahl J, et al. Variants in CPA1 are strongly associated with early onset chronic pancreatitis. Nat Genet. 2013;45:1216–1220.
Masamune A. Genetics of pancreatitis: the 2014 update. Tohoku J Exp Med. 2014;232:69–77.
Kume K, Masamune A, Mizutamari H, et al. Mutations in the serine protease inhibitor Kazal Type 1 (SPINK1) gene in Japanese patients with pancreatitis. Pancreatology. 2005;5:354–360.
Masamune A, Nakano E, Kume K, Kakuta Y, Ariga H, Shimosegawa T. Identification of novel missense CTRC variants in Japanese patients with chronic pancreatitis. Gut. 2013;62:653–654.
Masamune A, Nakano E, Kume K, Takikawa T, Kakuta Y, Shimosegawa T. PRSS1 c.623G>C (p.G208A) variant is associated with pancreatitis in Japan. Gut. 2014;63:336.
Sharer N, Schwarz M, Malone G, et al. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N Engl J Med. 1998;339:645–652.
Cohn JA, Friedman KJ, Noone PG, Knowles MR, Silverman LM, Jowell PS. Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N Engl J Med. 1998;339:653–658.
Bombieri C, Claustres M, De Boeck K, et al. Recommendations for the classification of diseases as CFTR-related disorders. J Cyst Fibros. 2011;10:S86–S102.
Riordan JR, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073.
Ratjen F, Döring G. Cystic fibrosis. Lancet. 2003;361:681–689.
Dequeker E, Stuhrmann M, Morris MA, et al. Best practice guidelines for molecular genetic diagnosis of cystic fibrosis and CFTR-related disorders–updated European recommendations. Eur J Hum Genet. 2009;17:51–65.
Metzker ML. Sequencing technologies-the next generation. Nat Rev Genet. 2009;11:31–46.
Gilissen C, Hoischen A, Brunner HG, Veltman JA. Unlocking Mendelian disease using exome sequencing. Genome Biol. 2011;12:228.
Do R, Kathiresan S, Abecasis GR. Exome sequencing and complex disease: practical aspects of rare variant association studies. Hum Mol Genet. 2012;21:R1–R9.
Myllykangas S, Ji HP. Targeted deep resequencing of the human cancer genome using next-generation technologies. Biotechnol Genet Eng Rev. 2010;27:135–158.
Berglund EC, Lindqvist CM, Hayat S, et al. Accurate detection of subclonal single nucleotide variants in whole genome amplified and pooled cancer samples using HaloPlex target enrichment. BMC Genom. 2013;14:856.
Mertes F, Elsharawy A, Sauer S, et al. Targeted enrichment of genomic DNA regions for next-generation sequencing. Brief Funct Genomics. 2011;10:374–386.
Etemad B, Whitcomb DC. Chronic pancreatitis: diagnosis, classification, and new genetic developments. Gastroenterology. 2001;120:682–707.
Howes N, Lerch MM, Greenhalf W, et al. European Registry of Hereditary Pancreatitis and Pancreatic Cancer (EUROPAC). Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin Gastroenterol Hepatol. 2004;2:252–261.
Nishimori I, Kamakura M, Fujikawa-Adachi K, et al. Mutations in exons 2 and 3 of the cationic trypsinogen gene in Japanese families with hereditary pancreatitis. Gut. 1999;44:259–263.
Sunyaev S, Ramensky V, Koch I, Lathe W III, Kondrashov AS, Bork P. Prediction of deleterious human alleles. Hum Mol Genet. 2001;10:591–597.
Dorfman R, Nalpathamkalam T, Taylor C, et al. Do common in silico tools predict the clinical consequences of amino-acid substitutions in the CFTR gene? Clin Genet. 2010;77:464–473.
Steiner B, Rosendahl J, Witt H, et al. Common CFTR haplotypes and susceptibility to chronic pancreatitis and congenital bilateral absence of the vas deferens. Hum Mutat. 2011;32:912–920.
Audrézet MP, Chen JM, Le Maréchal C, et al. Determination of the relative contribution of three genes-the cystic fibrosis transmembrane conductance regulator gene, the cationic trypsinogen gene, and the pancreatic secretory trypsin inhibitor gene-to the etiology of idiopathic chronic pancreatitis. Eur J Hum Genet. 2002;10:100–106.
Noone PG, Zhou Z, Silverman LM, Jowell PS, Knowles MR, Cohn JA. Cystic fibrosis gene mutations and pancreatitis risk: relation to epithelial ion transport and trypsin inhibitor gene mutations. Gastroenterology. 2001;121:1310–1319.
Schneider A, Larusch J, Sun X, et al. Combined bicarbonate conductance-impairing variants in CFTR and SPINK1 variants are associated with chronic pancreatitis in patients without cystic fibrosis. Gastroenterology. 2011;140:162–171.
Rosendahl J, Landt O, Bernadova J, et al. CFTR, SPINK1, CTRC and PRSS1 variants in chronic pancreatitis: is the role of mutated CFTR overestimated? Gut. 2013;62:582–592.
Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73:1251–1254.
Castellani C, Cuppens H, Macek M Jr, et al. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. J Cyst Fibros. 2008;7:179–196.
Ngiam NS, Chong SS, Shek LP, et al. Cystic fibrosis transmembrane conductance regulator (CFTR) gene mutations in Asians with chronic pulmonary disease: a pilot study. J Cyst Fibros. 2006;5:159–164.
Watson MS, Cutting GR, Desnick RJ, et al. Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genet Med. 2004;6:387–391.
Yamashiro Y, Shimizu T, Oguchi S, Shioya T, Nagata S, Ohtsuka Y. The estimated incidence of cystic fibrosis in Japan. J Pediatr Gastroenterol Nutr. 1997;24:544–547.
Fujiki K, Ishiguro H, Ko SB, et al. Genetic evidence for CFTR dysfunction in Japanese: background for chronic pancreatitis. J Med Genet. 2004;41:e55.
Wang W, Sun XT, Weng XL, et al. Comprehensive screening for PRSS1, SPINK1, CFTR, CTRC and CLDN2 gene mutations in Chinese paediatric patients with idiopathic chronic pancreatitis: a cohort study. BMJ Open. 2013;3:e003150.
Lee JH, Choi JH, Namkung W, et al. A haplotype-based molecular analysis of CFTR mutations associated with respiratory and pancreatic diseases. Hum Mol Genet. 2003;12:2321–2332.
Ko S, Zeng W, Fujiki K, et al. Functional characterization of L1156F CFTR: a newly identified mutation in Japanese patients with chronic pancreatitis. J Physiol Sci. 2006;56:S71. (abstract).
Chen JM, Férec C. Chronic pancreatitis: genetics and pathogenesis. Annu Rev Genomics Hum Genet. 2009;10:63–87.
Acknowledgments
The authors are grateful to Ms. Yoko Tateda for the excellent technical assistance. This work was supported in part by the Pancreas Research Foundation of Japan (to E. Nakano), the HIROMI Medical Research Foundation (to A. Masamune), the Mother and Child Health Foundation (to A. Masamune), the Smoking Research Foundation (to A. Masamune), and by the Ministry of Health, Labour, and Welfare of Japan.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Nakano, E., Masamune, A., Niihori, T. et al. Targeted Next-Generation Sequencing Effectively Analyzed the Cystic Fibrosis Transmembrane Conductance Regulator Gene in Pancreatitis. Dig Dis Sci 60, 1297–1307 (2015). https://doi.org/10.1007/s10620-014-3476-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-014-3476-9