
Abstract
Background and Aims
Hepatoportal sclerosis (HPS) is a clinicopathologic condition that is clinically characterized by portal hypertension (varices and portosystemic collateral vessels), splenomegaly and pancytopenia, in the absence of cirrhosis. Although the etiology is obscure, a number of theories such as immunologic and vascular endothelial cellular abnormalities have been put forward to explain the underlying pathophysiology. Angiotensin-converting enzyme (ACE), an important molecule of the renin–angiotensin system (RAS), is also known as a regulatory molecule in systemic and portal circulation in distinct disorders. The aim of the present study was to investigate the possible role of the ACE in the context of RAS in HPS pathogenesis.
Materials and Methods
The study was conducted on 30 HPS patients (16 men, 14 women; median age 36 years, range 18–63) and 20 healthy controls. The clinical features of HPS patients including demographics, laboratory, and ultrasonography findings were summarized. Serum ACE levels were measured by using commercially available kits.
Results
Serum median ACE levels were 36 (8–174) U/l and 16 (8–43) U/l for the HPS patients and controls, respectively. Serum ACE levels were significantly higher in patients with HPS compared to the control group (P < 0.05).
Conclusion
ACE in the context of RAS may be associated with pathological endothelial occlusive events in the microenvironment of the portal circulation in HPS. Revealing the interactions between circulating and local RAS within the hepatic microenvironment would enlighten the biologic basis and clinical management of liver diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Idiopathic hepatoportal sclerosis (HPS) is a clinical disorder of unknown etiology characterized by splenomegaly, anemia, and portal hypertension in the absence of cirrhosis, blood disease, parasites in the hepatobiliary system, and occlusion of the hepatic and portal veins [1, 2]. Several different synonyms have been used, other than HPS, for describing this entity, such as Banti’s disease, idiopathic portal hypertension, non-cirrhotic portal fibrosis with portal hypertension, benign intrahepatic portal hypertension, obliterative portal venopathy, nodular regenerative hyperplasia, and incomplete septal cirrhosis [3–10]. The principal pathologic changes of HPS are devastation of the intrahepatic terminal portal radicles with considerable portal fibrosis and secondary atrophy, sclerosis of portal vein branches and dilatation of sinusoidal spaces, resulting in non-cirrhotic portal hypertension [11–13]. Vascular endothelial cellular abnormalities contribute to the prothrombotic state of HPS [14–16].
Angiotensin-converting enzyme (ACE), an important molecule of the renin–angiotensin system (RAS) is also considered as a regulatory molecule in systemic and portal circulation in distinct disorders [17–20]. Demonstration of the local RASs at the tissue levels [18, 21, 22] further implicated the importance of ACE in the pathobiology of a wide variety of diseases. The aim of this study is to assess circulating ACE concentrations in HPS. The hypothesis was that circulating ACE may reflect ongoing pathological endothelial occlusive events in the microenvironment of the portal circulation. Elucidation of the associations between ACE in the context of RAS and HPS may help toward a better understanding the enigmatic pathogenesis of HPS.
Patients and Methods
The study was carried out in our tertiary reference center (Turkiye Yuksek Ihtisas Training and Research Hospital, Department of Gastroenterology) between June 2008 and January 2010. The study population included 30 HPS patients and 20 healthy controls. The diagnosis of HPS was considered when the following criteria were fulfilled:
-
1.
Evidence of portal hypertension (any one of the following features: esophageal varices on endoscopy, ascites, hypersplenism, or increased hepatic venous pressure gradient)
-
2.
Exclusion of obstruction of splenoportal axis on doppler ultrasound at the time of diagnosis
-
3.
Exclusion of conditions causing cirrhosis of the liver (chronic viral hepatitis, autoimmune hepatitis, alcoholic liver disease, hemochromatosis, Wilson’s disease or non-alcoholic steatohepatitis) on clinical, biochemical and ultrasonographic findings. Ultrasound findings included the absence of irregularities of intrahepatic portal vein radicles, thickening of the portal vein wall ≥3 mm (Fig. 1), a sudden narrowing of intra-hepatic second degree portal vein branches and smooth surface of the liver.
-
4.
Exclusion of chronic vitamin A intake, professional exposure to copper sulphate, arsenic salts or other toxic substrates
-
5.
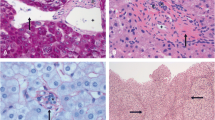
Liver biopsy was not thought to be mandatory except for the patients whose clinical and laboratory evaluation did not provide a definitive diagnosis. Liver biopsy was performed in just nine patients, while in the remaining patients, clinical, laboratory and radiological findings helped to exclude cirrhosis of the liver. Figure 2 shows the typical histologic appearance of HPS in one of the patients that underwent liver biopsy.
-
6.
If necessary, granulomatous and metabolic liver disease, Budd-Chiari syndrome, congenital hepatic fibrosis and other known liver disease were excluded by using appropriate diagnostic work-up.

Ultrasonographic view of a patient with HPS showing portal sclerosis at right hepatic lobe (arrow)

a Significant fibrosis surrounding hepatic central vein (arrow) and structural destruction due to fibrosis in portal vein (arrowhead; Masson’s trichrome stain, ×100); b fibrosis around hepatic central vein and herniation of central vein to hepatic parenchyme (arrow), significant fibrosis in portal vein (arrowhead; Masson’s trichrome stain, ×100)
None of the patients with HPS had any co-existing acute or chronic inflammatory diseases including hypertension, sarcoidosis, renal failure, diabetes mellitus or any other accompanying diseases that could influence ACE levels. None of the cases were receiving ACE inhibitors/Angiotensin receptor blockers or any other drug that could affect the renin–angiotensin (RAS) system. Twenty healthy controls were recruited from the healthy adults without any history of acute/chronic inflammatory disorders with no drug use history. The clinical features of HPS patients including demographics, clinical history, laboratory, ultrasonography and liver biopsy when needed were recorded. Serum ACE levels were measured and compared with healthy controls. The study was conducted in accordance with the guidelines of the Helsinki Declaration and written informed consents were obtained from each of the patients studied.
Serum ACE activity was measured by monitoring the alteration in absorbance at 340 nm of the hydrolysis of furylacrylolylphenylalanylglycylglycine (FAPGG) to FAP and GG (Sigma–Aldrich, Poole, UK) on an analyzer (Roche MIRA Analyser; Roche Diagnostic Systems, Welwyn Garden City, UK). The ACE activity in the sample was determined by comparing the sample reaction rate to that obtained with the ACE calibrator. Peripheral blood samples were also taken from each patients for complete blood counts and biochemical analysis.
Statistical analyses were performed via using PASW Statistics 18 (SPSS, Chicago, IL, USA). Statistically significant differences were analyzed by the χ 2 test for categorical variables. Continuous variables were tested for normality by the Kolmogorov–Smirnov test. Normally distributed data are presented as mean and standard deviations (SDs). Mann–Whitney U test was utilized for the independent subgroups. Two-tailed P values <0.05 was considered as statistically significant.
Results
Thirty patients with HPS and 20 control subjects were enrolled in the present study. There were 16 men and 14 women in the HPS group and 10 men and 10 women in the control group. The median age of HPS and control patients was 36 (18–63) and 41 years (19–65), respectively. There were no statistically significant differences between the ages of the study participants.
Clinical characteristics and complete blood cell counts values of study participants are summarized in Table 1. Biochemical values of the cases with HPS and controls are depicted in Table 2. There were no statistically significant differences between the two study groups with respect to routine biochemical levels.
The levels of serum ACE expressed as median and range. Serum ACE levels were 36 (8–174) and 16 (8–43) U/l for the HPS patients and controls, respectively. Serum ACE levels were significantly higher in patients with HPS compared to the control group (P < 0.05) (Fig. 3).

Serum ACE levels in patients with HPS and controls
Discussion
In this study, circulating ACE concentrations were significantly higher in patients with HPS in comparison to the control group. These results added a clue to the original hypothesis that ACE in the context of RAS may be associated with pathological endothelial occlusive events in the microenvironment of the portal circulation in HPS (Fig. 4).

Illustration of circulating renin–angiotensin system (RAS), local liver RAS acting in a paracrine fashion, regulating inflammation, fibrosis, angiogenesis, cell proliferation, apoptosis and thrombogenesis
HPS is caused by the endothelial abnormalities of the intra-hepatic vasculature. Intracellular processes that regulate the release/expression of vasoactive substances such as angiotensin peptides may promote the development of vasculopathy in portal hypertension [17]. The status of RAS in liver diseases has been investigated in several previous studies. Hepatic conversion of angiotensin I and the portal hypertensive response (PHR) to angiotensin II in normal and regenerating liver has been demonstrated by Carvalho et al. [18]. Angiotensin I is converted into Angiotensin II by the ACE and the sequential action of Angiotensin II on the Angiotensin II receptor type 1 located in the hepatic periportal zone. Angiotensin II induced PHR pattern changes during liver regeneration [18]. Increments in circulating ACE detected in our study may affect PHR in the portal circulation in patients with HPS. Angiotensin-converting enzyme (ACE), an important molecule of the renin–angiotensin system (RAS) is also considered as a regulatory molecule in systemic and portal circulation in distinct disorders [17–20]. In another study [23], ACE inhibition in the liver recipient by enalapril attenuated hepatic ischemia/reperfusion damage after experimental liver transplantation. ACE inhibitors were therefore suggested as an alternative approach to reduce ischemia/reperfusion injury in clinical liver transplantation [23]. After hepatic ischemia, ACE inhibitors reduced reperfusion injury. The treatment of reperfusion injury by Ramiprilat-mediated sinusoidal dilation and therefore blunting hepatic inflammation [24]. Small liver grafts generated angiotensin II after experimental liver transplantation, associated with increased angiotensinogen and ACE messenger RNA expression [25]. Therefore, this issue is not just academic as elucidation of RAS in HPS may open new avenues for the clinical management of this difficult patient population. Studies have also shown that the blockade of the RAS pathway stimulates liver regeneration and inhibits tumor progression. An understanding of the role of RAS in liver regeneration and tumorigenesis may enable alternative strategies to improve patient outcome and survival after liver resection [26].
The RAS system is overactivated in most cirrhotic patients with ascites due to a reduced effective arterial blood volume, a compensatory response to the marked splanchnic arterial vasodilatation, and has long been implicated in the pathogenesis of portal hypertension. Angiotensin II is linked with dynamic and static increases in intrahepatic resistance due to the contraction and proliferation of human hepatic stellate cells and the deposition of fibrous tissue [27–30]. Moreover, in addition to its well-known action in increasing portal venous inflow via sodium and water retention, the activation of aldosterone has been associated with increases in inflammation, oxidative stress, endothelial dysfunction, and fibrosis (Fig. 5) [31, 32].

Contribution of angiotensin II and aldosterone to the pathogenesis of portal hypertension as a part of renin–angiotensin–aldosterone system
RAS could also play a major role in liver fibrosis since the circulatory RAS is frequently activated in patients with chronic liver disease. Bataller et al. [33] investigated the contribution of RAS to the fibrosis progression. To test this hypothesis, they increased circulatory angiotensin II levels in rats undergoing biliary fibrosis. In their study, angiotensin II infusion increased serum levels of angiotensin II and augmented bile duct ligation-induced liver injury, as assessed by elevated liver serum enzymes. Moreover, Angiotensin II increased the hepatic concentration of inflammatory proteins (tumor necrosis factor alpha and interleukin 1beta) and the infiltration of CD43-positive inflammatory cells [33]. In the Bataller study [33], Ang II infusion also favored the development of vascular thrombosis and increased the procoagulant activity of tissue factor in the liver. Increased systemic Ang II augmented hepatic fibrosis and promoted inflammation, oxidative stress, cellular proliferation, and thrombogenic events. HPS is characterized by varying degrees of portal fibrosis, sclerosis of portal vein branches and dilatation of sinusoidal spaces, resulting in non-cirrhotic portal hypertension [11, 12]. Vascular endothelial cellular abnormalities contribute to the prothrombotic state of HPS [14–16]. Increments in circulating ACE detected in our study may be related to the portal fibrosis in our studied patients with HPS.
Portal hypertension may be associated with a hyperdynamic circulatory syndrome with high cardiac output and reduced systemic vascular resistance in patients with end-stage liver disease [19]. The hyperdynamic circulatory syndrome is due to arterial vasodilation that mainly occurs in the splanchnic circulation, while vascular resistance in the other circulatory districts is normal or increased, according to the degree of portal hypertension, liver fibrosis and circulatory RAS activation [19]. Enhanced circulating ACE detected in our study may be a ‘cause or effect’ of hemodynamic alterations in our patients with advanced-stage HPS.
Hemostatic abnormalities of primary hemostasis (interaction between platelets and vessel wall), coagulation (thrombin generation) and fibrinolysis in patients with chronic liver disease are evident [20]. The hemostatic profile of a patient with liver failure typically includes thrombocytopenia, reduced levels of coagulation factors and inhibitors, reduced levels of fibrinolytic proteins, and increased plasma levels of coagulation factor VIII and VWF [34]. Systemic activation of endothelial cells results in increased release or production of hemostatic factors in liver disease [27]. ACE is an integral part of the hematopoietic system and hemostasis [22, 35, 36]. Increased ACE level in our study could be a result of the systemic endothelial activation of the complex pathobiological course of HPS.
Further experimental and clinical studies are needed to determine the exact status of circulating and local RAS and ACE in the pathobiology of the expanding spectrum of distinct chronic liver diseases including HPS. Revelation of the interactions between circulating and local RAS within the hepatic microenvironment would enlighten the biological basis and clinical management of liver diseases.
References
Benhamou JP, Valla DC. Intrahepatic portal hypertension. In: Bircher J, Benhamou JP, McIntyre N, et al., eds. Oxford Textbook of Clinical Hepatology. 2nd ed. Oxford: Oxford University Press; 1999:661–670.
Hillaire S, Bonte E, Denninger MH, et al. Idiopathic non-cirrhotic intrahepatic portal hypertension in the West: a re-evaluation in 28 patients. Gut. 2002;51:275–280.
Banti BC. Ueber Morbus Banti. Folia Haematol. 1910;10:33–74.
Nayak NC, Ramalingaswami V. Obliterative portal venopathy of the liver. Associated with so-called idiopathic portal hypertension or tropical splenomegaly. Arch Pathol. 1969;87:359–369.
Villeneuve JP, Huet PM, Joly JG, et al. Idiopathic portal hypertension. Am J Med. 1976;61:459–464.
Sama SK, Bhargava S, Gopi-Nath N, et al. Non-cirrhotic portal fibrosis. Am J Med. 1971;51:160–171.
Levison DA, Kingham JG, Dawson AM, Stansfeld AG. Slow cirrhosis—or no cirrhosis? A lesion causing benign intrahepatic portal hypertension. J Pathol. 1982;137:253–272.
Wanless IE. Micronodular transformation (nodular regenerative hyperplasia) of the liver: a report of 64 cases among 2,500 autopsies and a new classification of benign hepatocellular nodules. Hepatology. 1990;11:787–797.
Kameda H, Yamazaki K, Imai F, et al. Obliterative portal venopathy: a comparative study of 184 cases of extrahepatic portal obstruction and 469 cases of idiopathic portal hypertension. J Gastroenterol Hepatol. 1986;1:139–149.
Mikkelsen WP, Edmondson HA, Peters RL, Redeker AG, Reynolds TB. Extra- and intrahepatic portal hypertension without cirrhosis (hepatoportal sclerosis). Ann Surg. 1965;162:602–620.
Bioulac-Sage P, Le Bail B, Bernard PH, et al. Hepatoportal sclerosis. Semin Liver Dis. 1995;15:329–339.
Schiano TD, Kotler DP, Ferran E, et al. Hepatoportal sclerosis as a cause of noncirrhotic portal hypertension in patients with HIV. Am J Gastroenterol. 2007;102:2536–2540.
Okudaira M, Ohbu M, Okuda K. Idiopathic portal hypertension and its pathology. Semin Liver Dis. 2002;22:59–72.
Isabel Fiel M, Thung SN, Hytiroglou P, et al. Liver failure and need for liver transplantation in patients with advanced hepatoportal sclerosis. Am J Surg Pathol. 2007;31:607–614.
Koksal AS, Koklu S, Ibis M, et al. Clinical features, serum interleukin-6, and interferon-gamma levels of 34 Turkish patients with hepatoportal sclerosis. Dig Dis Sci. 2007;52:3493–3498.
Sozer S, Fiel MI, Schiano T, et al. The presence of JAK2V617F mutation in the liver endothelial cells of patients with Budd-Chiari syndrome. Blood. 2009;113:5246–5249.
Cahill PA, Redmond EM, Sitzmann JV. Endothelial dysfunction in cirrhosis and portal hypertension. Pharmacol Ther. 2001;89:273–293.
Carvalho LT, Nascimento EA, Teixeira FO, et al. Hepatic conversion of angiotensin I and the portal hypertensive response to angiotensin II in normal and regenerating liver. J Gastroenterol Hepatol. 2007;22:1543–1548.
La Villa G, Gentilini P. Hemodynamic alterations in liver cirrhosis. Mol Aspects Med. 2008;29:112–118.
Tripodi A, Mannucci PM. Abnormalities of hemostasis in chronic liver disease: reappraisal of their clinical significance and need for clinical and laboratory research. J Hepatol. 2007;46:727–733.
Cobankara V, Ozturk MA, Kiraz S, et al. Renin and angiotensin-converting enzyme (ACE) as active components of the local synovial renin-angiotensin system in rheumatoid arthritis. Rheumatol Int. 2005;25:285–291.
Haznedaroglu IC, Beyazit Y. Pathobiological aspects of the local bone marrow renin-angiotensin system: a review. J Renin Angiotensin Aldosterone Syst. 2010;11:205–213.
Anthuber M, Farkas S, Rihl M, et al. Angiotensin-converting enzyme inhibition by enalapril: a novel approach to reduce ischemia/reperfusion damage after experimental liver transplantation. Hepatology. 1997;25:648–651.
Freise H, Palmes D, Spiegel HU. Inhibition of angiotensin-converting enzyme reduces rat liver reperfusion injury via bradykinin-2-receptor. J Surg Res. 2006;134:231–237.
Padrissa-Altes S, Franco-Gou R, Boillot O, et al. Effect of angiotensin II and bradykinin inhibition in rat reduced-size liver transplantation. Liver Transpl. 2009;15:313–320.
Koh SL, Ager EI, Christophi C. Liver regeneration and tumour stimulation: implications of the renin-angiotensin system. Liver Int. 2010;30:1414–1426.
Bataller R, Gines P, Nicolas JM, et al. Angiotensin II induces contraction and proliferation of human hepatic stellate cells. Gastroenterology. 2000;118:1149–1156.
Debernardi-Venon W, Martini S, Biasi F, et al. AT1 receptor antagonist Candesartan in selected cirrhotic patients: effect on portal pressure and liver fibrosis markers. J Hepatol. 2007;46:1026–1033.
Rimola A, Londono MC, Guevara G, et al. Beneficial effect of angiotensin-blocking agents on graft fibrosis in hepatitis C recurrence after liver transplantation. Transplantation. 2004;78:686–691.
Yoshiji H, Noguchi R, Kojima H, et al. Interferon augments the anti-fibrotic activity of an angiotensin-converting enzyme inhibitor in patients with refractory chronic hepatitis C. World J Gastroenterol. 2006;12:6786–6791.
Garcia-Pagan JC, Salmeron JM, Feu F, et al. Effects of low-sodium diet and spironolactone on portal pressure in patients with compensated cirrhosis. Hepatology. 1994;19:1095–1099.
Sowers JR, Whaley-Connell A, Epstein M. Narrative review: the emerging clinical implications of the role of aldosterone in the metabolic syndrome and resistant hypertension. Ann Intern Med. 2009;150:776–783.
Bataller R, Gabele E, Parsons CJ, et al. Systemic infusion of angiotensin II exacerbates liver fibrosis in bile duct-ligated rats. Hepatology. 2005;41:1046–1055.
Lisman T, Porte RJ. Rebalanced hemostasis in patients with liver disease: evidence and clinical consequences. Blood. 2010;116:878–885.
Acar K, Beyazit Y, Sucak A, et al. Alterations in the ‘local umbilical cord blood renin-angiotensin system’ during pre-eclampsia. Acta Obstet Gynecol Scand. 2007;86:1193–1199.
Haznedaroglu IC, Ozturk MA. Towards the understanding of the local hematopoietic bone marrow renin-angiotensin system. Int J Biochem Cell Biol. 2003;35:867–880.
Acknowledgment
We would like to thank to Dr. Sureyya Altunay for providing us an excellent illustration of circulating RAS within the hepatic microenvironment.
Conflict of interest
Authors declare no conflict of interest related to this article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Beyazit, Y., İbis, M., Purnak, T. et al. Elevated Levels of Circulating Angiotensin Converting Enzyme in Patients with Hepatoportal Sclerosis. Dig Dis Sci 56, 2160–2165 (2011). https://doi.org/10.1007/s10620-011-1580-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-011-1580-7