
Abstract
Peutz–Jeghers syndrome (PJS) is an autosomal-dominant inherited disorder characterized by mucocutaneous pigmentation, hamartomatous polyposis of the gastrointestinal tract, and an increased risk for the development of both gastrointestinal and extraintestinal malignancies. Germline mutation of the STK11 gene, which encodes a serine-threonine kinase, is responsible for PJS. We collected blood samples from a Chinese PJS family consisting of a total of four individuals (one male and three females) including one PJS patient. The whole coding region of STK11 was amplified by polymerase chain reaction and products analyzed by direct sequencing. Molecular analysis of the STK11 gene in this case of PJS revealed a substitution of thymine 217 for adenine (C.217T > A) in exon 1, resulting in a change of codon 73 from cysteine to serine (C73S). The point mutation was not found in normal individuals in this PJS family or in 100 control individuals. The results presented here enlarge the spectrum of mutations of the STK11 gene by identifying a de novo mutation in a PJS patient and further support the hypothesis that STK11 mutations are disease-causing mutations for PJS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Peutz–Jeghers syndrome (PJS; OMIM 175200) is a rare cancer predisposition, which is characterized by the presence of hamartomatous polyposis and mucocutaneous pigmentation. The first report describing Peutz–Jeghers syndrome (PJS) was probably by a London surgeon, Jonathan Hutchinson, in 1896. The association of mucocutaneous pigmentation with multiple gastrointestinal polyps was first described in 1921 by Peutz who reported this association in several members of a family. In 1949, Jeghers et al. [1] established the syndrome as an entity appearing to be inherited as an autosomal-dominant condition. The main clinical symptoms of the syndrome include abdominal pain, rectal blood loss, anemia, small bowel obstruction, and intussusception leading to a high laparotomy rate. Patients with PJS are at an increased risk of developing gastrointestinal cancer and extraintestinal neoplasms involving organs such as the ovaries, testes, breasts, pancreas, lungs, or uterine cervix [2]. The incidence of the syndrome is estimated to be between 1 in 8,300 and 1 in 120,000 [3].
In 1997, Hemminki et al. [4] utilized comparative genomic hybridization of hamartoma from a single PJS patient to detect loss of the telomeric region of chromosome 19p13.3, and subsequently mutations in a gene encoding a serine-threonine kinase, STK11 (other aliases LKB1, OMIM 602216) have been found in the majority of the patients ([5], [6]). STK11 is a known tumor suppressor and several studies reported that somatic mutations have been found at a low frequency in sporadic tumors of the colon, stomach, ovary, testes and lungs [7–10]. STK11 consists of nine coding exons with a 433-amino acid coding sequence and one non-coding exon 10. Codons 49–309 encode the catalytic kinase domain. Most of different mutations in the STK11 gene which have been reported are small insertions/deletions or missense/nonsense mutations. Here, we report a novel STK11 mutation in a Chinese PJS patient.
Materials and Methods
Patient and Sample Collection
The proband was a 25-year-old Chinese female. Her past medical history revealed that the mucocutaneous pigmentation (Fig. 1a) had appeared for 20 years and she had developed abdominal pain at the age of 12 years. Endoscopy had been performed when she was 25 years old and showed gastrointestinal tract multiple polypi (Fig. 1b). Some polypi with low-grade dysplasia were removed by polypectomy under endoscopy. The polypi in the jejunum were surgically removed and confirmed as hamartomatous polyp (Fig. 2). Her relatives had no known medical conditions, including mucocutaneous pigmentation or malignancies. Her normal sister and parents confirmed no polyps by endoscopic examination.

a Subtle perioral melanin pigmentation spots of the lips. b Double-balloon enteroscopy identified the jejunum polyp

Pathologic findings of the Peutz–Jeghers polyp. The polyp of jejunum shows Hamartomatous polyp of the jejunum (Hematoxylin-eosin stain, 100×)
After obtaining informed consent, blood samples were collected from four family members including the patient.
Genomic DNA Isolation
The genomic DNA was isolated from peripheral blood leukocytes using a Kit (Promega, USA) according to the manufacturer’s instructions.
DNA Sequencing
The STK11 gene was amplified via PCR, using the appropriate primers as according to the literature [11]. PCR was performed in 50 μl reaction volume containing 50 ng of genomic DNA, 0.3 mM dNTPs, 0.3 μM of each primer, 3.0 mM MgCl2 and 0.3 units of Hotstar®Taq DNA polymerase (Qiagen). The PCR conditions were: Hotstar®Taq activation at 95°C for 12 min, followed by 40 cycles, each having denaturation at 94°C for 40 s, annealing at 58°C for 60 s and extension at 72°C for 55 s, except that in the first 10 cycles the annealing temperature decreased from 63 to 58°C by 0.5°C per cycle, and the final extension was 72°C for 10 min. After the amplification, PCR products were purified using a QIA quick PCR Purification Kit (Qiagen) and sequenced using ABI PRISM® 3700 automated sequencer (Applied Biosystems). Sequence comparisons and analysis were performed using Phred-Phrap-Consed Version 12.0 program. Samples from 100 unrelated controls were sequenced for missense to exclude the possibility that these are polymorphism in the STK11 gene.
Results
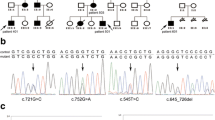
Direct sequencing analysis of the DNA from the proband showed a substitution of thymine 217 for adenine (C.217T > A) in exon 1 of the STK11 gene, resulted in a heterozygous missense mutation of cysteine(C) to serine(S) substitution at the 73rd codon. This mutation was absent in her family members and 100 control chromosomes (Fig. 3).

a Identification of the STK11 gene mutation: direct sequencing of the proband showed a heterozygous missense mutation of C.217T > A in exon 1 of the STK11 gene, resulting in a heterozygous missense mutation of C73S. b The proband’s family members did not have the mutation
Discussion
PJS has been associated with germline mutations in the STK11/LKB1 gene [5, 11–14]. Initial reports suggested the presence of sequence alterations in STK11/LKB1 in most PJS patients. However, recent studies suggest that STK11/LKB1 mutations only account for 40–60% of families with PJS [13]. This suggests the presence of significant genetic heterogeneity for PJS and the involvement of other loci in this syndrome, and another study suggest a second locus is suspected on chromosome 19q13.4 in a minority of families [15].
The STK11/LKB1 protein is ubiquitously expressed in all human fetal and adult tissues [16]. Homozygous deletion of STK11/LKB1 in mice leads to embryonic lethality at midgestation (E11.0), indicating that STK11/LKB1 plays an important role in embryogenesis [17, 18]. Interestingly, most of the STK11/LKB1 (+/−) mice developed intestinal polyps by the age of 45 weeks, identical to those observed in patients with PJS [10, 19]. STK11/LKB1 protein is mainly comprised of three major domains: the N-terminal non-catalytic domain containing the nuclear localization signal, the catalytic kinase domain important for ATP binding, and the carboxyterminal non-catalytic regulatory domain containing prenylation motif (CAAX box). Codons 49–309 encode the catalytic kinase domain. The C-terminal non-catalytic region of the STK11/LKB1 protein is encoded by exon 8 and 9 and encompasses amino acids 309–433. STK11/LKB1 has known to mediate its cellular functions through interactions with a number of proteins [20]. Although the exact function of STK11/LKB1 remains largely unknown, studies suggest its role in cell signaling and apoptosis [21]. STK11/LKB1 is proposed to induce cell cycle arrest through p21, be involved in p53-dependent apoptosis pathways and vascular endothelial growth factor (VEGF) signaling, as well as in BRG1-dependent chromatin remodeling and growth arrest [18, 22–24]. STK11/LKB1 is also known to have effects on metabolism, polarity, and proliferation by phosphorylating and activating the AMPK and AMPK-related kinases [25].
In this study, we identified a point mutations C.217T > A in exon 1 of STK11 gene, which resulted in a cysteine to serine substitution at the 73rd codon. This de novo mutation located in the catalytic kinase domain, encoded by the codons 49–309. We enquired about STK11 homology from GenBank, and found that the nucleotide/amino acid identities in Homo sapiens (human), Pan troglodytes (chimpanzee), Canis lupus familiaris (dog), Bos taurus (cattle), Mus musculus (house mouse), Rattus norvegicus (Norway rat), Gallus gallus (chicken), Danio rerio (zebrafish), and Drosophila melanogaster (fruit fly) were 99.2/99.3, 89.6/93.5, 84.0/87.7, 85.2/91.0, 85.5/91.4, 80.2/90.5, 78.1/83.1, and 61.3/54.7, respectively. The 73rd amino acids residue is cysteine, which is conserved absolutely among the above-mentioned species, as shown in Fig. 4. We speculated that the conserved amino acid residue is important to the enzyme and that this missense mutation may defect the catalytic activity of the enzyme.

Amino acid alignment of various species around the one missense mutation: human STK11 amino acid residues 62–112 are shown aligned to known STK11 orthologues. Residues C, at 73, is conserved in all species compared in the figure. Absolutely conserved and partly conserved amino acid residues among all available sequences are shown with red and yellow backgrounds, respectively. All data were cited from GenBank. (Color figure online)
In conclusion, we identified a de novo mutation of STK11 in a Chinese patient with PJS and enlarged the spectrum of mutations of the STK11 gene. Because of the increased risk of multi-organ cancers in PJS patients [2], molecular diagnosis will be an important factor in genetic counseling, prenatal diagnosis, clinical management of patients, and tumor screening.
References
Jeghers H, Mckusick VA, Katz KH. Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits; a syndrome of diagnostic significance. N Engl J Med. 1949;241:1031–1036.
Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz–Jeghers syndrome. Gastroenterology. 2000;119:1447–1453. doi:10.1053/gast.2000.20228.
Launonen V. Mutations in the human LKB1/STK11 gene. Hum Mutat. 2005;26:291–297. doi:10.1002/humu.20222.
Hemminki A, Tomlinson I, Markie D, et al. Localization of a susceptibility locus for Peutz–Jeghers syndrome to 19p using comparative genomic hybridization and targeted linkage analysis. Nat Genet. 1997;15:87–90. doi:10.1038/ng0197-87.
Hemminki A, Markie D, Tomlinson I, et al. A serine/threonine kinase gene defective in Peutz–Jeghers syndrome. Nature. 1998;391:184–187. doi:10.1038/34432.
Jenne DE, Reimann H, Nezu J, et al. Peutz–Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998;18:38–43. doi:10.1038/ng0198-38.
Avizienyte E, Roth S, Loukola A, et al. Somatic mutations in LKB1 are rare in sporadic colorectal and testicular tumors. Cancer Res. 1998;58:2087–2090.
Dong SM, Kim KM, Kim SY, et al. Frequent somatic mutations in serine/threonine kinase 11/Peutz–Jeghers syndrome gene in left-sided colon cancer. Cancer Res. 1998;58:3787–3790.
Avizienyte E, Loukola A, Roth S, et al. LKB1 somatic mutations in sporadic tumors. Am J Pathol. 1999;154:677–681.
Bardeesy N, Sinha M, Hezel AF, et al. Loss of the Lkb1 tumour suppressor provokes intestinal polyposis but resistance to transformation. Nature. 2002;419:162–167. doi:10.1038/nature01045.
Nakagawa H, Koyama K, Miyoshi Y, et al. Nine novel germline mutations of STK11 in ten families with Peutz–Jeghers syndrome. Hum Genet. 1998;103:168–172. doi:10.1007/s004390050801.
Hemminki A. The molecular basis and clinical aspects of Peutz–Jeghers syndrome. Cell Mol Life Sci. 1999;55:735–750. doi:10.1007/s000180050329.
Westerman AM, Entius MM, Boor PP, et al. Novel mutations in the LKB1/STK11 gene in Dutch Peutz–Jeghers families. Hum Mutat. 1999;13:476–481. doi:10.1002/(SICI)1098-1004(1999)13:6<476::AID-HUMU7>3.0.CO;2-2.
Scott RJ, Crooks R, Meldrum CJ, et al. Mutation analysis of the STK11/LKB1 gene and clinical characteristics of an Australian series of Peutz–Jeghers syndrome patients. Clin Genet. 2002;62:282–287. doi:10.1034/j.1399-0004.2002.620405.x.
Mehenni H, Gehrig C, Nezu J, et al. Loss of LKB1 kinase activity in Peutz–Jeghers syndrome, and evidence for allelic and locus heterogeneity. Am J Hum Genet. 1998;63:1641–1650. doi:10.1086/302159.
Rowan A, Churchman M, Jefferey R, Hanby A, Poulsom R, Tomlinson I. In situ analysis of LKB1/STK11 mRNA expression in human normal tissues and tumours. J Pathol. 2000;192:203–206. doi:10.1002/1096-9896(2000)9999:9999<::AID-PATH686>3.0.CO;2-J.
Jishage K, Nezu J, Kawase Y, et al. Role of Lkb1, the causative gene of Peutz–Jegher’s syndrome, in embryogenesis and polyposis. Proc Natl Acad Sci USA. 2002;99:8903–8908.
Ylikorkala A, Rossi DJ, Korsisaari N, et al. Vascular abnormalities and deregulation of VEGF in Lkb1-deficient mice. Science. 2001;293:1323–1326. doi:10.1126/science.1062074.
Nakau M, Miyoshi H, Seldin MF, Imamura M, Oshima M, Taketo MM. Hepatocellular carcinoma caused by loss of heterozygosity in Lkb1 gene knockout mice. Cancer Res. 2002;62:4549–4553.
Marignani PA. LKB1, the multitasking tumour suppressor kinase. J Clin Pathol. 2005;58:15–19. doi:10.1136/jcp.2003.015255.
Shaw RJ, Kosmatka M, Bardeesy N, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA. 2004;101:3329–3335. doi:10.1073/pnas.0308061100.
Tiainen M, Ylikorkala A, Makela TP. Growth suppression by Lkb1 is mediated by a G(1) cell cycle arrest. Proc Natl Acad Sci USA. 1999;96:9248–9251. doi:10.1073/pnas.96.16.9248.
Karuman P, Gozani O, Odze RD, et al. The Peutz–Jegher gene product LKB1 is a mediator of p53-dependent cell death. Mol Cell. 2001;7:1307–1319. doi:10.1016/S1097-2765(01)00258-1.
Marignani PA, Kanai F, Carpenter CL. LKB1 associates with Brg1 and is necessary for Brg1-induced growth arrest. J Biol Chem. 2001;276:32415–32418. doi:10.1074/jbc.C100207200.
Boudeau J, Scott JW, Resta N, et al. Alessi DR: Analysis of the LKB1-STRAD-MO25 complex. J Cell Sci. 2004;117:6365–6375. doi:10.1242/jcs.01571.
Acknowledgment
We are most grateful to the family with PJS who have so willingly participated and encouraged us in this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gao, Y., Zhang, FM., Huang, S. et al. A De Novo Mutation of STK11 Gene in a Chinese Patient with Peutz–Jeghers Syndrome. Dig Dis Sci 55, 1032–1036 (2010). https://doi.org/10.1007/s10620-009-0837-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-009-0837-x