
Abstract
Resistance to chemotherapeutic agents is one of the distinct features of cancer cells. We evaluate the role of activated MEK-ERK signaling in Camptotecin/irinotecan (CPT-11)-induced cell death using constitutively activated MEK1-transfected normal rat intestinal epithelial cells (IEC-caMEK cells). A CPT-11-induced inhibitory concentration of 50% was determined by WST assay. Apoptosis was evaluated by DNA staining and fragmented DNA analysis. Protein expressions were analyzed by western blotting. We also examined the role of cyclooxygenase-2 in the cell systems. IEC-caMEK cells possessed survival advantages compared to control cells. Apoptosis was remarkably suppressed in IEC-caMEK cells. Western blot analysis revealed increased expression of Bcl-2, Bcl-xL, Mcl-1, and COX-2 and decreased expression of Bak in IEC-caMEK cells. The COX-2 selective inhibitor ameliorated the antiapoptotic nature of IEC-caMEK cells. MEK activation suppressed CPT-11-induced apoptosis in IEC-caMEK cells via a COX-2- dependent mechanism. Therefore, MEK-ERK signaling may contribute to the drug-resistant nature of cancer cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mitogen-activated protein kinase (MEK) and extracellular signal-regulated protein kinase (ERK) are serine/threonine kinases mainly activated by mitogens and growth factors and involved in the regulation of various cellular responses, such as cell proliferation and differentiation [1–3]. Several studies have provided evidence for the activation of these kinases in colorectal cancer, renal cancer, and lung cancer mediated by function of these kinases [4, 5]. Therefore, it is considered that MEK-ERK signaling might bestow a survival advantage in cancer cells [6].
Likewise, several reports have provided evidence that inhibition of MEK-ERK signaling induces cell growth arrest in some cancer cells [7, 8]. And several combination therapies have been reported, such as MEK inhibitor with PPARγ ligands [9], nonsteroidal anti-inflammatory drugs (NSAIDs) [10], and some anticancer drugs [11, 12]. These observations predict that MEK inhibitors may increase the sensitivity of cancer cells to some chemotherapeutic agents.
Resistance to chemotherapeutic agents is one of the distinct features of cancer cells. Since chemotherapeutic agents usually function as inducers of apoptosis [13], chemoresistance in cancer cells is assumed to acquire resistance to chemotherapeutic agent-induced apoptosis [14, 15]. The MEK-ERK cascade has been reported to possess resistance to some chemotherapeutic agents [16]. Therefore, this signaling pathway may contribute to the drug-resistant mechanism in cancer cells.
Recent reports have shown that the activation of this signaling pathway protects certain cancer cells from undergoing apoptosis in response to a variety of agents [17–19]. However, the exact role of MEK-ERK signaling in chemosensitivity has not been addressed well.
On the other hand, overexpression of cyclooxygenase-2 (COX-2) frequently occurs in a variety of human malignancies, including those of colon, lung, breast, skin, and esophagus [20]. Several studies using cultured cells derived from colorectal, pancreatic, prostatic, or lung cancer have demonstrated that NSAIDs significantly inhibit cell proliferative activity [21, 22]. MEK-ERK signaling has also been associated with COX-2 [23, 24]. In normal rat intestinal cells, forced expression of COX-2 provides the cells with a survival advantage [25]. We previously reported that the constitutive activation of MEK1 (CAMEK) signaling results in transformation of RIE and IEC-6 rat intestinal epithelial cells [23]. In this report, MEK-ERK signaling possessed some oncogenic potential, including increased pro-cell cycle properties and an antiapoptosis effect via COX-2 expression, in RIE rat intestinal epithelial cells [23, 26, 27].
Several studies on chemotherapeutic agents have been performed using cancer cells. However, it is possibile that many other antiapoptotic signaling pathways could be activated and differ from normal cells. The exact role of MEK-ERK signaling in chemo-sensitivity in normal cells has not been addressed well. The current study sought to evaluate the role of activated MEK-ERK signaling in sensitivity to chemotherapeutic agents using CAMEK-transfected rat normal intestinal epithelial cells (IEC-caMEK cells). These cells may be a better model to evaluate the real role of MEK-ERK signaling than experimental models using cancer cells.
Materials and Methods
Cells and Culture Conditions
IEC-6 cells were purchased from Japan Health Sciences (Tokyo) and were grown in Dulbecco's minimal essential medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (Hyclone, Logan, UT), 2 mM l-glutamine, and 1% penicillin-streptomycin (Invitrogen, Carlsbad, CA) in a humidified 5% CO2 atmosphere at 37°C
Stable Transfection
IEC-caMEK cells contain constitutively expressed MEK1 phosphorylation site mutant complementary DNA under the control of a cytomegalovirus promoter (pCMV-CA-MEK), and IEC-mock cells contain an empty vector (pcDNA3.1Zeo; Invitrogen). The details of expression of plasmid pCMV- CA-MEK were described previously. IEC-6 cells were transfected with pcDNA3.1Zeo or pCMV-CA-MEK using FuGENE 6 transfection reagent (Roche, Indianapolis, IN) according to the manufacturer's protocol, and clones were selected by growth in culture medium containing 100 μg/ml Zeocin (Invitrogen).
Chemicals
Camptothecin/irinotecan (CPT-11) was purchased from Sigma (St. Louis, MO), and NS-398, a selective COX-2 inhibitor, was purchased from Cayman (Ann Arbor, MI).
Cell Viability Assay
Cells were seeded into 96-well plates at a density of 8 × 104 cells/well at 48 hr before serum starvation from media. Following changing the media without serum, cells were additionally cultured up to 72 hr and then measured viability. Cell viability was determined using the cell proliferation reagent WST-1 assay kit (Roche) according to the manufacturer's protocol. This assay is based on the cleavage of 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(disufopheneyl)-2h-tetrazodium, monosodium salt (WST-1), by mitochondorial dehydrogenase in variable cells. The percentage of cells surviving is expressed as a percentage relative to that obtained from untreated controls.
Drug Sensitivity Assay
Cells were seeded into 96-well plates at a density of 8 × 104 cells/well at 48 hr before exposure to CPT-11. Following changing the media with various concentrations of CPT-11, cells were additionally cultured up to 24 hr and then viability was measured by WST assay as described above. The concentration of CPT-11 yielding 50% inhibition of cell growth (IC50) was calculated using a curve-fitting algorithm.
4,6-Diamidino-2-phenylindole (DAPI) Staining
To distinguish apoptotic cells among dead cells, DAPI (Roche) staining was performed. Cells were seeded into six-well plates at a density of 6 × 105 cells/well and were cultured for 24 hr. Then cells were treated with 15 mM CPT-11 for 1, 3, or 6 hr and fixed in ice-cold acetone and methanol (1:1). After washing with PBS, the cells were stained with DAPI for 60 min. The stained cells were examined under a digital confocal microscopy with a WU filter and photographed.
Apoptosis Assay
Apoptotic cells were quantified using a Cell Death ELISA kit (Roche). Cells were seeded into 96-well plates at a density of 8 × 104 cells/well and cultured for 48 hr. The medium was replaced with 15 mM CPT-11-containing medium, and the cells were cultured for up to 6 hr. Then the cells were harvested and 1 μg of cell lysate was applied for the assay. The assay procedure was done according to the manufacturer's protocol.

Establishment of caMEK-expressing IEC-6 rat intestinal epithelial cells. (A) Morphological findings of constitutively activated MEK1 (caMEK)-expressing IEC-6 cells (IEC-caMEK cells) under phase-contrast microscopy. Pictures show IEC-6 cells transfected with the empty vector (Mock) or caMEK expression vector (IEC-caMEK cells; clones CAMEK1 and CAMEK2). (B) Western blot analysis of EE-tagged caMEK, phosphorylated-ERK1/2, ERK1/2, and β-actin protein levels in cultured cells as described previously. (Original magnifications: ×40.)
Immunoblot Analysis and Antibodies
Cells were washed with ice-cold PBS and lysed with cell lysis buffer [50 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.1% Triton X-100, Complete protease inhibitor cocktail (Roche), and Phoaphates inhibitor cocktail I and II (Sigma)] at 4°C for 10 min, then cell debris was removed by centrifugation at 16,000 rpm for 2 mins. The protein concentration of the supernatant of cell lysates was measured with the Bio-Rad Protein Assay kit (Bio-Rad, Hercules, CA) according to the manufacturer's instructions. The supernatant of cell lysates (10–30 μg/lane) was denatured and fractionated by sodium dodecyl sulfate/polyacrylamide (SDS-PAGE) gel electrophoresis [28]. After electrophoresis, the proteins were transferred electrophoretically to a polyvinylidene difluoride transfer membrane (Nihon Millipore Kogyo, Tokyo) and the filters then probed with the indicated antibodies and developed by enhanced chemiluminescence (Amersham, Piscataway, NJ) [29]. Anti-GluGlu (EE) antibody was purchased from Chemicon (Temecula, CA). Anti-phospho-ERK (Thr202/Tyr204), anti-ERK, and anti-Bcl-XL antibodies were purchased from Cell Signaling Technology (Beverly, MA). Anti-Bax, anti-Bcl-2, anti-Bak, anti-COX-2, and anti-Mcl-1 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-β-actin antibody was purchased from Sigma.
Statistical Analysis
Data were analyzed by Student's t test using the Stat-View software program (SAS Institute, Inc., Cary, NC). Data were considered significant if P < 0.05, and individual P values are indicated in the figure legends.

caMEK-expressing cells possess survival advantages following serum starvation from medium. Each 8 × 104 cells were seeded into 96-well microplates and cultured with 10% fetal bovine serum-containing medium. After 48 hr, the medium was replaced without serum. The cells were cultured for an additional 72 hr, and then the cells were supplied for the WST assay according to the manufacture's protocol. The cell viability was determined by values that were expressed as a percentage relative to those obtained from serum unstarved controls. Values are the mean ± SD of three separate experiments performed in triplicate. * P < 0.05 compared with control cells (Mock).

Concentrations of CPT-11 yielding 50% inhibition of cell viability (IC50) in IEC-caMEK cells. Cells were seeded at a density of 8 × 104 cells/well in 96-multiwell plates and exposed to various concentrations (0–50 mM) of CPT-11 for 24 hr. The number of viable cells was measured by WST assay. The IC50 was calculated using a curve-fitting algorithm, and the data indicate the mean ± SD for triplicate determinations. * P < 0.05 compared with control cells (Mock).
Results
Establishment of CAMEK-Expressing IEC-6 Rat Intestinal Epithelial Cells
To address the role of MEK-ERK signaling in intestinal epithelial cell survival, we generated IEC-6 cells that stably express caMEK, and used two clones for further study (IEC-caMEK cells; clones CAMEK1 and CAMEK2). Distinct features of IEC-caMEK cells were that the cells showed a fibroblast-like shape, were highly retractile, and could pile up to the other cells as seen in transformed cells (Fig. 1A). Western blot analysis revealed elevated levels of EE-tagged caMEK and phosphorylated ERK1/2 expression in IEC-caMEK cells compared with Mock control cells (Fig. 1B). Because we generated these cells by stably expressing an epitope-tagged caMEK, elevated levels of EE signal represented the level of cellular transgene expression. Thus, IEC-caMEK cells showed higher MEK activity and changed their shape compared to empty vector-transfected control cells (Mock) or IEC-6 parental cells (data not shown).
IEC-caMEK Cells Possess Survival Advantages Following Serum Starvation
To determine whether or not MEK-ERK signaling possesses a survival benefit, we examined the cell viability of IEC-6 cells following serum starvation from media. At 72 hr after serum starvation, the percentage of surviving cells was determined by the WST assay. The percentage of surviving cells was significantly higher in IEC-caMEK cells (clone CAMEK1 and CAMEK2), whereas the majority of Mock control cells were dead (Fig. 2).
MEK Signaling Suppresses CPT-11-Mediated Apoptosis
As the next step, we evaluated the sensitivity of the cells to an antitumor agent, camptotecin/irinotecan (CPT-11). The IC50 values of CPT-11 were examined. After 24-hr exposure to CPT-11, caMEK-expressing cells showed a significant increase in the IC50 of CPT-11 compared to Mock control cells: sixfold in CAMEK1 cells and threefold in CAMEK2 cells (Fig. 3). Thus, activated caMEK signaling enhanced the resistance of intestinal cells to CPT-11-mediated cytotoxity.

IEC-caMEK cells are resistant to CPT-11-induced apoptosis. (A) Evaluation of apoptosis using fluorochrome staining. IEC-caMEK cells (clones CAMEK1 and CAMEK2) and control cells (Mock) were cultured with 15 mM CPT-11 for 3 hr, fixed in acetone/methanol (1:1), and stained with the DNA-specific fluorochrome 4,6-diamidino-2-phenylindole (DAPI). (Original magnification, ×400.) (B) Evaluation of apoptosis. IEC-caMEK cells and control cells were treated with 15 mM CPT11 for up to 6 hr. Then 1 μg of cell lysate was applied for the assay. The percentage of apoptotic cells was determined using a Cell Death ELISA kit as described previously. The results are shown as mean ± SD. * P < 0.05 compared with Mock (control). Each experimental or control treatment was performed in triplicate.
To investigate whether the protective effect seen in caMEK-cells during CPT-11-induced cytotoxity is mediated via an antiapoptotic mechanism, we performed nuclear staining and quantitation of DNA fragmentation following CPT-11 treatment of the cells. At 3 hr following treatment with 15 mM CPT-11, Mock control cells resulted in the appearance of typical morphological changes of apoptosis upon staining the cells with the DNA-specific fluorochrome DAPI, as demonstrated in Fig. 4A (left). These changes included condensation of chromatin, its compaction along the periphery of the nucleus, and segmentation of the nucleus. On the other hand, only a few IEC-caMEK cells showed these morphological changes of apoptosis following treatment with CPT-11 (Fig. 4A, middle and right). Quantitative analysis of fragmented DNA, which was seen in the process of apoptotic cell death, revealed that a significant number of Mock control IEC cells cultured under these conditions became apoptotic (Fig. 4B). However, contrary to the results from morphological observation, very few apoptotic IEC-caMEK cells (clones CAMEK1 and CAMEK2) were detected following CPT-11 treatment. These results represent additional evidence that caMEK signaling inhibits the apoptosis induced by CPT-11 treatment.
MEK Activation Modulates Bcl-Family Homologues in CPT-11-Mediated Apoptosis
In light of the above results, we investigated the expression profile of the Bcl-2 family proteins in IEC-caMEK cells following treatment with CPT-11. caMEK expression did not alter the expression levels of Bcl-2 and Bax but did induce Bcl-XL and Mcl-1 (Fig. 5). Notably, the proapoptotic protein Bak was significantly induced in the Mock control IEC cells following CPT-11 treatment, but only nominally in IEC-caMEK cells. Therefore, overexpressed Bcl-XL in IEC-caMEK cells could block Bak induction, and that could result in the prevention of apoptosis in IEC-caMEK cells induced by CPT-11 treatment.
COX-2 Expression Increases Resistance to CPT-11-Mediated Cell Death in IEC-caMEK Cells
We also investigated the role of COX-2 in IEC-caMEK cells following treatment with CPT-11. Consistent with our previous report, COX-2 protein was overexpressed in IEC-caMEK cells compared to Mock control cells (Fig. 6A). The addition of a COX-2 selective inhibitor (NS-398) did not alter cell viability in Mock control IEC cells following CPT-11 treatment (Fig. 6B). However, COX-2 inhibition significantly enhanced cell death in IEC-caMEK cells following CPT-11 treatment (Figs. 6C and D).

Expression profiling of Bcl-homologue proteins during CPT-11-induced apoptosis. Western blot analysis of Bcl-2 family proteins in IEC-caMEK cells and Mock control cells at 0, 3, and 6 hr following treatment with 15 mM CPT-11. β-Actin indicates equal loading of protein in each samples.

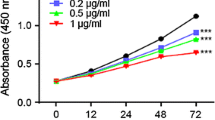
Inhibition of COX-2 ameliorates caMEK-madiated antipoptotic effect in IEC-caMEK cells following treatment of CPT-11. (A) Expression levels of COX-2 protein in cultured cells (IEC-caMEK cells [clones CAMEK1 and CAMEK2], Mock control cells). Cells were exposed to CPT11 (15 mM) for 0, 3, and 6 hr, then the cells were harvested and cell lysates were subjected to Western blot analysis. (B–D) Relative absorbance of cells following treatment of various concentration of NS398 with 15 mM CPT-11 detected by WST assay. Cells were prepared in 96-well plates, treated with vehicle (0.1% DMSO) or various concentrations of NS-398 for 12 hr, and then exposed to medium containing NS-398 and 15 mM CPT-11 for 24 hr. Relative absorbance shows mitochondrial dehydrogenase activity in viable cells determined by WST assay. (B) Mock control cells; (C) IEC-caMEK cells (clone CAMEK1); (D) IEC-caMEK cells (clone CAMEK2). Values are the mean ± SD of three separate experiments performed in triplicate. * P < 0.05 compared with vehicle (0 μM).
Discussion
The MEK-ERK signaling pathway is known to be an important mediator of cell growth and differentiation [1–3]. In this study, to address the role of this signaling pathway in CPT-11-induced cell death, we generated caMEK permanently expressing IEC-6 rat intestinal epithelial cells (IEC-caMEK cells). Recent reports have shown that the activation of this signaling pathway protects certain cancer cells from undergoing apoptosis in response to a variety of agents [17–19]. Consistent with those reports, our data indicate that caMEK signaling bestows a survival advantage and inhibits the cytotoxity induced by CPT-11 treatment in IEC-caMEK cells. CPT-11 is well known to induce apoptosis in a wide variety of cells and is used as an anticancer reagent clinically. These results represent additional evidence that the caMEK signaling pathway may also play a role in drug-resistant activity to some antitumor reagents in cancer cells.
Induction of apoptosis following exposure to chemotherapeutic agents in cancer cells was generated through modulation of Bcl-2 family proteins [30]. Several reports suggests that ERK-stimulated enhancement of cell survival might be mediated through its effects on the expression of Bcl-2 or other Bcl-2 family members [17, 31, 32]. Also, Bcl-2 family members represent critical checkpoints in most apoptotic pathways acting upstream of irreversible damage to cellular constituents [17, 18]. In addition, up-regulation of antiapoptotic proteins such as Bcl-2 and Bcl-XL contributes to tumorigenesis and resistance to drug treatment in certain type of cancers [33, 34].
Therefore we investigated the expression profile of the Bcl-2 family of proteins in IEC-caMEK cells following treatment with CPT-11. In this study, caMEK expression did not alter the levels of Bcl-2 and Bax but did induce Bcl-XL and Mcl-1 (Fig. 5), consistent with our previous report. Notably, the proapoptotic protein Bak was significantly induced in Mock control IEC cells following CPT-11 treatment, but only nominally in IEC-caMEK cells.
Bcl-2 homology (BH) domains are critical for their activities including the induction or suppression of cell death and the ability to heterodimerize with other family members [35, 36]. In particular, the BH3 domain plays a critical role in mediating the cell death and protein-binding functions of Bak and related proapoptotic proteins [37]. These peptides can bind directly to death suppressors such as Bcl-XL [36] and block their subsequent heterodimerization with death promoters in vitro [38]. Likewise, the BH3 peptide may antagonize Bcl-XL and promote apoptosis by preventing Bcl-XL/Apaf-1 heterodimerization [39], leaving Apaf-1 free to participate in the activation of caspases [37]. Bcl-2 and Bcl-XL sequester BH3 domain-only molecules in stable mitochondrial complexes, preventing the activation of Bax, Bak [40]. The ratio between the antiapoptotic and the multidomain proapoptotic Bcl-2 members provides the susceptibility of cells to a death signal [41]. Therefore, overexpressed Bcl-XL in IEC-caMEK cells may block Bak function, and that may result in the prevention of apoptosis in IEC-caMEK cells following CPT-11 treatment.
Many recent studies have shown that COX-2 inhibition induced apoptosis in human prostate cancer cells [21], gastric cancer xenografts [42], and intestinal epithelial cells [25]. It has also been reported that the combined use of COX inhibitors and cisplatin (CDDP) increases the antiproliferative effect in lung cancer, and that a COX inhibitor, sulindac sulfide, increases the sensitivity of non-small cell and small cell lung cancer cell lines to CDDP and paclitaxel [43, 44].
In our previous study, we showed that caMEK signaling prevented apoptotic cell death via COX-2 expression in RIE rat intestinal epithelial cells [23]. We also investigated the role of COX-2 in IEC-caMEK cells following treatment with CPT-11. In this study, consistent with our previous report, COX-2 protein was overexpressed in IEC-caMEK cells compared to Mock control cells (Fig. 6A) following treatment with CPT-11. To investigate whether the protective effect seen in caMEK-cells during CPT-11-induced cytotoxity is mediated via COX-2 overexpression, we examined the additional effect of a COX-2 selective inhibitor (NS-398). As COX-2 inhibition enhanced cell death in IEC-caMEK cells following CPT-11 treatment, the protective effect seen in IEC-caMEK cells was dependent on COX-2.
In conclusion, MEK activation led to suppression of CPT-11-induced apoptosis in rat intestinal epithelial cells through COX-2-dependent mechanisms. This result suggests that MEK-ERK signaling bestows a survival advantage and may contribute to the drug-resistant nature of cancer cells as well as carcinogenesis. Also, COX-2 may play an important role in this process. Further, MEK-ERK signaling may represent an important target for developing a new therapy for treatment of colorectal cancer.
This report is the first demonstrating the effect of MEK-ERK signaling activation in normal intestinal epithelial cells. Therefore, our results may be important for understanding the role of MEK-ERK signaling not only in drug resistance but also in carcinogenesis of intestinal epithelial cells.
References
Babyastsky MW, Podolsky DK (1999) Growth and development of the gastrointestinal tract. In: Textbook of gastroenterology. Vol. 1. 3rd ed. Yamada T (ed). J. B. Lippincott, Philadelphia, pp 547–584
Taupin D, Podolsky DK (1999) Mitogen-activated protein kinase activation regulates intestinal epithelial differentation. Gastroenterology 116:1071–1080
Ding Q, Wang Q, Evers BM (2001) Alterations of MAPK activities associated with intestinal cell differentiation. Biochem Biophys Res Commun 284:282–288
Oka H, Chatani Y, Hoshino Y, Ogawa O, Kakehi Y, Terauchi T, Okada Y, Kawaichi M, Kohno M, Yoshida O (1995) Constitutive activation of mitogen-activated protein (MAP) kinases in human renal cell carcinoma. Cancer Res 55:4182–4187
Gioeli D, Mandell JW, Petroni GR, Frierson HF, Weber MJ (1999) Activated mitogen-activated protein kinase associated with prostate cancer progression. Cancer Res 59:279–284
Marshall CJ (1995) Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80:179–185
Sebolt-Leopold JS, Dudley DT, Herrera R, Becelaere KV, Wiland A, Gowan RC, Tecle H, Barrett SD, Bridges A, Przybranowski S, Leopold WR, Saltiel AR (1999) Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat Med 5:810–816
Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR (1995) A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA 92:7686–7689
Motomura W, Tannno S, Takahashi N, Nagamine M, Fukuda M, Kohgo Y, Okumura T (2004) Involvement of MEK-ERK signaling pathway in the inhibition of cell growth by troglitazone in human pancreatic cancer cells. Biochem Biophys Res Commun 39:461–468
Gao J, Niwa K, Takemura M, Sun W, Onogi K, Wu Y, Seishima M, Mori H, Tamaya T (2005) Significant anti-proliferation of human endometrial cancer cells by combined treatment with selective COX-2 inhibitor NS398 and specific MEK inhibitor U0126. Int J Oncol 26:737–744
Kohno M, Pouyssegur J (2003) Pharmacological inhibitors of the ERK signaling pathway: application as anticancer drugs. Prog Cell Cycle Res 5:219–224
Mackeigan JP, Collins TS, Ting JPY (2000) MEK inhibition enhances paclitaxel-induced tumor apoptosis. J Biol Chem 275:38953–38956
Susin SA, Zamzami N, Kroemer G (1998) Mitochondria as regulators of apoptosis: doubt no more. Biochim Biophys Acta 1366:151–165
Helmbach H, Kern MA, Rossmann E, Renz K, Kissel C, Gschwendt B, Schadendorf D (2002) Drug resistance towards etoposide and cisplatin in human melanoma cells is associated with drug-dependent apoptosis deficiency. J Invest Dermatol 118:923–932
Soengas MS, Capodieci P, Polsky D, Mora J, Esteller M, Opitz-Araya X (2001) Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature 409:207–211
Dent P, Grant S (2001) Pharmacologic interruption of the mitogen-activated extracellular regulated kinase/mitogen-activated protein kinase signal transduction pathway: potential role in promoting cytotoxic drug action. Clin Cancer Res 7:775–783
Boucher M-J, Morisset J, Vachon PH, Reed JC, Laine J, Rivard N (2000) MEK/ERK signaling pathway regulates the expression of Bcl-2, Bcl-XL, and Mcl-1 and promotes survaival of human pancreatic cancer cells. J Cell Biochem 79:355–369
Adams JM, Cory S (1998) The Bcl-2 protein family: arbiters of cell survival. Science 281:1322–1326
Jost M, Huggett TM, Kari C, Boise LH, Rodeck U (2001) Epidermal growth factor receptor-dependent control of keratinocyte survival and Bcl-XL, expression through a MEK-dependent pathway. J Biol Chem 276:6320–6326
Gupta RA, Dubois RN (2001) Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer 1:11–21
Liu XH, Yao S, Kirschenbaum A, Levine AC (1998) NS398, a selective cyclooxygenase-2 inhibitor, induces apoptosis and down-regulates bcl-2 expression in LNCaP cells. Cancer Res 58:4245–4249
Hida T, Kozaki K, Muramatsu H, Masuda A, Shimizu S, Mitsudomi T, Sugita T, Ogawa M, Takahashi T (2000) Cyclooxygenase-2 inhibitor induces apoptosis and enhances cytotoxicity of various anticancer agents in non-small cell lung cancer cell lines. Clin Cancer Res 6:2006–2011
Komatsu K, Buchanan FG, Katkuri S, Morrow JD, Inoue H, Otaka M, Watanabe S, Dubois RN (2005) Oncogenic potential of MEK1 in Rat intestinal epithelial cells is mediated via cyclooxygenase-2. Gastroenterology 129(2):577–590
Sheng H, Shao J, DuBois RN (2001) K-Ras-mediated increase in cyclooxygenase-2 mRNA stability involves activation of the protein kinase B. Cancer Res 61:2670–2675
Tsujii M, Dubois RN (1995) Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell 83:493–501
Taniura S, Nomura K, Ozaki K-I, Tsujimoto M, Kondo T, Kohno M (2002) Prolonged unclear retension of activated extracellular signal-regulated kinase 1/2 is required for hepatocyte growth factor-induced cell motility. J Biol Chem 277:28256–28264
Taniura S, Asato K, Fujishiro S-H, Kohno M (2003) Specific blockade of the ERK pathway inhibits the invasiveness of tumor cells: down-regulation of matrix metalloproteinase-3/-9/-14 and CD44. Biochem Biophys Res Commun 304:801–806,
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Towbin H, Staehelin T, Gordon J (1992) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Biotechnology 24:145–149
Simonian PL, Grillot DA, Nunez G (1997) Bcl-2 and Bcl-XL can differentially block chemotherapy-induced cell death. Blood 90:1208–1216
Townsend KJ, Zhou P, Qian L, Bieszczad CK, Lowrey CH, Yen A, Craig RW (1999) Regulation of MCL1 through a serum response factor/ELK-1 mediated mechanism links expression of a viability-promoting member of the BCL family to the induction of hematopoietic cell differentiation. J Biol Chem 274:1801–1813
Kinoshita T, Yokota T, Arai K, Miyajima A (1995) Regulation of Bcl-2 expression by oncogenic Ras protein in hematopoietic cells. Oncogene 10:2207–2212
Strasser A, Huang DC, Vaux DL (1997) The role of the bcl-2/ced-9 gene family in cancer and general implications of defects in cell death control for tumourigenesis and resistance to chemotherapy. Biochim Biophys Acta 1333:F151–F178
Reed JC, Miyashita T, Takeyama S, Wang HG, Sato T, Krajewski S, Aime-Sempe C, Bodrug S, Kitada S, Hanada M (1996) BCL-2 family proteins: regulators of cell death involved in the pathogenesis of cancer and resistance to therapy. J Cell Biochem 60:23–32
Yin XM, Oltvai ZN, Korsmeyer SJ (1994) BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature 369:321–323
Sattler M, Liang H, Nettesheim D, Meadows RP, Harlen JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, Thompson CB, Fesik SW (1997) Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science 275:983–986
Eric PH, Thomas C, Robert JL (1997) Bak BH3 peptide antagonize Bcl-XL function and induce apoptosis through cytochrome c-independent activation of caspases. J Biol Chem 274:13298–13304
Diaz JL, Oltersdorf T, Horne W, McConnel M, Wilson G, Weeks S, Garcia T, Fritz LC (1997) A common binding site mediates heterodimerization and homodimerization of Bcl-2 family members. J Biol Chem 272:11350–11355
Hu Y, Benedict MA, Wu D, Inohara N, Nunez G (1998) Bcl-XL interacts with Apaf-1 and inhibits Apaf-1-dependent caspase-9 activation. Proc Natl Acad Sci USA 95:4386–4391
Chen EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ (2001) Bcl-2, Bcl-XL sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell 8:705–711
Oltvai ZN, Milliman CL, Korsmeyer SJ (1993) Bcl-2heterodimerrizes in vivo with a conserved homolog, BAX, that accelerates programmed cell death. Cell 74:609–619
Takei Y, Nagano K, Hori M (1998) Cyclooxygenase-2 inhibitors suppress the growth of gastric cancer xenografts via induction of apoptosis in nude mice. Am J Physiol 274:G1061–G1067
Teicher BA, Korbut TT, Menon K, Holden SA, Ara G (1994) Cyclooxygenase and lipoxygenase inhibitors as modulators of cancer therapies. Cancer Chemother Pharmacol 33:515–522
Soriano AF, Helfrich B, Chan DC, Heasley LE, Bunn PA Jr, Chou TC (1999) Synergistic effects of new chemopreventive agents and conventional cytotoxic agents against human lung cancer cell lines. Cancer Res 59:6178–6184
Acknowledgment
This work was supported in part by Grant-in-Aid No. 17590610 for Scientific Research from the Ministry of Education, Science, Sports and Culture, Japan, to M.O.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Horikawa, Y., Otaka, M., Komatsu, K. et al. MEK Activation Suppresses CPT11-Induced Apoptosis in Rat Intestinal Epithelial Cells Through a COX-2-Dependent Mechanism. Dig Dis Sci 52, 2757–2765 (2007). https://doi.org/10.1007/s10620-007-9798-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-007-9798-0