
Abstract
Gills cells of the freshwater mussel Lasmigona costata and the seawater clam Mesodesma mactroides were isolated (mussel: chemical dissociation; clam: mechanical dissociation) and fractionated (Percoll gradient) into Fractions I and II. Mitochondrial dyes (DASPEI: mussel; MitoTracker®: clam) and Na+, K+-ATPase activity measurement were used to distinguish between cells of Fractions I and II. For mussel and clam, 80.5 ± 1.5 and 48.3 ± 3.2 % of cells were in Fraction II, respectively. For both species, cells of Fraction II had higher fluorescence emission and higher enzyme activity than those of Fraction I, being characterized as ‘cells rich in mitochondria’. Cells of Fraction II were kept in saline solutions approximating the ionic composition of hemolymph either under control conditions (no Cu addition) or exposed (3 h) to copper (Cu: 5, 9 and 20 μg Cu/L). Cell viability and Cu and Na+ content were measured. For both species, Cu content was higher and Na+ content was lower in cells exposed to 20 μg Cu/L. Furthermore, a strong negative correlation was observed between cell Na+ and Cu content in the two bivalve species, indicating a possible competition between Cu and Na+ for ion-transporting mechanisms or binding sites at gill cells of Fraction II. Considering that Cu is an ionoregulatory toxicant in aquatic invertebrates, these preliminary toxicological data support the idea of using isolated gill cells rich in mitochondria to study the mechanisms underlying the acute toxicity of waterborne Cu in freshwater and marine bivalves.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In vitro studies in bivalves have been largely performed with digestive glands, mantle, haemocytes and gills (Le Pennec and Le Pennec 2001; Chelomin et al. 2005; Lopes et al. 2011a, b). However, studies involving the identification of cells present in these tissues are generally scarce. Studies examined the gill tissue and analysed the cells of freshwater mussels, Pyganodon cataracta, Utterbackia imbecillis and Ligumia substrata (Unionid mussels), and reported different cellular types, including the ‘cells rich in mitochondria’, which play a significant role in active uptake of ions and solute transport into and out of the gills (Kays et al. 1990; Schwartz and Dimock, 2001). In vivo and in vitro gill epithelium analyses were also performed in the seawater mussel Mytilus galloprovincialis (Gómez-Mendikute et al. 2005). Findings reported the presence of ciliated (58 %) and non-ciliated (42 %) cells, including epithelial cells and haemocytes (4.3 %).
Although morphological studies in bivalves describe the gill epithelium as being heterogeneous, the specific types of cells and their physiological functions are not fully known. Moreover, there is considerable interest in the development of in vitro models due to the action of aquatic pollutants on cell membranes via the outer epithelial layer of the gills, as well as other organs located in the paleal cavity, and the consequent toxicity to cells (Bigas et al. 2001; Le Pennec and Le Pennec 2001; Lopes et al. 2011a, b). Therefore, the first goal of the present study was to isolate and fractionate cells from the gills of the freshwater mussel Lasmigona costata and seawater clam Mesodesma mactroides. For isolation, two different techniques (mechanical and enzymatic) were performed according to the gill tissue morphology of the different bivalves. Fractionation was performed using discontinuous Percoll gradient and confirmed by fluorescence microscopy using specific mitochondrial dyes and Na+, K+-ATPase activity measurement, as earlier applied for teleost fish (Fletcher et al. 2000; Kelly et al. 2000; Wong and Chan 2001; Tse et al. 2006). Two distinct fractions were identified, one of which (Fraction II) was rich in mitochondria and Na+, K+-ATPase activity.
In teleost fish, separation of different isolated cell types has been useful for biochemical and cytological analyses, e.g. functional characterization of cell membrane transporters related to Na+ uptake (Goss et al. 2001; Galvez et al. 2002; Reid et al. 2003), and particularly in analyzing the ionoregulatory disrupting effects of metals such as Cu (Goss et al. 2011). Cu is now generally recognized to be an ionoregulatory toxicant in aquatic invertebrates (Pinho et al. 2007; Lopes et al. 2011a, b) as well as in fish. Therefore, we exposed gill cells of Fraction II isolated from the freshwater mussel L. costata and the seawater clam M. mactroides to three environmentally relevant concentrations of copper (Cu) (5, 9 and 20 μg Cu/L) for 3 h and analysed the cellular content of both Cu and Na+. Results obtained were compared between species and the methodology used to obtain gill cells of bivalves for in vitro toxicological studies was evaluated. While the results were encouraging, the toxicological data obtained must be considered preliminary as cell exposures were performed in saline solutions which were rich in salts approximating hemolymph composition rather than external water composition and lacking in energy sources (e.g. amino acids, glucose).
Materials and methods
Experimental animals
Wild adults of the fluted-shell mussel L. costata (Rafinesque, 1820) were collected from a reference site in the Grand River (southern Ontario, Canada). In turn, adults of the yellow clam M. mactroides (Deshayes, 1854) were collected from a reference site at the Mar Grosso beach (southern Rio Grande do Sul, Brazil). Both species were acclimated to laboratory conditions for at least 1 week prior to experimentation. Mussels were held in aerated reconstituted moderately-hard water (composition in mM: 2.28 NaHCO3, 0.78 CaSO4.H-2O, 1 MgSO4, 0.1 KCl; pH 7.8–8.0; ASTM 2006) under a 16 h:8 h light:dark cycle at 14 °C. The water was renewed once a week when the mussels were fed with a commercial shellfish diet (Instant Algae Shellfish Diet 1800®, Richmond Hill, Ontario, Canada) at the rate of approximately 1.2 × 1010 algal cells per mussel. Clams were held in natural filtered (45 μm-mesh filter) sea water (salinity 30 ppt) continuously aerated and in the absence of sediment. Room temperature (20 °C) and photoperiod (12L:12D) were fixed. The acclimation medium was renewed three times a week, when clams were fed with the diatom Thalassiosira weissflogii at the rate of approximately 2 × 102 cells per clam.
Cell isolation
Initially, cell isolation from the gill epithelium of the mussel L. costata was performed using mechanical dissociation as this approach was thought to be less aggressive. However, the maintenance of small pieces of gill tissue in freshwater phosphate buffer solution (PBS) for 40 min caused massive cell death (data not shown). Therefore, we subsequently employed enzymatic dissociation as described by Quinn et al. (2009) with modifications. Briefly, gill tissue from two mussels were dissected, pooled, and held in calcium-free phosphate buffer solution (freshwater PBS; composition in mM: 9 NaCl, 5 NaHCO3, 0.5 KCl, 5 Na2PO4; pH 7.6; 20 °C) for 10 min to remove the excess mucus. Using forceps and scissors, tissues were sliced into small pieces and incubated (120 rotations per minute) with pronase from Streptomyces griseus (0.025 % in freshwater PBS) (Sigma-Aldrich, Oakville, Ontario, Canada), for 15 min at room temperature (21 °C). After enzymatic digestion, isolated cells were filtered using a 30 μm-mesh nylon filter into stop buffer (1:10 fetal bovine serum/FBS; Sigma-Aldrich, Oakville, Ontario, Canada), centrifuged (600×g for 4 min; Sorvall Legend X1, Thermo Scientific, Walthan, MA, USA) and resuspended in freshwater PBS for 30 min before the start of experiments.
Isolation of gill cells from the seawater clam M. mactroides was performed following the procedures previously described by Lopes et al. (2011a, b) with modifications. Briefly, gill tissue from three clams (three rather than two, as the clam gills were smaller than the mussel gills) was dissected, pooled, sliced into small pieces, incubated in calcium-free phosphate buffer solution (seawater PBS; composition in mM: 342 NaCl, 20 Na2HPO4, 1.7 K2HPO4, 16 KCl, 5.5 EDTA; pH 7.6; 20 °C), and shaken (120 rotations per minute; Certomat-MO-II; Sartorius Stedim Biotech GmbH, Göttingen, Germany) for 30 min. After incubation, dissociated cells were filtered (30 μm-mesh nylon filter) to remove the non-dissociated tissue and large debris. The filtered solution containing isolated cells was transferred to plastic tubes, centrifuged (360×g) for 3 min (Hettich Zentrifugen, Model Mikro 22 R, Global Medical Instrumentation, Ramsey, MN, USA). The pellet obtained was resuspended in seawater PBS for 30 min before the start of experiments.
Fractionation of isolated gill cells
Isolated gill cells from both species were layered onto a discontinuous Percoll gradient (Sigma-Aldrich, St. Louis, MO, USA). The gradient for L. costata was obtained by diluting the Percoll stock solution to obtain the following densities: 1.03 g/mL (18.6 % Percoll stock solution, 71.4 % bi-distilled H2O, 10 % 1.5 M NaCl), 1.05 g/mL (34.0 % Percoll stock solution, 56.0 % bi-distilled H2O, 10 % 1.5 M NaCl); 1.09 g/mL (64.8 % Percoll stock solution, 25.2 % bi-distilled H2O, 10 % 1.5 M NaCl). The gradient for M. mesodesma was obtained as described for L. costata, but replacing bi-distilled H2O with seawater PBS.
Each layer of the Percoll gradient (1 mL) was placed into a plastic falcon-type tube (15 mL) in the following sequence: 1.09; 1.05; and 1.03 g/mL. Finally, the pool of isolated gill cells (1 mL) was added and centrifuged at 2000×g for 10 min as described by Galvez et al. (2002), with modifications. Briefly, gill cells from both species were separated into Fraction I (1.03–1.05 g/mL Percoll interface) and Fraction II (1.05–1.09 g/mL Percoll interface). These Fractions were washed with 10 mL of the respective PBS for each species and centrifuged at 600×g for 4 min. Finally, the new pellet obtained for each species was resuspended in 1 mL of saline solution of similar composition to the freshwater mussel hemolymph (composition in mM: 18 NaCl, 1.6 CaCl2, 0.9 KCl, 0.4 MgCl2, 2 NaHCO3, and 1.4 glucose; pH 7.6; 20 °C; Dietz 1979) and to the seawater clam hemolymph (composition in mM: 350 NaCl, 9 KCl, 30 MgSO4, 9 CaCl2, 2 NaHCO3; pH 7.6; 20 °C; Lopes et al. 2011a, b).
Cell viability
Cell viability was determined using the trypan blue exclusion assay (0.08 % trypan blue). Cells from each sample were counted under a light microscope. Cell viability was determined by dividing the number of non-stained (living) cells by the number of total cells counted. Measurements were performed immediately after cell isolation, after 5 h of maintenance in the corresponding saline solution, after cell fractionation (Percoll gradient), and the 3-h period of Cu exposure (Fraction II cells only; see below).
Cell distinction
For both species, a specific mitochondrial dye was used to distinguish between cells from Fractions I and II (Lin and Hwang 2004; Lin et al. 2006; Buhariwalla et al. 2012). The fluorescent dye DASPEI (2-(4-dimethylaminostyryl)-1-methylpyridinium iodide; Sigma-Aldrich, St. Louis, MO, USA) was used in isolated gill cells of L. costata. Cells from Fractions I and II were incubated with DASPEI (0.2 μM) for 20 min, washed with freshwater PBS, and held in the corresponding saline solution for image processing. Images were analyzed using a fluorescent microscope (Leica DMR microscope; Leica Microsystems, Wetzlar, Germany) equipped with a Qimaging FAST 1394 digital camera. In turn, fractionated gill cells of M. mactroides were incubated with the fluorescent dye MitoTracker Green® (1 μM) for 10 min, washed with seawater PBS, and held in the corresponding saline solution for image processing. Images were analyzed using a fluorescent microscope (Olympus IX 81, Markham, Ontario, Canada) equipped with a DP72 digital camera. For both species, gills cells from Fractions I and II were evaluated for the presence or the absence of fluorescence emission.
Gill cells present in Fractions I and II were also distinguished through measurements of the cell Na+, K+-ATPase activity. Following cell isolation and Percoll gradient fractionation, gill cells of L. costata were homogenized in buffer solution (composition in mM: 150 sucrose; 5 EDTA; 50 imidazole; pH 7.5). Na+, K+-ATPase activity was measured based on the difference in the amount of inorganic phosphate (Pi) released from ATP in the presence of K+ (medium A) and in the absence of K+ (medium B) and the presence of ouabain as described by Bianchini and Castilho (1999). For each cell fraction, 10 μL of the cell homogenate was incubated in 500 μL of medium A (composition in mM: 100 NaCl; 6 MgCl2; 50 imidazole; 20 KCl; 3 ATP; pH 7.5) and in 500 μL of medium B (composition in mM: 100 NaCl; 6 MgCl2; 50 imidazole; 3 ATP; 2 ouabain; pH 7.5) at 30 °C for 30 min. The reaction was stopped by adding 0.2 mL trichloroacetic acid (50 %) to the reaction medium. Phosphate concentration was determined using a colorimetric method (Fiske and Subbarow, 1925). Protein concentration in the cell homogenate was determined using the method described by Bradford (1976). The specific enzyme activity was expressed as μmol Pi/mg protein/h.
Isolated gill cells of M. mactroides were homogenized in buffer solution (composition in mM: 300 sucrose; 20 EDTA; 100 imidazole; pH: 7.4). Na+, K+-ATPase activity was measured based on the ADP production in the presence of K+ (medium A) and in the absence of K+ and the presence of ouabain (medium B) as described by McCormick (1993). An aliquot (10 μL) of the cell homogenate was added to 500 μL of medium A (composition in mM: 100 NaCl; 10.5 MgCl2.H2O; 30 KCl; 50 imidazole) and to 500 μL of medium B (composition in mM: 100 NaCl; 10.5 MgCl2.H2O; 50 imidazole; 1 ouabain). The reaction solution (composition in mM: 2.8 phosphoenolpyruvate; 3.5 ATP; 0.22 NADH; 50 mM imidazole; composition in μL/ml: 4 lactate dehydrogenase; 5 pyruvate kinase) was then added to the mixture, which was incubated for 30 min at room temperature. Protein concentration in the cell homogenate was determined using the method described by Bradford (1976). The specific enzyme activity was expressed as μmol ADP/mg protein/h.
Copper accumulation and Na+ content after Cu exposure
For both species, isolated cells (106 cells) present in Fraction II (cells rich in mitochondria, see Results section) were kept in their respective saline solution without Cu addition (control) or exposed (3 h) to Cu (5, 9 and 20 μg/L) at room temperature, as previously described for mantle cells of the clam M. mactroides (Lopes et al. 2011a, b) with some modification. The 5 and 9 μg Cu/L concentrations used in our study correspond to the Brazilian water quality criteria for Cu in sea water and fresh water, respectively (CONAMA 2005). They are also in consonance with the Canadian water quality guidelines (CCME 2005). In turn, 20 μg Cu/L was selected as a non-conforming concentration according to the Brazilian and Canadian regulations. Briefly, Cu as CuCl2 (Merck, St. Louis, MO, USA) was added to the respective saline solution for L. costata and M. mactroides. After exposure, cells rich in mitochondria were centrifuged 360×g, 3 min at room temperature (Hettich Zentrifugen, Model Mikro 22 R, Global Medical Instrumentation, Ramsey, MN, USA). The supernatant was discarded and the pellet was quickly washed with the respective saline solution containing EDTA (12 mM) to remove the loosely Cu bound on the cell surface. The washing and centrifugation procedures were repeated. The pellet was resuspended in the respective (freshwater or seawater) PBS solution. An aliquot of the suspension was collected to determine cell viability using the Trypan Blue exclusion method. Remaining cells were then split into two aliquots. The first aliquot was dried in an incubator (50 °C; 24 h), completely digested with HNO3 (Suprapur, Merck, St. Louis, MO, USA), diluted with Milli-Q water, and analyzed for Cu concentration. The second aliquot was sonicated (Sonozap, Ultrasonic Processor, New York, NY, USA) and diluted with Milli-Q water for Na+ concentration analysis.
For L. costata samples, Cu and Na+ concentration was analyzed by graphite furnace atomic absorption spectrometry (GFAAS, Varian Spectra AA-20 equipped with graphite tube atomizer [GTA-110], Mulgrave, Australia) and flame atomic absorption spectrometry (SpectrAA 220FS Atomic Absorption Spectrometer), respectively. For M. mactroides samples, Cu and Na+ concentration was analyzed by atomic absorption spectrophotometry (AAS 932 Avanta Plus, GBC, Hampshire, IL, USA), as previously described (Pinho et al. 2007; Lopes et al. 2011b). Cell Cu or Na+ content was expressed as μmol Cu or Na+/106 cells, considering the respective cell viability in each experimental condition.
Statistical analyses
Data are expressed as mean ± standard error (n = 5), where each measurement represents a pool of cells (Fraction I or II) prepared with gills from two (L. costata) and three (M. mactroides) animals. Each pool of cells was analyzed at least in triplicate and a mean value was calculated for each pool of cells. Na+, K+-ATPase activity, as well as Na+ and Cu cell content data were compared using one-way analysis of variance (ANOVA) followed by the Fisher least significant difference (LSD) test. ANOVA assumptions (data normality and homogeneity of variances) were checked. Correlations between cell Cu content and Cu concentration in the incubation medium or between cell Cu content and Na+ content were evaluated through the Spearman correlation index. Statistical analyses were performed using the software Statistica 7.0 (Stat Soft, Tulsa, OK, USA). The significance level adopted was 95 % (α = 0.05).
Results
Cell dissociation, fractionation and distinction
The enzymatic cellular dissociation employed for the freshwater mussel L. costata showed satisfactory results when cell viability data were considered. After isolation, cell viability measured by the trypan blue exclusion assay was 87.4 ± 2.7 % and no significant change (as determined by ANOVA) was observed after 5 h of cell maintenance in saline solution of similar composition to the mussel hemolymph. Also, the mechanical dissociation of gill cells applied to the gill epithelium of the seawater clam M. mactroides proved to be satisfactory with respect to the number of isolated cells acquired and the cell viability observed (82.4 ± 1.0 %).
For both species, the discontinuous Percoll gradient technique separated gill cells into two different groups of cells (Fraction I: cells retained in the 1.03–1.05 g/mL Percoll interface; Fraction II: cells retained in the 1.05–1.09 g/mL Percoll interface). For both fractions, cell viability did not differ from that observed for the total pool of cells. Cell separation was confirmed through two different approaches (mitochondrial dyes and Na+, K+-ATPase activity measurement), as described below.
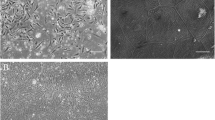
For L. costata, cells collected from the 1.03–1.05 (Fraction I) and the 1.05–1.09 g/mL (Fraction II) Percoll interfaces were incubated separately in the mitochondrial fluorescent dye DASPEI. Cells present in Fraction II (Fig. 1c) showed higher levels of fluorescence emission (Fig. 1d) than those from Fraction I (Fig. 1a, b). For M. mactroides, each cell fraction was incubated separately in the mitochondrial fluorescent dye MitoTracker Green®. Cells from Fraction I (Fig. 2a) showed lower fluorescence emission (Fig. 2b) than those present in Fraction II (Fig. 2c, d).

Fluorescent images of isolated gill cells from the freshwater mussel Lasmigona costata after layering over a discontinuous Percoll gradient (1.03–1.05–1.09 g/mL) and separation by centrifugation. Cells from each interface were collected, incubated with a specific mitochondrial dye (DASPEI) and examined under a fluorescence microscope (×200 magnification). a and c phase contrast image; b and d fluorescent images of the same fields from a and c. a and b 1.03–1.05 g/L Percoll density interface (Fraction I); c and d 1.05–1.09 g/L Percoll density interface (Fraction II). Scale bars 40 μm

Fluorescent images of isolated gill cells from the seawater clam Mesodesma mactroides after layering over a discontinuous Percoll gradient (1.03–1.05–1.09 g/mL) and separation by centrifugation. Cells from each interface were collected, washed, incubated with a mitochondrial dye (MitoTracker®) and examined under a fluorescence microscope (×400 magnification). a and c phase contrast images; b and d fluorescent images of the same fields from a and c. a and b = 1.03–1.05 g/L Percoll density interface (Fraction I); c and d 1.05–1.09 g/L Percoll density interface (Fraction II). Scale bars 20 μm
For both the freshwater mussel (Fig. 3a) and seawater clam (Fig. 3b), Na+, K+-ATPase activity was significantly higher in cells from Fraction II than in those from Fraction I. Cell counting data indicated that 80.5 ± 1.5 and 48.3 ± 3.2 % of the isolated cells were actually cells showing high fluorescence emission and Na+, K+-ATPase activity in L. costata and M. mactroides, respectively.

Na+, K+-ATPase activity in cells of Fraction I (1.03–1.05 g/L Percoll density interface) and Fraction II (1.05–1.09 g/L Percoll density interface) isolated from gills of the freshwater mussel Lasmigona costata (a) and seawater clam Mesodesma mactroides (b). Data are expressed as mean ± standard error (n = 5). Different letters indicate significant different mean values among treatments (p < 0.05)
Copper accumulation and Na+ content after Cu exposure
For both species, Cu exposure (3 h) did not significantly affect viability of gill cells present in Fraction II. For L. costata, control cells showed 87.4 ± 2.7 % viability while those exposed to 5, 9 and 20 μg Cu/L had 89.9 ± 2.2, 88.0 ± 2.5, and 88.0 ± 4.5 % viability, respectively. Cell Cu content in control cells was 1.02 ± 0.06 μmol/106 cells which did not significantly differ from cells exposed to 5 and 9 μg Cu/L (1.14 ± 0.08 and 1.30 ± 0.08 μmol/106cells, respectively). However Cu content significantly increased in those exposed to 20 μg Cu/L (1.53 ± 0.21 μmol/106 cells). Notably, a significant positive correlation between Cu content in gill cells and Cu concentration in the incubation medium was observed (R = 0.72; p < 0.05). Notably, the Na+ content was significantly lower in cells exposed to 20 μg Cu/L (3.44 ± 0.16 μmol/106 cells) than in control cells (5.02 ± 0.43 μmol/106 cells). In addition, a significant negative correlation between cell Cu content and cell Na+ content was observed (R = −0.57; p < 0.05) (Fig. 4a).

Copper and Na+ content in isolated gill cells from Fraction II of the freshwater mussel Lasmigona costata (a) and the seawater clam Mesodesma mactroides (b) after exposure to copper (5, 9 and 20 μg Cu/L). Data are expressed as mean ± standard error (n = 5). Different letters indicate significant different mean values among treatments (p < 0.05). Closed circle: Cu content; open circle: Na+ content
For M. mactroides, viability in control cells was 81.28 ± 2.75 % while it was 78.81 ± 3.22 %, 75.81 ± 3.05 % and 76.42 ± 2.87 % in those exposed to 5, 9 and 20 μg Cu/L, respectively. As for L. costata, a significant increase in Cu content was also observed in cells exposed to 20 μg Cu/L (0.048 ± 0.004 μmol/106 cells) when compared to that observed in control cells (0.037 ± 0.001 μmol/106 cells) and 5 and 9 μg Cu/L exposure (0.040 ± 0.004 and 0.044 ± 0.001 μmol/106 cells, respectively). Also, there was a positive correlation between Cu content in gill cells and Cu concentration in the incubation medium (R = 0.44; p < 0.05). The Na+ content was also significantly lower in cells exposed to 20 μg Cu/L (5.77 ± 0.88 μmol/106 cells) than in control cells (12.31 ± 1.22 μmol/106 cells). Finally, a significant negative correlation between cell Cu content and cell Na+ content was also observed (R = −0.59; p < 0.05) (Fig. 4b).
Discussion
Two different techniques were employed to dissociate and isolate gill cells from the freshwater mussel L. costata (enzymatic technique) and the seawater clam M. mactroides (mechanical technique). Although the mechanical dissociation approach is considered to be less aggressive for cells during the isolation process, it proved to be inappropriate for dissociating gill cells of the freshwater mussel L. costata. The massive cell mortality observed using this technique (during preliminary trials) may have resulted from the high sensitivity of freshwater bivalves to the low K+ concentration in the external medium (Fisher et al. 1991). In contrast, our data indicate that the enzymatic method was an excellent option to obtain isolated cells from gills of L. costata. Pronase, the enzyme used in the present study, has been described as the most satisfactory in cell dissociation in the freshwater mussel Dreissena polymorpha (Quinn et al. 2009). However, mechanical shaking was included and the time of tissue incubation with the enzyme was improved (reduced from 16 h to 20 min) in the protocol applied for L. costata in the present study, thus resulting in a satisfactory number of isolated cells (~107 cells) at a high percentage of viability in a shorter time. On the other hand, the mechanical cell isolation technique applied to the seawater clam M. mactroides proved very effective, liberating a satisfactory number of isolated cells with a higher percent viability. In addition, gill cells showed a high viability even 5 h after being isolated. According to Lilius et al. (1995), cells are able to maintain their membrane polarity and normal functions up to 5 h after being isolated and then they likely lose this ability after that period.
Following the successful application of the different techniques for isolation of gill cells in the two bivalve species, a three-step Percoll gradient was applied to separate the pool of gill cells obtained into two major fractions. The gill cells of L. costata and M. mactroides present in Fraction II (1.05–1.09 g/L) of the Percoll gradient showed a markedly higher fluorescence emission in the presence of the DASPEI and MitoTracker Green®, respectively. To confirm our finding in the freshwater mussel, MitoTracker Green® was also used with L. costata and similar results were obtained (data not shown). According to Galvez et al. (2002), isolated gill cells from fish retained in the first interface of the Percoll gradient (1.03–1.05 g/L) were pavement cells (PVCs), while those retained in the second interface of the Percoll gradient (1.05–1.09 g/L) were mitochondria-rich cells. Therefore, we could assume that these gill cells of bivalves would correspond also to cells rich in mitochondria.
Cells rich in mitochondria are known to have a high metabolic potential due to their high ability for energy production, thus usually exhibiting high Na+, K+-ATPase activity. Therefore, enzyme activity analysis was the second approach used to differentiate the two pools of cells isolated from gills of the freshwater mussel L. costata and seawater clam M. mactroides. As in other animal species (Wong and Chan 1999; Galvez et al. 2008), both methodologies used in our studies were also satisfactory and cells collected from the Fraction II (1.05–1.09 g/L Percoll gradient interface) showed a markedly higher Na+, K+-ATPase activity than those collected from the Fraction I (1.03–1.05 g/L Percoll gradient interface). In the freshwater mussel, the Na+, K+-ATPase activity was twofold higher in Fraction II than Fraction I while in the seawater clam, the difference was sixfold. This finding was in accord with Na+, K+-ATPase activity in gill cells isolated from Japanese eels where a greater relative difference between Fractions was observed in the seawater animal (Wong and Chan 1999).
In these bivalves, the ratio of gill cells rich in mitochondria to the other cell types present after Percoll gradient separation was very different from that observed for other animals. For example, around 85 % of gills cells in rainbow trout (Oncorhynchus mykiss) were characterized as pavement cells and only 15 % as mitochondria-rich cells after using the Percoll gradient technique to fractionate isolated gill cells (Galvez et al. 2008). In the present study, 80.5 ± 1.5 % of cells isolated from gills of the freshwater mussel L. costata were characterized as showing high mitochondrial density and Na+, K+-ATPase activity, while 48.3 ± 3.2 % of gill cells isolated from the seawater clam M. mactroides exhibited the same properties.
As previously mentioned, Unionid mussels have a high amount of cells rich in mitochondria in their gill epithelium, but pavement cells are present in a greater proportion as in fish (Schwartz and Dimock 2001). However, the higher percentage of cells rich in mitochondria found in the present study suggests that there could have been enrichment associated with the cellular dissociation technique employed. Although it is regarded as the best method for cell isolation in the freshwater bivalve Dreissena polymorpha (Quinn et al. 2009), enzymatic digestion is considered to be an “aggressive” method to perform cell isolation. Therefore, we suspect that a large proportion of the debris and dead cells found after applying the enzymatic method in L. costata, i.e., tissue digestion with pronase, may have been derived from cells showing low mitochondrial density and low Na+, K+-ATPase activity. The two cell fractions were more equal in M. mactroides (Fraction I: 51.7 ± 3.2 %; Fraction II: 48.3 ± 3.2 %) but still not comparable with fish results (Reid et al., 2003; Galvez et al. 2008). Probably, this is a consequence of the less aggressive mechanical method applied for cell dissociation. Regardless, the methods employed in both bivalve species in the current study produced an adequate abundance of viable cells (>80 %) for in vitro experiments.
Following separation and fractionation, gill cells of Fraction II from both species of bivalves were exposed to different Cu concentrations, and analysis of cellular Cu and Na+ content were performed. Cell Cu content in the freshwater mussel L. costata as well as the seawater clam M. mactroides increased linearly with the increasing concentration of Cu in the incubation medium. Also, a significant decrease in cell Na+ content was observed after exposure to the highest concentration of Cu tested (20 μg Cu/L). As a cation, Cu would outcompete with Na+ present in the incubation medium for active sites of ion transport at the membrane cell surface. Therefore, an increase in Cu concentration in the incubation medium could interfere with cell metabolism and function through disturbance of the regulation of essential ions, especially Na+ (Kültz 2001; Grosell et al. 2002; Blanchard and Grosell 2006). In the present study, the rate of cell survival was not significantly affected by exposure to any Cu concentration tested. Therefore, the ionic (Na+) disturbance observed after Cu exposure was not enough to induce cell death. However, a possible lethal effect after exposure to higher concentrations of Cu (>20 μg Cu/L) or for a longer period of time (>3 h) to those tested (5, 9, 20 μg Cu/L) in the present study cannot be ruled out.
Our data indicate that cells isolated from gills of bivalves are a suitable model for in vitro toxicological studies in both freshwater and seawater bivalves. This statement is based on two facts: (1) the significant and positive correlation observed between Cu content in isolated gill cells and the Cu concentration in the incubation medium; (2) the significant and negative correlation observed between the Na+ content and the Cu content in isolated cells present in Fraction II. It is interesting to note that the negative correlations in the freshwater mussel L. costata (R = −0.57) and the seawater clam M. mactroides (R = −0.59) are very similar. This finding is likely associated with the relatively high concentration of Na+ employed in the freshwater saline solution (similar to that in freshwater mussel hemolymph—18 mM NaCl), when compared to that generally found in fresh water (~1 mM). Presumably, 18 mM NaCl was sufficient to induce the maximum cellular Na+ influx in isolated gills cells of both L. costata and M. mactroides, thus limiting the competitive effect of Cu on cellular Na+ uptake and its consequent toxicity to isolated cells.
Although we did not observe any lethal toxicity of Cu to isolated gill cells, studies at the cellular level like the one described here allow the investigation and a better understanding of the mechanisms involved in Cu toxicity because they simulate in vitro, at least in part, the exposure of aquatic animals to contaminants (Castaño et al. 2003). Nevertheless, it is important to recognize the limitations of the current approach which mean that conclusions must be interpreted cautiously. Notably, the saline incubation media employed approximate the ionic composition of hemolymph, whereas in vivo gill cells will be bathed apically with either seawater or freshwater (bearing waterborne Cu) and basolaterally with hemolymph (bearing blood-borne Cu). This polarity of exposure in vivo is lost in vitro. In addition, the saline solutions used in our experiments may not be appropriate for long-term cellular homeostasis due the lack of amino acids (an important component of the mollusc diet; Renault et al. 1995) and other energy sources (e.g. glucose). Therefore, our toxicological data should be considered with caution, since the cells rich in mitochondria could be less sensitive to the toxic conditions if they are in an optimal homeostasis, or more sensitive if they are particularly susceptible to waterborne Cu. Thus, future research should be directed at developing more complete incubation media for the maintenance of cells rich in mitochondria of the freshwater and seawater bivalves in culture, as well as the possible development of cultured gill epithelia on filter membranes. The latter have been successful in allowing apical water and basolateral blood exposures in fish gill cell culture systems (e.g. Fletcher et al. 2000; Kelly et al. 2000; Galvez et al. 2008). These improved in vitro approaches may provide new insights into the transport and metabolic pathways which make bivalves so sensitive to many chemical contaminants found in aquatic ecosystems.
Conclusion
The methodologies employed for isolation and fractionation of cells from the gills of the freshwater bivalve L. costata and the seawater clam M. mactroides were found to be effective, as previously reported for fish (Goss et al. 2001; Galvez et al. 2002; Tse et al. 2006). Also, the application of two different approaches (mitochondria density and Na+, K+-ATPase activity) proved to be reliable in characterizing different sub-types of gill cells in both bivalve species analyzed. Finally, our findings support the idea that Cu outcompetes with Na+ for sites of ion transport on the plasma membrane, as previously observed in the gills of intact crustaceans and fish. Taken altogether, these findings indicate that cells isolated from gills of freshwater and seawater bivalves are a useful tool for studying the cellular mechanisms of metal uptake and toxicity.
References
ASTM (2006) Standard Guide for Conducting Laboratory Toxicity Tests with Freshwater Mussels (E2455-05). ASTM International, West Conshohocken, PA, USA
Bianchini A, Castilho PC (1999) Effects of zinc exposure on oxygen consumption and gill Na+, K+-ATPase of the estuarine crab Chasmagnathus granulata Dana, 1851 (Decapoda, Grapsidae). Bull Environ Contam Toxicol 62:63–69
Bigas M, Durfort M, Poquet M (2001) Cytological effects of experimental exposure to Hg on the gill epithelium of the European flat oyster Ostrea edulis: ultrastructural and quantitative changes related to bioaccumulation. Tissue Cell 33:178–188
Blanchard J, Grosell M (2006) Copper toxicity across salinities from freshwater to seawater in the euryhaline fish Fundulus heteroclitus: is copper an ionoregulatory toxicant in high salinities? Aquat Toxicol 80:131–139
Bradford M (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Buhariwalla HEC, Osmond EM, Barnes KR, Cozzi RRF, Robertson GN, Marshall WS (2012) Control of ion transport by mitochondrion-rich chloride cells of eurythermic teleost fish: cold shock vs. cold acclimation. Comp Biochem Physiol A Mol Integr Physiol 162:234–244
Castaño A, Bols N, Braunbeck T, Dierickx P, Halder M, Isomaa B, Kawahara K, Lee LEJ, Mothersill C, Pärt P, Repetto G, Sintes JR, Rufli H, Smith R, Wood C, Segner H (2003) The use of fish cells in ecotoxicology: the report and recommendations of ECVAM Workshop 47. ATLA 31:317–351
CCME (2005) Canadian water quality guidelines. Canadian council of ministers of the environment, Environment Canada
Chelomin VP, Zakhartsev MV, Kurilenko AV, Belcheva NN (2005) An in vitro study of the effect of reactive oxygen species on subcellular distribution of deposited cadmium in digestive gland of mussel Crenomytilus grayanus. Aquat Toxicol 73:181–189
CONAMA, 2005. Conselho Nacional do Meio Ambiente. Resolução no 357, de 17 de março de 2005
Dietz TH (1979) Uptake of sodium and chloride by freshwater mussels. Can J Zool 57:156–160
Fisher SW, Stromberg P, Bruner KA, Boulet LD (1991) Molluscicidal activity of potassium to the zebra mussel, Dreissena polymorpha: toxicity and mode of action. Aquat Toxicol 20:219–234
Fiske CH, Subbarow Y (1925) The colorimetric determination of phosphorus. J Biol Chem 66:375–400
Fletcher M, Kelly SP, Part P, O’Donnell MJ, Wood CM (2000) Transport properties of cultured branchial epithelia from freshwater rainbow trout: a novel preparation with mitochondria-rich cells. J Exp Biol 203:1523–1537
Galvez F, Reid SD, Hawkings G, Goss GG (2002) Isolation and characterization of mitochondria-rich cell types from the gill of freshwater rainbow trout. Am J Physiol Regul Integr Comp Physiol 282:R658–R668
Galvez F, Tsui T, Wood CM (2008) Cultured trout gill epithelia enriched in pavement cells or in mitochondria-rich cells provides insights into Na+ and Ca2+ transport. In Vitro Cell Dev Biol Anim 44:415–425
Gómez-Mendikute A, Elizondo M, Venier P, Cajaraville MP (2005) Characterization of mussel gill cells in vivo and in vitro. Cell Tissue Res 321:131–140
Goss GG, Adamia S, Galvez F (2001) Peanut lectin binds to a subpopulation of mitochondria-rich cells in the rainbow trout gill epithelium. Am J Physiol Regul Integr Comp Physiol 281:R1718–R1725
Goss G, Gilmour K, Hawkings G, Brumbach JH, Huynh M, Galvez F (2011) Mechanism of sodium uptake in PNA negative MR cells from rainbow trout, Oncorhynchus mykiss as revealed by silver and copper inhibition. Comp Biochem Physiol A Mol Integr Physiol 159:234–241
Grosell M, Nielsen C, Bianchini A (2002) Sodium turnover rate determines sensitivity to acute copper and silver exposure in freshwater animals. Comp Biochem Physiol C Toxicol Pharmacol 133:287–303
Kays WT, Silverman H, Dietz TH (1990) Water channels and water canals in the gill of the freshwater mussel, Ligumia subrostrata: ultrastructure and histochemistry. J Exp Zool 254:256–269
Kelly SP, Fletcher M, Part P, Wood CM (2000) Procedures for the preparation and culture of ‘reconstructed’ rainbow trout branchial epithelia. Methods Cell Sci 22:153–163
Kültz D (2001) Cellular osmoregulation: beyond ion transport and cell volume. Zoology 104:198–208
Le Pennec G, Le Pennec M (2001) Acinar primary cell culture from the digestive gland of Pecten maximus (L): an original model for ecotoxicological purposes. J Exp Mar Biol Ecol 259:171–187
Lilius H, Sandbacka M, Isomaa B (1995) The use of freshly isolated gill epithelial cells in toxicity testing. Toxicol In Vitro 9:299–305
Lin LY, Hwang PP (2004) Mitochondria-rich cell activity in the yolk-sac membrane of tilapia (Oreochromis mossambicus) larvae acclimatized to different ambient chloride levels. J Exp Biol 207:1335–1344
Lin LY, Horng JL, Kunkel JG, Hwang PP (2006) Proton pump-rich cell secretes acid in skin of zebrafish larvae. Am J Physiol Cell Physiol C371-C378
Lopes TM, Barcarolli IF, Oliveira CB, Souza MM, Bianchini A (2011a) Effect of copper on ion content in isolated mantle cells of the marine clam Mesodesma mactroides. Environ Toxicol Chem 30:1582–1585
Lopes TM, Barcarolli IF, Oliveira CB, Souza MM, Bianchini A (2011b) Mechanism of copper accumulation in isolated mantle cells of the marine clam Mesodesma mactroides. Environ Toxicol Chem 30:1586–1592
McCormick SD (1993) Method for non-lethal gill biopsy and measurement of Na+, K+-ATPase activity. Can J Fish Aquat Sci 50:656–658
Pinho GLL, Pedroso MS, Rodrigues SC, Souza SS, Bianchini A (2007) Physiological effects of copper in the euryhaline copepod Acartia tonsa: waterborne versus waterborne plus dietborne exposure. Aquat Toxicol 84:62–70
Quinn B, Costello MJ, Dorange G, Wilson JG, Mothersill C (2009) Development of an in vitro culture method for cells and tissues from the zebra-mussel (Dreissena polymorpha). Cytotechnology 59:121–134
Reid SP, Hawkings GS, Galvez F, Goss GG (2003) Localization and characterization of phenamil-sensitive Na+ influx in isolated rainbow trout gill epithelial cells. J Exp Biol 206:551–559
Renault T, Flaujac G, Le Deuff R-M (1995) Isolation and culture of heart cells from the European flat oyster, Ostrea edulis. Methods Cell Sci 17:199–205
Schwartz ML, Dimock RV Jr (2001) Ultrastructural evidence for nutritional exchange between brooding unionid mussels and their glochidia larvae. Invertebr Biol 120:227–236
Tse WKF, Au DWT, Wong CKC (2006) Characterization of ion channel and transporter mRNA expressions in isolated gill chloride and pavement cells of seawater acclimating eels. Biochem Biophys Res Commun 346:1181–1190
Wong CKC, Chan DKO (1999) Isolation of viable cell types from the gill epithelium of Japanese eel Anguilla japonica. Am J Physiol 276:R363–R372
Wong CKC, Chan DKO (2001) Effects of cortisol on chloride cells in the gill epithelium of Japanese eel, Anguilla japonica. J Endocrinol 168:185–192
Acknowledgments
We thank Rodney McInnis, Tina Hooey, Jim Bennett, Mark McMaster (Environment Canada) and Robert Boyle and Ana Cristina Kalb (Universidade Federal do Rio Grande, Brazil), and two anonymous reviewers for constructive comments. This research was supported by awards from the International Development Research Centre (IDRC, Ottawa, ON, Canada) to A. Bianchini and C.M. Wood, the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brasília, DF, Brazil), and the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brasília, DF, Brazil). A. Bianchini is a research fellow from the Brazilian CNPq (Proc. #304430/2009-9) and is supported by the International Research Chair Program from IDRC. C.M. Wood is supported by the Canada Research Chair Program. PL. Gillis is supported by Environment Canada.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nogueira, L.S., Wood, C.M., Gillis, P.L. et al. Isolation and fractionation of gill cells from freshwater (Lasmigona costata) and seawater (Mesodesma mactroides) bivalves for use in toxicological studies with copper. Cytotechnology 65, 773–783 (2013). https://doi.org/10.1007/s10616-013-9647-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10616-013-9647-2