
Abstract
Pedicularis dudleyi (Dudley’s Lousewort, Orobanchaceae) is an extremely rare plant endemic to the redwood forests of Central California. Until recently, the species was known only from three extant natural populations. However, in 2019, one of those populations was described as a novel species (P. rigginsiae D.J. Keil) based on morphological and ecological data leaving only two populations described as P. dudleyi. While little is known about the past distribution of the species, historical records have led to speculation that the species was once more widespread and may have suffered from habitat destruction as a result of widespread logging during the early twentieth century. We utilized a combination of ddRAD SNP and Sanger sequencing data to: (1) Test the morphological hypothesis that P. rigginsiae is distinct from P. dudleyi; (2) Describe the genetic diversity and population structure of P. dudleyi and; (3) Test the hypothesis that the species underwent a bottleneck corresponding with increased logging of redwood forests in the early twentieth century. Our results support the recognition of P. rigginsiae as distinct from P. dudleyi, increasing the conservation priority of both species. Genetic diversity statistics and analyses of genetic structure suggest that both populations of P. dudleyi are highly differentiated from each other with one population exhibiting unexpected substructure. Finally, demographic modeling supports a scenario where the contemporary rarity of the species is explained by a recent bottleneck.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rare species with small population sizes are at high risk for extinction due to environmental and demographic stochasticity and genetic drift. These forces can lead to a loss of allelic diversity and increased homozygosity (Barrett and Kohn 1991; Ellstrand and Elam 1993), which can increase the risk of inbreeding depression, reduce survival and fecundity, and limit the ability of species to adapt to changing environmental conditions (Booy et al. 2000; Oostermeijer et al. 1994, 1996; Spielman et al. 2004; Reed and Frankham 2003). Still, rarity can arise through a variety of evolutionary, ecological, and demographic processes, each of which are likely to influence the extent to which species are at risk for negative outcomes associated with low genetic diversity (Habel and Schmitt 2012).
Previously widespread species can become rare due to environmental change. For example, anthropogenic factors such as habitat loss, introduction of novel pathogens, and competition with invasive species have all contributed to recent species declines (Anagnostakis 1987; Tilman et al 1994; Rizzo and Garbelotto 2003; Bellard et al. 2016). In many of these cases, initial differentiation among populations is low and genetic diversity within is high. Upon reduction of population size, these species are expected to suffer from inbreeding depression and stochastic shifts leading to the loss of adaptive alleles (Frankham 2005; Habel and Schmitt 2012).
Alternatively, species may be innately or historically rare, maintaining small effective population sizes for much of their existence. This may be due to specialization on restricted or patchy habitats (Brown 1984; Gaston and Lawton 1990), a combination of metapopulation dynamics and life history traits (i.e., low dispersal ability or breeding system) that may increase the probability of local extinction and reduce recolonization (Hanski 1982; Gaston and Lawton 1990; Hanski and Gyllenberg 1993), or evolutionary history, where recently evolved species may not have had time to expand their range (Stebbins 1979). These species often exhibit high differentiation among, and low genetic diversity within populations and are able to persist over time as such. Over successive generations, small populations are expected to purge the majority of their deleterious alleles such that further reductions in population size may not lead to inbreeding depression (Crnokrak and Barrett 2002; Taylor and Jamieson 2008; Habel et al. 2009). As a result, rare species with historically low levels of genetic diversity may be less susceptible to negative effects associated with loss of genetic diversity than rare species that were more recently widespread (Frankham et al. 2001; Crnokrak and Barrett 2002; Habel and Schmitt 2012). While many conservation genetic studies of rare or threatened plant species focus on quantifying the distribution of genetic diversity and gene flow within and among populations, insight into the ecological, demographic and evolutionary processes that underlie rarity can complement traditional conservation genetic studies and inform effective management and restoration programs (Frankham 2005; Maschinski and Albrecht 2017).
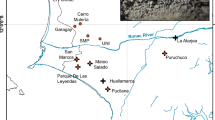
Pedicularis dudleyi Elmer (Dudley’s Lousewort, Orobanchaceae) is endemic to the redwood forests of Central California (Fig. 1A and B) and is state listed as “rare” (CDFW 2021) under the California Endangered Species Act (CESA) (California Fish and Game Code §§2050, et seq.) with a California Native Plant Society Rare Plant Rank of 1B.1 (seriously threatened) (CNPS 2021). Until recently, the species was known from three extant populations: one occurring in and around Portola Redwoods State Park in San Mateo County, the second along the North Fork Little Sur River in Monterey County and the third in San Luis Obispo County (Fig. 1A). However, in 2019, the southern-most population was described as a novel species, P. rigginsiae D.J. Keil (Arroyo de la Cruz lousewort), based on morphological and ecological data (Keil 2019). While little is known about the past distribution of the species, the existence of herbarium specimens from locations between the two extant populations has led to speculation that it was once more widespread and may have suffered from habitat destruction and fragmentation in the early twentieth century when redwood forests were heavily logged (Gujral et al. In Press).

A Map with county delimitations for localities of the two known P. dudleyi populations and one known P. rigginsiae population. B Photo of P. dudleyi. Photo credit: Benjamin Carter
Pedicularis dudleyi is hemiparasitic and is thought to form haustorial connections with multiple host species including Vaccinium ovatum, Notholithocarpus densiflorus and Ceanothus thyrsiflorus (Sprague 1959, 1962). The species is primarily outcrossing and is chiefly pollinated by two native bees, Bombus sitkensis and Bombus edwardsii (Gujral et al. In Press). Seeds are relatively large (ca. 1–1.5 mm × 2–3 mm) and experience passive dispersal in areas immediately surrounding the maternal plants. However, the wasp Vespula alascensi is a frequent visitor to P. dudleyi fruiting stalks and may be an important vector for longer distance seed dispersal (Gujral et al. In Press).
Population sizes at the two known populations, Portola Redwoods State Park and the North Fork of the Little Sur River, are small. A 2019 census of the species at Portola Redwoods found 468 reproductive adults (Gujral et al. In Press) scattered around the campgrounds and visitor’s center with up to a few hundred more potentially existing in satellite populations on surrounding private land. A 2020 census of the species along the Little Sur River counted approximately 200 reproductive adults (Hauser unpublished data) distributed linearly along the river and likely represent the full extent of the population in that area. The species is perennial and individuals reach reproductive maturity after 5–6 years and can live for up to 50 years. Long-term surveys suggest that populations may be declining, and recent research has found that low germination and seedling establishment may be the biggest barriers to population growth (Gujral et al. In Press). To date there have been no studies investigating population genetic diversity or differentiation within the species.
We utilized a combination of ddRAD SNP and Sanger sequencing data to: (1) Test the morphological hypothesis that P. rigginsiae is distinct from P. dudleyi; (2) Describe the genetic diversity and population structure of P. dudleyi and; (3) Test the hypothesis that the species underwent a bottleneck corresponding with increased logging of redwood forests in the early twentieth century.
Methods
Sample collection
We sampled 146 Pedicularis dudleyi individuals from the two known populations (n = 75, Portola Redwoods; and n = 71, Little Sur River, North Branch; Figs. 1 and 2) in the summer of 2018 under California Department of Fish and Wildlife permit 2081(a)-19–004-RP. We sampled individuals that were at least 2 m apart to avoid obtaining multiple samples from the same genet. Approximately 2 cm2 of leaf material was collected per plant and dried and stored on silica gel. Voucher specimens were deposited in the Carl W. Sharsmith Herbarium, San Jose State University (SJSU) and the Robert F. Hoover Herbarium at California Polytechnic State University, San Luis Obispo (OBI). Pedicularis rigginsiae leaf material was obtained from two herbarium specimens at the Hoover Herbarium, including one from an Isotype. Due to the rarity of the species we have omitted coordinates for individual plants.

A Evolutionary clusters (K = 2 and 3) inferred from STRUCTURE analysis of 118 P. dudleyi individuals from the two known populations. Each color represents an inferred character, and each individual is represented by a vertical line colored according to the probability of its assignment to a given population. B Map of individuals from the Little Sur Location. Gray points represent locations at which multiple individuals were sampled along the Little Sur River. Large circles represent individual plants collected at each location and are colored according to the proportion admixture inferred by STRUCTURE (K = 3). The Little Sur River flows from East to West
Phylogenetic inference
Genomic DNA was extracted from the leaf material using a Qiagen DNeasy Plant Mini Kit (Valencia, CA, USA). To test the morphological and ecological hypothesis that P. rigginsiae is distinct from P. dudleyi we generated nuclear ribosomal internal transcribed spacer (ITS) and matK-5’trnK spacer sequences for P. dudleyi (N = 5) and P. rigginsiae (N = 2) to reconstruct the phylogeny. ITS was sequenced in two parts, ITS1 and ITS2 using the primer pairs described in Daly and Fine (2011). The matK-5’trnK spacer was amplified using the matK5 and matK6 primers described in Shaw et al. (2005). Polymerase chain reactions were carried out in 25 μl reactions containing approximately 25 ng genomic DNA, 1.5 μM each of forward and reverse primer, 12.5 μl Kapa Hifi HotStart ReadyMix (Roche Sequencing Solutions, Indianapolis, IN, USA) and water. PCR thermocycling conditions for both ITS 1 and 2 consisted of an initial denaturation step at 95 °C for 2:30 m followed by 34 cycles of 95 °C for 30 s, 56 °C for 30 s, and 72 °C for 30 s and a final extension step of 72 °C for 10 m. Thermocycling conditions for matK-5’trnK spacer consisted of an initial denaturation at 94 °C for 3 m followed by 40 cycles of 94 °C for 30 s, 48 °C for 40 s, and 72 °C for 1 m with a final extension at 72 °C for 10 min. PCR products were visualized on a 1% TBE agarose gel and purified using SPRI beads prior to sequencing. PCR products were cycle sequenced in both directions using the original PCR primers on an ABI 3730 automated DNA Analyzer (Applied Biosystems, Foster City, CA, USA) at the UC Berkeley DNA Sequencing Facility. Geneious Pro 5.3.3 (Drummond et al. 2010) was used to analyze and edit chromatograms, and to assemble double-stranded consensus sequences. Only high quality, unambiguous reads were assembled. ITS and matK-5’trnK spacer sequences for closely related taxa as determined by previous phylogenetic analysis of the genus (Robart et al. 2015) were downloaded from GenBank and are listed in Table 1. All sequences for each locus were aligned with MAFFT 7 (Katoh and Standley 2013) using the L-INS-I strategy. Trees were constructed for each locus and the concatenated dataset, using RAxML v8.2.9 (Stamatakis 2014) with the “-f a” option, GTRGAMMA model and 100 bootstrap replicates. Alignment summary statistics were calculated using AMAS (Borowiec 2016).
Library preparation and sequencing
Libraries were constructed following a modified version of the protocol described in Peterson et al. (2012), where amplification was performed prior to size selection. Briefly, 500 – 150 ng of genomic DNA were digested using the restriction enzymes EcoRI and SphI-HF (New England Biolabs, Ipswich, MA, USA) following NEB guidelines. P1 and P2 “flex” adapters were ligated to the digested DNA and ligation products were combined into pools containing 10–24 samples. Each pool was dual indexed using PCR primers as described in Meyer and Kircher (2010). After amplification, indexed libraries were quantified using qPCR and then combined in equimolar ratios. The final library was size selected for 750 bp (including internal adapters) using tight mode on a Pippin Prep System (Sage Science, Beverly, MA, USA) at the Functional Genomics Laboratory at the University of California, Berkeley. The final Illumina library was sequenced by Novogene at the UC Davis Sequencing Center on a single lane of Illumina HiSeq X Ten (150 bp, paired end) (Illumina, Inc., San Diego, CA, USA).
Data processing and SNP calling
Raw sequences were demultiplexed, quality filtered, and assembled using Stacks v2.54 (Rochette et al. 2019) at the University of Oklahoma Supercomputing Center for Education and Research (OSCER). Sequences were sorted and filtered using the process_radtags script. Clustering, assembly, and filtering parameters were optimized with a subset of 24 individuals from both study sites using the r80 method as described in Paris et al. (2017). The following parameters were used to call the final dataset: minimum number of raw reads required to form a stack, m = 3; maximum number of mismatches between stacks within individuals, M = 1; maximum number of mismatches allowed between stacks among individuals n = 2; minimum percentage of individuals across populations required to process a locus, R = 0.75; minimum minor allele frequency, min-maf = 0.01; and maximum observed heterozygosity, max-obs-het = 0.7. All 146 individuals were used to create the locus catalog (cstacks) for the final Stacks analysis. Because SNPs found on the same locus are expected to be tightly linked (Rochette and Catchen 2017) we filtered our dataset to one SNP per locus using the –write-random-snp flag in the STACKS script populations. Our data was further refined using VCFtools v0.1.17 (Danecek et al. 2011) to remove individuals with > 30% missing data and loci sequenced at > 2 × the SD of coverage as they may represent paralogous loci. Finally, we used BayeScan v.2.1, which measures the dissonance between global and population level allele frequencies, to identify loci that may be under selection in each population. Prior odds for this analysis was set to 100. We used a burn-in of 50,000, thinning interval of 10, sample size of 50,000 and 20 pilot runs. Pilot run length was set to 50,000 and a false discovery rate of 0.05 was used (Foll and Gaggiotti 2008).
Population structure and differentiation
We sought to understand how populations are genetically structured using a combination of Bayesian clustering analysis and descriptive statistics. Population genetic structure was assessed using Bayesian Markov chain Monte Carlo clustering implemented in STRUCTURE 2.3.3 (Pritchard et al. 2000). STRUCTURE was run using the admixture model and assuming correlated allele frequencies with a burn-in period of 500,000 generations followed by 105 Markov chain Monte Carlo generations for each value of K = 1–4. Simulations were repeated twenty times for each value of K. Structure Harvester (Earl and vonHoldt 2012) was used to interpret the output as described by Evanno et al. (2005) and Pritchard et al. (2000). Admixture proportions were averaged over all runs using CLUMPP (Jakobsson and Rosenberg 2007), and DISTRUCT 1.1 (Rosenberg 2004) was used to visualize the final matrix. Principal components analysis (PCA) was carried out using the R package adegenet v2.1.1 (Jombart 2008) to identify differences in allele frequencies among populations and confirm our STRUCTURE results. Finally, genetic differentiation among all three populations was assessed using population-level pairwise comparisons of FST calculated in GenoDive v3.03 (Meirmans and Tienderen 2004) with confidence intervals being generated over 105 permutations.
Genetic diversity
Initial analysis of population structure suggested the presence of fine scale structuring in the Little Sur population (Fig. 2). As a result, estimates of genetic diversity were carried out with individuals being assigned to one of three populations (Portola, Little Sur 1, and Little Sur 2). To characterize genetic diversity within populations, we calculated summary statistics including the number of private alleles, percentage of polymorphic loci, the average frequency of the major allele (P), the average observed heterozygosity (HO), expected heterozygosity (HE), and the average Wright’s inbreeding coefficient (FIS) for each population using the populations script in STACKS.
Population history
To test the hypothesis that P. dudleyi populations underwent a bottleneck in the early twentieth century we utilized an Approximate Bayesian Computation (ABC) approach implemented in DIYABC Random Forest v1.0 (Collin et al. 2021). Briefly, ABC implements model-based inference in a Bayesian setting to simulate coalescence of the observed populations given different demographic scenarios. Parameter values are drawn from prior distributions and calculated summary statistics. Simulated datasets are then compared to the observed dataset using the machine learning tool Random Forest (RF) (Breiman 2001) to determine the best supported model and estimate its posterior probability. Because P. dudleyi is monoecious, sex ratio was set to 1. Minimum allele frequency was set to 0.01. Three scenarios were considered to investigate whether populations of P. dudleyi underwent a bottleneck that corresponds with early twentieth century logging of the redwood forests. We chose the Little Sur 2 population to represent the ancestral population because it has significantly greater levels of genetic diversity than the Portola population (Table 2, Fig. 3). All demographic models included three populations (Portola, Little Sur 1, and Little Sur 2) where Portola and Little Sur initially diverge from the ancestral population followed by the subsequent divergence of Little Sur 1 and 2 (Fig. 3). In scenario 1 population sizes remain stable over time. In scenario 2 all populations undergo a bottleneck after the divergence of Little Sur 1 and 2. In scenario 3 a bottleneck occurs in all populations when Portola diverges from Little Sur but prior to the divergence of Little Sur 1 and 2 (Fig. 3).

Schematic representations of models tested for P. dudleyi demographic history using approximate Bayesian computation. N1, N2 and N3 represent the effective population sizes for Portola, Little Sur 1 and Little Sur 2 respectively, t represents time, and db represents bottleneck duration. Prior distributions and descriptions are listed in supplementary Table S2
Demographic and historical parameters are illustrated in Fig. 3 and include effective population sizes (N1, N2, and N3 for populations Portola 1, Little Sur 1 and Little Sur 2 respectively) and time events at which divergence and/or bottleneck events occur (t1, t2, t3). For scenarios that included a bottleneck, parameter db represents the duration of the bottleneck in generations. For time events DIYABC RF utilizes generation time as opposed to years. Thus, all priors for time events are in generations where one generation for P. dudleyi is equivalent to approximately 5 years (Gujral et al. In Press). All prior values were drawn from uniform distributions and are listed in Supplementary Table S2. Additional conditions were used to specify that t3 > t2 > t1 and that N1c > N1b, N2c > N2b, N3c > N3b, N3e > N3d and N3e > N1d.
In our Random Forest analysis each scenario was considered separately. Prior values were drawn from the distributions described above and from a reference table computed using 1964 SNPs and 304 simulated data sets per scenario. All summary statistics as well as the optional axes of a linear discriminant analysis (LDA) were used in the RF analyses. Model choice is a two-step process where the best scenario is first chosen based on the number of classification votes and then followed by the calculation of the posterior probability for that scenario as well as global and local error rates. For model choice we utilized 5000 RF trees.
Independent RF analyses were conducted for each parameter of interest. Parameter values were estimated for all time events, bottleneck duration, and effective population sizes in our best supported scenario, scenario 3. The training set included 105 datasets and utilized 2000 RF trees. For each parameter, we inferred point estimates and computed global and local accuracy indices corresponding to global and local normalized mean absolute error (NMAE), which is the absolute difference between the point estimate and the true simulated value divided by the true simulated value, using out-of-bag estimators from 50,000 data randomly chosen from the training set.
Results
Phylogenetic inference
The complete datasets for the matK-5’trnK spacer gene contained sequences from 22 individuals and 849 characters of which 5.9% were variable and 3.3% were parsimony informative. The ITS data set (ITS 1 and 2 combined) contained sequences from 22 individuals and 618 characters, of which 27% were variable and 12.6% were parsimony-informative. Results of our ML analysis support the morphological and ecological hypothesis that P. rigginsiae is distinct from P. dudleyi. The maximum likelihood analyses for individual genes as well as the concatenated dataset all yielded similar topologies with the clade formed by P. dudleyi, P. rigginsiae, and P. densiflora highly supported as monophyletic. Pedicularis dudleyi and P. rigginsiae each formed their own clades with P. rigginsiae sister to P. densiflora (Fig. 4 and Figs. S1 and S2). No nucleotide variation was observed in either the ITS or matK-5’trnK spacer when comparing P. dudleyi individuals from the Little Sur and Portola populations.

Phylogeny of Pedicularis inferred from Maximum Likelihood (ML) methods using the concatenated nuclear ribosomal internal transcribed spacer (ITS) and the matK-5’trnK spacer. Sample ID for individuals sequenced as part of this study are included in parentheses following the taxon name. Values below the branches indicate ML bootstrap values ≥ 70
Data processing
After all quality filters were implemented in the Stacks program process_radtags, 6,324,404 reads that were missing a barcode, 7,396,631 reads which had no rad tag and 244,114 reads identified as low quality were discarded. A total of 1,030,346,369 reads from the initial 1,044,311,518 raw reads were retained, with an average of 6,291,033 reads per sample. Mean average coverage per locus after ustacks filters, was 52.6 × (± 49.3x). Twenty-eight individuals and 116 sites were subsequently dropped due to missing data and potential paralogy. Average coverage per locus for individuals included in the final dataset can be found in Supplementary Table S1. No loci were identified as being under selection. The final genetic marker dataset consisted 2,026 variant sites with no more than 25% missing data for 118 individuals.
Population structure and differentiation
Analysis of population structure uncovered unexpected structure within the Little Sur population and revealed near complete genetic differentiation between the Portola and Little Sur populations. Using the method of Pritchard et al. (2000), we found K = 3 to be best supported model where LnP(D) begins to asymptote (results not shown). Clustering for K = 2 separated the Portola and Little Sur populations while the identified clusters for K = 3 distinguish additional structure between upstream and downstream portions of the Little Sur population (Fig. 2A and B). Results from the PCA are also consistent with the result of K = 3 with PC1 representing 72% of the variation and accounting primarily for differentiation between Portola and the two Little Sur populations while PC2 accounts for 6% of the variation, primarily differentiating the two Little Sur populations from each other (Fig. 5). Pairwise FST values between Portola and the two genetic groups in the Little Sur population (Little Sur 1 and 2) were 0.903 and 0.891 respectively suggesting that the Portola and Little Sur populations share very few alleles. FST for the Little Sur populations was 0.348 which, while lower than FST values observed for the Portola population, still suggests significant population differentiation.

Principal component (PC) plot of SNP data for PC1 and PC2. Each point represents a single individual from one of the three populations (Portola, Little Sur 1, or Little Sur 2)
Genetic diversity
Genetic diversity across the two Little Sur populations were comparable for all indices and they exhibited greater overall genetic diversity compared to the Portola population (Table 2). The proportion of polymorphic loci ranged from 29% to 48%. Average major allele frequency (P) ranged from 0.987 for Portola to 0.915 for Little Sur 2. Expected heterozygosity, a measure of gene diversity, ranged from 0.017 to 0.121 and was significantly greater in both the Little Sur populations than the Portola population. The inbreeding coefficient, which measures the reduction in observed heterozygosity compared to expected heterozygosity, did not significantly differ from zero in Portola (FIS = 0.013) but was significantly greater than zero in the Little Sur populations (Little Sur 1 FIS = 0.229 and Little Sur 2 FIS = 0.254). Inbreeding values greater than zero indicate higher homozygosity often attributable to inbreeding, assortative mating or cryptic population structure (Nei 1987; Holsinger and Weir 2009).
Population history
Three demographic scenarios were tested. In scenario 1, the three population sizes remain constant over time; in scenario 2, bottlenecks occur in all three populations after Little Sur 1 and 2 have diverged and in scenario 3, bottlenecks occur in all populations beginning when Portola and the ancestral Little Sur population diverge (Fig. 3). Our observed data were located within the simulated dataset of the LDA projection suggesting that our priors and scenarios were compatible with the observed dataset (Fig. S3A). The best supported demographic scenario was scenario 2, which received the highest number of classification votes (99.9%) and greatest posterior probability (PP = 1.00; Table 3). Scenarios 1 and scenario 3 were not supported, with each receiving less than 0.1% of the classification votes (Table 3). Table 4 shows point estimates with 95% confidence intervals of posterior distributions as well as NMAE accuracy measures for parameters of interest corresponding to onset of the bottleneck (t1), duration of bottleneck (db) and effective population size for the Portola, Little Sur 1 and Little Sur 2 populations (N1c, N2c and N3c) for scenario 2. The median estimate for the onset of the bottleneck was 39 generations ago (95% CI 5–117 generations ago) and the median duration was estimated to be 100 generations (95% CI 8–192 generations). The median pre-bottleneck population sizes for Portola, Little Sur 1 and Little Sur 2 were 1747, 10,574, and 14,743 respectively (95% CI 940–5197; 2986–18,708 and 7586–19,672). Estimation for parameter db was less supported than other parameters as reflected by higher local and global NMAE values (Table 4). Additional output from our demographic analysis can be found in Figs. S2 and S3.
Discussion
Pedicularis dudleyi is a rare wildflower endemic to the redwood forests of Central California and is currently known from only two naturally occurring populations. Phylogenetic analysis strongly supports the morphological hypothesis that P. rigginsiae is distinct from P. dudleyi and is instead sister to the common, widespread, and closely related P. densiflora.
Our results provide evidence of strong genetic structuring between Portola and Little Sur but also unexpected population structure within Little Sur. Overall, populations exhibited low genetic diversity. The Portola population was significantly less genetically diverse than the populations from Little Sur while the Little Sur populations were found to have higher levels of inbreeding than Portola. Demographic analyses suggest that the rarity of P. dudleyi is likely to be the result of a recent bottleneck and could be the result of habitat destruction during the early twentieth century.
Results from our phylogenetic analysis support the classification of P. rigginsiae as distinct from P. dudleyi. Pedicularis rigginsiae is known from only one population on private land. While once considered a population of P. dudleyi, Keil (2019) recently described it as a separate species and hypothesized that it was more closely related to the common and widespread P. densiflora. Pedicularis rigginsiae differs from P. dudleyi in both habitat and morphology. While P. dudleyi occupies the Southern Coastal Redwood forests, P. rigginsiae is found in dwarfed maritime chaparral on coastal terraces. Pedicularis rigginsiae is morphologically distinguished from P. dudleyi by its narrower and more densely lobed leaves as well as differences in corolla shape and length (Keil 2019). Whether P. dudleyi’s rarity is due to a young evolutionary age is equivocal. While the fact that P. dudleyi is sister to both the extremely rare P. rigginsiae and the very common and widespread P. densiflora suggests that the observed rarity is unlikely to be the result of young evolutionary age with insufficient time for range expansion, we cannot rule out a scenario where P. dudleyi evolved recently from an extinct sister species.
We found evidence of three distinct genetic clusters in P. dudleyi. One genetic cluster corresponds with individuals collected from Portola Redwoods. Individuals from this population showed no evidence of admixture with either Little Sur population (Fig. 2A) and FST values between Portola and Little Sur 1 and 2 were exceptionally high, suggesting that these populations have long been isolated and share very few alleles. Portola Redwoods State Park is separated from the Little Sur populations by nearly 110 km. Given that P. dudleyi is pollinated by native bumblebees and seeds are likely dispersed by a combination of gravity and wasps (Sprague 1959, 1962; Gujral et al. In Press) the lack of gene flow among these geographically distant populations is unsurprising. Moreover, small population sizes coupled with low genetic diversity and little to no gene flow makes these populations subject to high levels of genetic drift which is likely driving the genetic differentiation observed in this study (Allendorf 1986; Slatkin 1987).
More surprising was the genetic structure uncovered at Little Sur. The Little Sur population is distributed linearly along the North Fork of the Little Sur River, and the break in genetic structure occurs about halfway along the population’s distribution and shortly after the confluence of Skinner Creek with the Little Sur River. Geneflow appears to be directional to some extent with individuals immediately downstream from the genetic break exhibiting increased admixture with genetic contributions originating from the upstream population (Fig. 2B). FST between Little Sur 1 and Little Sur 2 was considerable (0.348) especially given that none of the plants sampled in this study are found more than 1 km from their nearest neighbor and bumblebees are known to forage over 1 km and up to 2.5 km from their nesting sites (Kreyer et al. 2004; Osborne et al. 2008; Hagen et al. 2011). One explanation could be that the two populations have been historically isolated along different parts of the river and are now coming into contact. Alternatively, there could be some indiscernible barrier to gene flow, although this seems unlikely given the close ecological and geographic proximity and lack of observable morphological differences between the two populations.
Overall estimates of genetic diversity for P. dudleyi populations were comparable to those found in other studies of rare and endangered plants employing ddRAD data. Estimations of HE for populations of the California endangered salt marsh plant Chloropyron maritimum ssp. maritimum, which is also a hemiparasitic plant in the Orobanchaceae, ranged from 0.004 to 0.059 (Milano et al. 2020). HE estimates for the Hawaiian lobelioids Clermontia and Cyania ranged from 0.127 to 0.138 (Jennings et al. 2016) and estimates for Rhododendron cyanocarpum ranged from 0.060 to 0.65 (Liu et al. 2020). While P. dudleyi populations from Portola and Little Sur both exhibit low levels of genetic diversity (0.017–0.121), the Portola population is significantly less genetically diverse than the Little Sur populations in spite of being larger. These differences could be explained by a combination of demography and life history. For example, a small number of progenitors may have given rise to the modern-day Portola population via a founder event. Given the long lifespan and low recruitment observed in the species today, a population arising from only a small number of individuals would be likely to remain small for a long period of time with little accumulation of new genetic diversity. The Little Sur populations on the other hand have substantially fewer individuals than the Portola population but significantly higher genetic diversity, suggesting that they may be the remnants of a once larger and more genetically diverse population. This hypothesis is supported by our demographic modeling results, where the estimate of pre-bottleneck effective population size for the Portola population is significantly smaller than the pre-bottleneck effective population size estimations for the Little Sur populations (Table 4).
Like all model-based methods, our demographic inferences cannot reveal the ‘true’ history of the species. Instead it allows us to choose the best scenario from among a necessarily limited set of options. Our results suggest that the current-day rarity of P. dudleyi can be attributed to a recent bottleneck that may correspond with the heavy logging of redwood forests in the early twentieth century. Given that heavy logging of the Central California redwood forests was most intense between 1850 and 1920 a bottleneck onset driven by logging would likely have begun between 101 and 171 years ago and lasted for approximately 70 years. These times fall well within the confidence intervals for our parameter estimations of the onset and duration of a bottleneck in our best supported scenario. Regardless of the exact timing or cause of the population decline, our data fail to support a hypothesis that P. dudleyi has simply persisted in small, naturally isolated populations over time and instead suggests that the observed rarity of the species is likely the result of a relatively recent bottleneck.
Conservation implications
Pedicularis dudleyi is at high risk for extinction from stochastic environmental and demographic events due to its extremely small population sizes. Currently, two potential threats are of concern. First, trampling and routine road and trail maintenance are potential issues as a large proportion of individuals are found along trails, roads and in campgrounds. Second, leaf litter buildup (presumably a result of fire suppression) may be negatively impacting seedling establishment, with the litter layer often exceeding 10 cm around established plants. Herbivory by deer has also been documented several times, including removal of flowering stalks, most leaves from a rosette or, in at least one case, removal of an entire plant with roots. Furthermore, P. dudleyi may not be able to withstand a changing climate and shifting habitats given the apparent low capacity for seed dispersal and establishment. This scenario is particularly concerning given predictions of near-term range contractions for redwood forests in Central CA as a result of climate change (Fernández et al. 2015). Both populations of P. dudleyi are small, strongly genetically differentiated from one another and likely to have their own site-specific adaptations. Conservation initiatives should not mix individuals from the Portola and Little Sur populations and should source seeds for seed banking initiatives from all three populations. The majority of P. dudleyi plants in the Portola region are found on public property at Portola Redwoods State Park and approximately one third of plants in the Little Sur region occur in Los Padres National Forest. While current landowners have been amenable to conservation efforts on their property, we recommend prioritizing permanent protection of private land along the upstream portion of the Little Sur River which houses the bulk of the Little Sur 1 population. Finally, given the extreme rarity of both P. dudleyi and P. rigginsiae, we recommend that both species be considered for recognition under the Endangered Species Act.
References
Allendorf FW (1986) Genetic drift and the loss of alleles versus heterozygosity. Zoo Biology 5:181–190. https://doi.org/10.1002/zoo.1430050212
Anagnostakis SL (1987) Chestnut blight: the classical problem of an introduced pathogen. Mycologia 79:23–37. https://doi.org/10.1080/00275514.1987.12025367
Barrett SC, Kohn JR (1991) Genetics and conservation of rare plants. Genetic and evolutionary consequences of small population size in plants: Implications for conservation. Oxford University Press, New York, pp 3–30
Bellard C, Cassey P, Blackburn TM (2016) Alien species as a driver of recent extinctions. Biol Let 12:20150623. https://doi.org/10.1098/rsbl.2015.0623
Booy G, Hendriks RJ, Smulders MJ, Van Groenendael JM, Vosman B (2000) Genetic diversity and the survival of populations. Plant Biol 2:379–395. https://doi.org/10.1055/s-2000-5958
Borowiec ML (2016) AMAS: a fast tool for alignment manipulation and computing of summary statistics. PeerJ 4:e1660. https://doi.org/10.7717/peerj.1660
Breiman L (2001) Random forests. Mach Learn 45:5–32. https://doi.org/10.1023/A:1010933404324
Brown JH (1984) On the relationship between abundance and distribution of species. Am Nat 124:255–279. https://doi.org/10.1086/284267
California Native Plant Society, Rare Plant Program (2021) Inventory of Rare and Endangered Plants of California (online edition, v9–01 0.0). Available from https://www.rareplants.cnps.org. Accessed Jun 22, 2021).
California Department of Fish and Wildlife (CDFW) (2021) State and Federally Listed Endangered, Threatened, and Rare Plants of California.
Collin FD, Durif G, Raynal L, Lombaert E, Gautier M, Vitalis R, Marin JM, Estoup A (2021) Extending approximate bayesian computation with supervised machine learning to infer demographic history from genetic polymorphisms using DIYABC random forest. Mol Ecol Resour 00:1–16. https://doi.org/10.1111/1755-0998.13413
Crnokrak P, Barrett SC (2002) Perspective: purging the genetic load: a review of the experimental evidence. Evolution 56:2347–2358. https://doi.org/10.1554/0014-3820(2002)056[2347:PPTGLA]2.0.CO;2
Daly DC, Fine PV (2011) A new Amazonian section of Protium (Burseraceae) including both edaphic specialist and generalist taxa. Studies in neotropical Burseraceae XVI. Syst Bot 36:939–949. https://doi.org/10.1600/036364411X604958
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G (2011) The variant call format and VCF tools. Bioinformatics 27:2156–2158. https://doi.org/10.1093/bioinformatics/btr330
Drummond AJ, Ashton B, Buxton S, Cheung M, Cooper A, Heled J, Kearse M, Moir R, Stones-Havas S, Sturrock S, Thierer T, Wilson A (2010) Geneious v5.3.3. Biomatters Ltd, Auckland, New Zealand
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Ellstrand NC, Elam DR (1993) Population genetic consequences of small population size: implications for plant conservation. Annu Rev Ecol Syst 24:217–242. https://doi.org/10.1146/annurev.es.24.110193.001245
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Fernández M, Hamilton HH, Kueppers LM (2015) Back to the future: using historical climate variation to project near-term shifts in habitat suitable for coast redwood. Glob Change Biol 21:4141–4152. https://doi.org/10.1111/gcb.13027
Foll M, Gaggiotti O (2008) A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180:977–993. https://doi.org/10.1534/genetics.108.092221
Frankham R (2005) Genetics and extinction. Biol Cons 126:131–140. https://doi.org/10.1016/j.biocon.2005.05.002
Frankham R, Gilligan DM, Morris D, Briscoe DA (2001) Inbreeding and extinction: effects of purging. Conserv Genet 2:279–284. https://doi.org/10.1023/A:1012299230482
Gaston KJ, Lawton JH (1990) Effects of scale and habitat on the relationship between regional distribution and local abundance. Oikos 58:329–335. https://doi.org/10.2307/3545224
AK Gujral TM Misiewicz C Hauser B Carter In press Natural history and demography of the imperiled redwood forest specialist Pedicularis dudleyi (Dudley’s Lousewort, Orobanchaceae). Madroño
Habel JC, Schmitt T (2012) The burden of genetic diversity. Biol Cons 147:270–274. https://doi.org/10.1016/j.biocon.2011.11.028
Habel JC, Zachos FE, Finger A, Meyer M, Louy D, Assmann T, Schmitt T (2009) Unprecedented long-term genetic monomorphism in an endangered relict butterfly species. Conserv Genet 10:1659. https://doi.org/10.1007/s10592-008-9744-5
Hagen M, Wikelski M, Kissling WD (2011) Space use of bumblebees (Bombus spp.) revealed by radio-tracking. PloS one 6:e19997. https://doi.org/10.1371/journal.pone.0019997
Hanski I (1982) Dynamics of regional distribution: the core and satellite species hypothesis. Oikos 38:210–221. https://doi.org/10.2307/3544021
Hanski I, Gyllenberg M (1993) Two general metapopulation models and the core-satellite species hypothesis. Am Nat 142:17–41. https://doi.org/10.1086/285527
Holsinger KE, Weir BS (2009) Genetics in geographically structured populations: defining, estimating and interpreting FST. Nat Rev Genet 10:639–650. https://doi.org/10.1038/nrg2611
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806. https://doi.org/10.1093/bioinformatics/btm233
Jennings H, Wallin K, Brennan J, Del Valle A, Guzman A, Hein D, Hunter S, Lewandowski A, Olson S, Parsons H, Scheidt S (2016) Inbreeding, low genetic diversity, and spatial genetic structure in the endemic Hawaiian lobeliads Clermontiafauriei and Cyaneapilosa ssp. longipedunculata. Conserv Genet 17:497–502. https://doi.org/10.1007/s10592-015-0785-2
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. https://doi.org/10.1093/molbev/mst010
Keil DJ (2019) Pedicularis rigginsiae (Orobanchaceae), a new species from an area of botanical endemism in central-coastal California. Phytoneuron 2:1–8
Kreyer D, Oed A, Walther-Hellwig K, Frankl R (2004) Are forests potential landscape barriers for foraging bumblebees? Landscape scale experiments with Bombus terrestris agg. and Bombus pascuorum (Hymenoptera, Apidae). Biol Cons 116:111–118. https://doi.org/10.1016/S0006-3207(03)00182-4
Liu D, Zhang L, Wang J, Ma Y (2020) Conservation genomics of a threatened Rhododendron: contrasting patterns of population structure revealed from neutral and selected SNPs. Front Genet 11:757. https://doi.org/10.3389/fgene.2020.00757
Maschinski J, Albrecht MA (2017) Center for plant conservation’s best practice guidelines for the reintroduction of rare plants. Plant Diversity 39:390–395. https://doi.org/10.1016/j.pld.2017.09.006
Meirmans PG, Van Tienderen PH (2004) GENOTYPE and GENODIVE: two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Notes 4:792–794. https://doi.org/10.1111/j.1471-8286.2004.00770.x
Meyer M, Kircher M (2010) Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb Protoc 6:prot5448
Milano ER, Mulligan MR, Rebman JP, Vandergast AG (2020) High-throughput sequencing reveals distinct regional genetic structure among remaining populations of an endangered salt marsh plant in California. Conserv Genet 21:547–559. https://doi.org/10.1007/s10592-020-01269-3
Nei M (1987) Molecular evolutionary genetics. Columbia University Press. https://doi.org/10.7312/nei-92038
Oostermeijer JGB, Van Eijck MW, Den Nijs JCM (1994) Offspring fitness in relation to population size and genetic variation in the rare perennial plant species Gentiana pneumonanthe (Gentianaceae). Oecologia 97:289–296. https://doi.org/10.1007/BF00317317
Oostermeijer JGB, Berholz A, Poschlod P (1996) Genetical aspects of fragmented plant populations. In: Settele J, Margules C, Poschlod P, Henle K (eds) Species survival in fragmented landscapes. Springer, Dordrecht, pp 93–101
Osborne JL, Martin AP, Carreck NL, Swain JL, Knight ME, Goulson D, Hale RJ, Sanderson RA (2008) Bumblebee flight distances in relation to the forage landscape. J Anim Ecol 77:406–415. https://doi.org/10.1111/j.1365-2656.2007.01333.x
Paris JR, Stevens JR, Catchen JM (2017) Lost in parameter space: a road map for stacks. Methods Ecol Evol 8:1360–1373. https://doi.org/10.1111/2041-210X.12775
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PloS one 7:e37135
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1111/j.1471-8286.2007.01758.x
Reed DH, Frankham R (2003) Correlation between fitness and genetic diversity. Conserv Biol 17:230–237. https://doi.org/10.1046/j.1523-1739.2003.01236.x
Rizzo DM, Garbelotto M (2003) Sudden oak death: endangering California and Oregon forest ecosystems. Front Ecol Environ 1:197–204. https://doi.org/10.1890/1540-9295(2003)001[0197:SODECA]2.0.CO;2
Robart BW, Gladys C, Frank T, Kilpatrick S (2015) Phylogeny and biogeography of North American and Asian Pedicularis (Orobanchaceae). Syst Bot 40:229–258
Rochette NC, Catchen JM (2017) Deriving genotypes from RAD-seq short-read data using Stacks. Nat Protoc 12:2640–2659. https://doi.org/10.1600/036364415X686549
Rochette NC, Rivera-Colón AG, Catchen JM (2019) Stacks 2: analytical methods for paired-end sequencing improve RADseq-based population genomics. Mol Ecol 28:4737–4754. https://doi.org/10.1111/mec.15253
Rosenberg NA (2004) DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138. https://doi.org/10.1046/j.1471-8286.2003.00566.x
Shaw J, Lickey EB, Beck JT, Farmer SB, Liu W, Miller J, Siripun KC, Winder CT, Schilling EE, Small RL (2005) The tortoise and the hare II: Relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am J Bot 92:142–166. https://doi.org/10.3732/ajb.92.1.142
Slatkin M (1987) Gene flow and the geographic structure of natural populations. Science 236:787–792. https://doi.org/10.1126/science.3576198
Spielman D, Brook BW, Frankham R (2004) Most species are not driven to extinction before genetic factors impact them. Proc Natl Acad Sci 101:15261–15264. https://doi.org/10.1073/pnas.0403809101
Sprague E (1962) Parasitism in Pedicularis. Madroño 16:192–200
Sprague E (1959) Ecological Life History of California species of Pedicularis. PhD Dissertation, Claremont Graduate School, Claremont, CA.
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. https://doi.org/10.1093/bioinformatics/btu033
Stebbins GL (1979) Rare species as examples of plant evolution. Great Basin Naturalist Memoirs 3:113–117
Taylor SS, Jamieson IG (2008) No evidence for loss of genetic variation following sequential translocations in extant populations of a genetically depauperate species. Mol Ecol 17:545–556
Tilman D, May RM, Lehman CL, Nowak MA (1994) Habitat destruction and the extinction debt. Nature 371:65–66. https://doi.org/10.1038/371065a0
Acknowledgements
We thank Abigail Moore and Leann Monaghan, Christine Edwards and two anonymous reviewers for their insightful comments on the manuscript. We also thank Lydia Smith, Austin Betancourt, Anjum Gujral, and Tracey Simmons for their invaluable assistance in the laboratory and field. The study benefited from the insights and logistics assistance of Portola Redwoods State Park staff Tyler Knapp, Ryan Diller, Tim Hyland and Joanne Kerbavaz. We are also grateful to Mujtaba Ghouse (Portola and Castle Rock Foundation) and Amy Patten (California Native Plant Society) for their valuable discussion and Kim Kuska, who shared many of his observations and insights with us. The computing for this project was performed at the OU Supercomputing Center for Education & Research (OSCER) at the University of Oklahoma (OU).
Funding
This study was funded through Research Grant #144 from Save the Redwoods League and the National Postdoctoral Research Fellowship in Biology Award #1810989.
Author information
Authors and Affiliations
Contributions
TMM, CH and BEC designed the study. TMM gathered and analyzed the data and wrote the manuscript. CH and BEC provided guidance on the study and commented on all drafts of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest.
Data availability
Raw sequence data were deposited at the National Center for Biotechnology Information Short Read Archive (http://www.ncbi.nlm.nih.gov/sra/) database under the accession number BioProject ID PRJNA742774. Processed SNP data (vcf, genepop, STRUCTURE and DIYABC-RF formats) and the concatenated ITS and matK-5’trnK intron sequence alignment are available from DataDryad (https://doi.org/10.6078/D1S415). Genbank accession numbers for the ITS and matK-5’trnK intron are listed in Table 1.
Code availability
Not Applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Misiewicz, T.M., Hauser, C. & Carter, B.E. Population genetics, demographic and evolutionary history of the Dudley’s lousewort (Pedicularis dudleyi), a rare redwood forest specialist. Conserv Genet 23, 513–525 (2022). https://doi.org/10.1007/s10592-022-01433-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-022-01433-x