
Abstract
The restricted range, taxonomic uncertainties and the apparent phylopatric behavior of Parotocinclus (dwarf plecos) make this fish group an excellent model to infer diversification patterns in Neotropical rivers. Thus, the aim of this study was to investigate the spatial–temporal relationships and the presence of cryptic forms in Parotocinclus cristatus from poorly known and human-impacted drainages in northeastern Brazil based on mitochondrial COI (589 bp) and Cyt-b (835 bp) and nuclear (Rhodopsin, 473 bp) markers. The phylogenetic reconstruction, haplotype network and species delimitation algorithms revealed three conspicuous and restricted molecular operational units (MOTUs) within P. cristatus. The samples from Cachoeira and Una basins represent a single MOTU dating back to the Early Pliocene, resulting in deep genetic divergence when compared to the other lineages. The MOTUs from Contas and Almada river basins diverged around 2.1 Ma, while the latter encompassed highly structured populations between upper and lower watersheds (0.6 Ma, Pleistocene). The closest relationship between more distant basins than nearby drainages indicates an intricate scenario of headwater captures that challenge traditional studies based on current geographic reliefs and ichthyofauna inventories. Therefore, caution when biogeographic inferences are carried out without further understanding about the genetic diversity and the geomorphological history, as commonly reported in fishes from eastern basins in Brazil. Finally, the conspicuous Linnean and Wallacean shortfalls in dwarf plecos, including unique lineages from unprotected and highly degraded areas, justifies a thorough review of their taxonomic status since valid species are the major source of information for defining conservation policies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Neotropical region, particularly in Brazil, harbors the highest diversity of freshwater fish worldwide (Reis et al. 2016) as a result of a long and complex biogeographic history driven by continental drift and geomorphological dynamics of hydrographic basins (Lundberg et al. 1998; Ribeiro 2006; Ribeiro et al. 2018). This scenario is conspicuously intricate for the coastal basins in eastern Brazil because of the several rearrangements of their drainages related to tectonic activities and sea level fluctuations (Bizerril 1994; Ribeiro 2006; Ribeiro et al. 2016; Thomaz and Knowles 2018). In this sense, both headwater capture events (Christofoletti 1975; Bishop 1995) and retraction of sea level could have determined the past connection in paleodrainages, followed by their separation into the isolated hydrographic basins observed at present (Dias et al. 2014; Thomaz and Knowles 2018). Most likely, these forces have played a major role in the high richness and endemism of ichthyofauna from coastal drainages in eastern Atlantic basins (Gery 1969; Bizerril 1994). Over the last decade, the number of endemic species (e.g. Zanata and Camelier 2009; Severi et al. 2010), cryptic forms (e.g. Bitencourt et al. 2011; Medrado et al. 2012, 2015; Oliveira et al. 2016; Souza et al. 2018) and undescribed or new taxa (e.g. Cetra et al. 2009, 2010; Trindade et al. 2010; Camelier and Zanata 2014; Barreto et al.2018) in rivers from northeastern Brazil have increased. In this sense, DNA barcode (Hebert et al. 2003) studies have been highly efficient in resolving taxonomic uncertainties and describing accurately the richness in Neotropical ichthyofauna (Pereira et al. 2013; Barreto et al. 2018; Anjos et al. 2020).
Nonetheless, little is known about the spatial–temporal diversification patterns in fish species from eastern coastal basins, properly recognized as areas of “insufficient knowledge” (Nogueira et al. 2010). To change this scenario, small-sized and/or phylopatric taxa can be used as potential models to infer biogeographic processes and speciation of freshwater fish in Neotropical rivers since they usually present restricted gene flow, being particularly susceptible to vicariance effects and allopatric isolation, thus accumulating high rates of endemism (Castro 1999; Montoya-Burgos 2003). This is particularly true for the dwarf plecos (Loricariidae: Hypoptopomatinae)—a group of benthonic and low-vagile species commonly found in habitats such as rapid waters and waterfalls from several eastern basins in South America (Roxo et al. 2017a).
The genus Parotocinclus is one of the most diversified taxon within Hypoptopomatinae, comprising 34 recognized species so far (Eschmeyer et al. 2019). However, their taxonomic and evolutionary relationships remain mostly unclear, including evidence of undescribed species and taxa that should be reallocated to other genera (Gauger and Buckup 2005; Roxo et al. 2014) or to distinct tribes (Roxo et al. 2019). Moreover, the “diagnostic” morphological features of several species in this genus are subtle and usually imprecise to differentiate them properly (Armbruster, 2004; Cramer et al. 2011). Taxonomic reports in isolated coastal basins from the state of Bahia, northeastern Brazil, identified five species of Parotocinclus: P. cristatus (Garavello 1977), P. jimi (Garavello 1977), P. minutus (Garavello 1988), P. bahiensis (Britski and Garavello 2009) and P. arandai (Sarmento-Soares et al. 2009).
Like most species from these overlooked river basins, P. cristatus lacks detailed biological information, being restricted to their taxonomic identification and distribution. Originally described only in Almada River basin, southern Bahia (Garavello 1977), the distribution of this species was further expanded to three adjacent drainages, all characterized by intensive human occupation and environmental degradation: Cachoeira and Una (southern range) and Contas (northern range) basins (Schaefer 2003; Cetra et al. 2009; Camelier and Zanata 2014). P. cristatus are restricted to four coastal hydrographic systems in Eastern Atlantic within the Northeastern Atlantic Forest (NAF) ecoregion, suggesting they share a common biogeographic history (Camelier and Zanata, 2014). However, recent studies based on genetic data in other taxa of Neotropical fish from these basins have detected putative new species and evolutionary relationships among lineages that contradict the expected pattern according to present hydrogeological configurations (Barreto et al. 2018; Souza et al. 2018).
The taxonomic uncertainties of Parotocinclus and their biological features (low vagility, small body size, abundance and site-fidelity) are appealing to test their potential in tracing phylogeographic history and to assess the actual diversity of regional species from isolated coastal basins in northeastern Brazil based on molecular markers. These data are particularly important because most rivers from this region combine high endemism, scarce information and increased human impacts (pollution, damming, deforestation of margins, and introduction of non-native species), being regarded as hotspots for the conservation of aquatic organisms (Menezes et al. 2007; Cetra et al. 2010; Nogueira et al. 2010; Gomes et al. 2012). Therefore, the first phylogeographic analysis was carried out on P. cristatus though their entire range, in order to test the hypothesis of cryptic diversity and to infer their genetic structure along the coastal drainages in NAF. Besides shedding some light on the intricate biogeographic processes of eastern Atlantic rivers, the present results also stress the importance of these areas to biodiversity conservation in times when environmental issues and support to science in Brazil have been neglected.
Methods
Sampling and data collection
We collected 108 specimens of Parotocinclus cristatus (4–19 specimens per locality) in the main river and tributaries from Upper, Middle and Lower Almada basin as well as Contas, Cachoeira and Una basins (Fig. 1a, b), encompassing the known range of this species. In addition, three specimens of Parotocinclus jimi were also sampled in a tributary from Middle Contas River basin (see Online Resource, ESM 1) for comparative analyses. The permission for collecting these taxa was granted by the Chico Mendes Institute of Biodiversity – ICMBio (license SISBIO n. 26752). The euthanasia (according to Blessing et al. 2010) and experimental procedures were approved by the Ethics Committee of Utilization of Animals from Universidade Estadual do Sudoeste da Bahia (CEUA/UESB, number 32/2013). Voucher specimens were identified and deposited in the fish collection at Universidade Estadual Paulista (UNESP) in Botucatu-SP, National Institute of Atlantic Forest in Santa Teresa-ES, and in the Zoology Museum at Universidade Federal da Bahia (UFBA) in Salvador-BA.

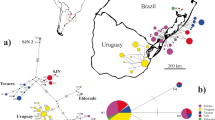
Map of sampled collection sites (a) of Parotocinclus cristatus (b) in northeastern Brazil. The detail in b highlights the tuft of denticles on occiput, a main diagnostic feature for this species. See online version for colored figure
The genomic DNA was isolated from muscle tissues (stored at − 20 °C in 100% ethanol) using the Wizard® Genomic DNA Purification (Promega) kit according to the manufacturer’s instructions. Two fragments of the mitochondrial DNA (mtDNA) genes Cytochrome c Oxidase subunit I (COI) and Cytochrome b (Cyt-b) were amplified using VF1_t1/VR1_t1 (Ivanova et al. 2007) and GluDG.L /H16460 (Perdices et al. 2002) primer sets, respectively. A nuclear fragment of the Rhodopsin gene was amplified using Rod-F2w/Rod-R4n primers (Sevilla et al. 2007). Each PCR (Polymerase Chain Reaction) comprised 1 × buffer, MgCl2 at 2 mM, 0.2 ng/μL of each primer, 0.2 mM of dNTPs, 50 ng of template DNA, 0.04 U/uL of Taq DNA polymerase (Invitrogen) and ultrapure water to a final volume of 15 μL. The annealing temperature tests for each of the primers after electrophoresis in 1.2% agarose gel revealed unique bands for all tested temperatures (50–60 °C). Thus, the PCR conditions were: a first denaturation step at 95 °C for 4 min; 35 cycles at 95 °C (40 s), 54 °C (40 s) and 72 °C (90 s), and a final extension step at 72 °C for 7 min.
The PCR products were purified in 20% polyethylene glycol (PEG) according to Paithankar and Prasad (1991) and the sequencing reactions were performed bidirectionally using BigDye Terminator v. 3.1 Cycle Sequencing (Applied Biosystems/Life Technologies, USA) according to the manufacturer’s instructions. After precipitation in EDTA (125 mM), 100% and 70% ethanol, the sequences were automatically detected in an ABI 3500 XL Genetic Analyzer sequencer (Applied Biosystems/Life Technologies).
DNA sequence analyses
The DNA fragments were checked using BLAST (Basic Local Alignment Search Tool) in NCBI (National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov) and compared to sequences available in GenBank (http://www.ncbi.nlm.nih.gov/genbank) for homologies. The consensus sequences were aligned using the Clustal W tool (Thompson et al. 1994) after edition in BioEdit Sequence Alignment Editor 7.2.6.1 (Hall 1999). In the case of Rhodopsin fragments, heterozygous sequences were manually edited according to IUPAC (International Union of Pure and Applied Chemistry) code.
All contigs were translated to check if they represent coding regions free of stop codons by using the software MEGA v. 10 (Kumar et al. 2018). The COI sequences as well as the fragments from other genes were uploaded in the BOLD (Barcode of Life Database) platform (project “Phylogeography of Parotocinclus cristatus—PCA” as part of the FISHBOL campaign). The final alignment and edition were performed along with a total of 36 DNA sequences from other Parotocinclus species, 19 sequences of closely related genera from Hypoptopomatinae and six representatives of Loricariidae representatives (used as outgroups) available in BOLD and GenBank (Online Resource, ESM 2, ESM 3).
Genetic structure
To infer the interrelationships among samples, a haplotype network was built for each locus based on median-network method (Bandelt et al 1999) in the software PopART (http://popart.otago.ac.nz). For the following analyses, we used only the mitochondrial markers since the phylogeographic signal of Rhodopsin sequences was weak (see Results).
The number of polymorphic sites (S), total number of mutations (Eta), haplotype (h) and nucleotide (π) diversity as well as the demographic history using Tajima’s D (Tajima et al. 1998), Fu’s Fs (Fu 1997) and R2 (Ramos-Onsins and Rozas 2002) tests based on 10,000 coalescent simulations were estimated using the software DNA Sequence Polymorphism (DnaSP) v. 6 (Rozas et al. 2017). The Extended Bayesian Skyline Plot (EBSP) (Heled and Drummond 2008) available in the software BEAST. 1.10.4 (Suchard et al. 2018) was used to infer putative changes in effective population size over time. Two independent EBSP runs were performed for each phylogroup with 10 million generation chains sampled every 1000 generations and 10% burn-in, following the strict-clock model, mutation rate of 1% under a normal distribution prior and GTR + G substitution model as estimated by jModelTest v. 2.1.10. The quality and the convergence of runs and the coalescence graph were verified using Tracer v. 1.7.1 (Rambaut et al. 2018).
The genetic structure was inferred by a spatial analysis of molecular variance (SAMOVA) in the software SAMOVA 2.0 (Dupanloup et al. 2002) with the number of groups (k) ranging from 2 to 7 with 1000 simulations for each k value. The most suitable k was established according to the highest structure among groups based on FCT and FST index values, as recommended by the abovementioned authors. In addition, a Bayesian Analysis of Population Structure (BAPS) was also carried out in the software BAPS v. 6.0 (Corander et al. 2008), assuming admixture model and 500 repetitions per individual.
The pairwise FST values and gene flow (Nm) were used to evaluate the genetic differentiation between pairs of populations and the partition of the genetic variation within and among populations. Both were determined by an analysis of molecular variance (AMOVA) (Excoffier et al. 1992) in the software Arlequin v. 3.5.2.2 (Excoffier and Lischer 2010) with 10,000 random permutations. The significance levels in pairwise FST values were adjusted using Bonferroni correction (Rice 1989). The Rcmd function available in Arlequin v. 3.5.2.2 was used to graph representation of pairwise FST values. The correlation between geographic (straight line between sites) and genetic distances was estimated by Mantel’s test (Mantel 1967) in the web server IBDWS 3.23 (http://ibdws.sdsu.edu/~ibdws/) (Jensen et al. 2005) for the samples from Almada River basin, since only one location was sampled along the Cachoeira, Una and Contas basins.
DNA barcoding and species delimitation methods
Following the standard procedure in DNA barcode analyses (Hebert et al. 2003), a pairwise distance matrix using the Kimura-2-parameter (K2P) model (Kimura 1980) based on COI fragments was obtained in MEGA v. 10 (Kumar et al. 2018) and used to generate a Neighbor-Joining (NJ) tree (Saitou and Nei 1987) with 1000 bootstrap replicates (Online Resource, ESM 4).
Considering the robustness of the COI marker for discriminating species (Ratnasingham and Hebert 2013) and that the use of combined species delimitation methods increases their reliability (e.g. Fujisawa and Barraclough 2013; Tang et al. 2014; Anjos et al. 2020), we selected five different methods based on distance and coalescence approaches to analyze the COI fragments. These sequences were stored in BOLD were validated as barcode tags, being automatically assigned to Barcode Index Numbers (BINs) through the algorithm RESL (Refined Single Linkage) (Ratnasingham and Hebert 2013). Additionally, ABGD (Automatic Barcode Gap Discovery) (Puillandre et al. 2012) was performed after inclusion of the pairwise genetic distance of COI sequences in a free online platform (http://wwwabi.snv.jussieu.fr/public/abgd/). In the case of the General Mixed Yule-Coalescent (GMYC) analysis (Fujisawa and Barraclough 2013), an ultrametric tree built in the software BEAST 1.10.4 (Suchard et al. 2018) was used as input through the web server http://species.h-its.org/gmyc/ after estimation of the best-fit substitution model by jModelTest 2.1.10 (Darriba et al. 2012). For the Bayesian Poisson Tree Process (bPTP) analysis (Zhang et al. 2013) (also available in https://species.h-its.org/) and Multi-rate Poisson Tree Process (mPTP) (Kapli et al. 2017) (https://mptp.h-its.org/#/tree) a Maximum Likelihood (ML) tree was built in the software RAxML 8.2.10 (Stamatakis 2014) using the CIPRES Science Gateway v. 3.3 (http://www.phylo.org/index.php) (Miller et al. 2010). The input file for bPTP included one individual per haplotype to avoid generating an unrealistic number of species (Blair and Bryson 2017) while the mPTP encompassed the complete sequence database, as recommended by Kapli et al. (2017).
Phylogenetic and divergence time inferences
In order to evaluate the evolutionary relationships among Parotocinclus representatives, phylogenetic trees based on ML and Bayesian inference (BI) were generated from the three amplified loci (COI, Cyt-b and Rhodopsin) using the platform CIPRES Science Gateway 3.3 (http://www.phylo.org/index.php) (Miller et al. 2010). The ML inference was carried out in RAxML 8.2.10 (Stamatakis, 2014) using the GTR + G model and 1000 bootstrap replicates, while BI was performed in Mr. Bayes v. 2.6.3 (Huelsenbeck and Ronquist 2001) using the best-fit substitution model for each locus (GTR + I + G for COI, GTR + G for Cyt-b and GTR + I + G for Rhodopsin). The BI phylogenies comprised two independent runs of 10 million generations with four Markov chains sampling a tree every 1000 generations and a burn-in of 10%. All models used were estimated by the jModelTest 2.1.10 (Darriba et al. 2012) using the Akaike Information Criterion (AIC) for ML and based on the Bayesian Information Criterion (BIC) for BI.
The divergence time was estimated from a calibrated tree in the software BEAST v. 1.10.4 (Suchard et al. 2018) based on the COI database since it encompassed the highest number of sequences and because the mutation rate for this locus (~ 1%) is well established for fishes (Strecker et al. 2004; Ornelas-García et al. 2008; Thomaz et al. 2015). We assumed a strict-clock model as supported by the ML clock test in the software MEGA v. 10 (Kumar et al. 2018). Therefore, we carried out two runs and four chains for 100 million generations sampling every 1000 steps, following a mutation rate of 1% per million years, GTR + I + G substitution model, and a normal distribution prior. The quality and the convergence or runs were evaluated in Tracer v. 1.7.1 (Rambaut et al. 2018) to check if the Effective Sample Size (ESS) values were above 200. The final tree was generated in TreeAnnotator v. 1.10.4 (Suchard et al. 2018) using 10% of burn-in and the branch support was based on posterior probabilities (PP). All trees were visualized and edited in the FigTree 1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/) and Inkscape v. 0.92.3 (https://inkscape.org/).
Results
Genetic structure
The haplotype network based on mitochondrial genes supported the high genetic differentiation between Contas, Almada and Cachoeira + Una groups as well as the structure between samples from Lower and Middle/Upper portions in Almada River basin. On the other hand, the haplotype network based on Rhodopsin sequences discriminated only two haplotypes corresponding to samples from Cachoeira + Una and Almada + Contas basins (Fig. 2a). For the Cyt-b, the group from Middle/Upper Almada River presented the highest levels of haplotype (h) and nucleotide (π) diversity, as well as the only significant signal of population expansion (Table 1). Moreover, the demographic history inferred from EBSP showed a subtle expansion in this group between 80 and 40 thousand years ago, followed by population stability ever since (Online Resource, ESM 5).

Summary analysis of population structure within Parotocinclus cristatus: (a) haplotype network based on COI, Cyt-b and Rhodopsin markers. The circles represent the haplotypes (their size is proportional to the frequency of each haplotype), while hatch marks indicate the number of mutations among haplotypes. Distinct colors are used to represent the collection sites. The boxes bounding the groups in the COI e Cyt-b haplotype network shower the clusters obtained after Bayesian Analysis of Population Structure (BAPS) based on mitochondrial DNA sequences; (b) spatial analysis of molecular variance (SAMOVA) in COI and Cyt-b fragments with estimation of fixation index values for each population cluster (k) after 1000 partition simulations. See online version for colored figure
According to SAMOVA, the grouping composed of four clusters recovered simultaneously the highest FST and FCT values (Fig. 2b). On the other hand, the highest posterior probability in, BAPS supported the formation of four and three clusters for the Cyt-b and COI markers, respectively (Fig. 2a). Indeed, the AMOVA considering three hierarchical levels revealed that about 89% to 95% of the genetic variation are explained by the differentiation among Contas, Almada and Cachoeira + Una lineages (Table 2).
The pairwise FST values and gene flow (Nm) were congruent with the structure pattern indicated by both mitochondrial markers (Fig. 3a, c). The non-significant values of FST (p ≥ 0.05) and high values of Nm (> 2) (according to Hebert 2004) presented by samples between the Middle and High Almada indicate active gene flow between these locations. On the other hand, the samples from Lower portions in Almada River basin were highly structured from the others in the same basin and with low gene flow (FST > 0.25; Nm < 1;p < 0.05) (according to Wright 1978). Similarly, high and significant FST (p < 0.05) values and a relatively low gene flow (Nm = 1.3) were observed between the samples from Cachoeira and Una River basins. A significantly positive correlation (p < 0.05) was observed between genetic and geographic distance among samples in the Almada River basin (Fig. 3b, d).

Heatmap highlighting the pairwise FST values and respective significant levels after Bonferroni correction (NS = non-significant, * = significant) (a, c) and graph of Mantel’s test (b, d) based on COI (a, b) and Cyt-b (c, d) fragments from samples of Parotocinclus cristatus. The Mantel’s test was restricted to the samples from Almada River basin. See online version for colored figure
DNA barcoding and species delimitation methods
The COI fragments (589 bp) revealed high genetic distances among samples (Online Resource, ESM 4). The mean genetic distance considering all collected specimens morphologically identified as P. cristatus was 4.4%. Nonetheless, three genetic groups were consistently recovered with intraspecific divergence below < 0.5%: (1) the samples from Almada River basin (0.4%), (2) from Cachoeira and Una River basins (0.2%) and the specimens from Contas River basin (0%). The genetic distance between the Cachoeira + Una and the other samples (Almada and Contas samples) was 9%. The genetic distance between samples from Almada and Contas River basins was 5%.
When the COI sequences of P. cristatus from the present study were compared to those available for congeneric species, the genetic divergence ranged from 10% (Parotocinclus sp1. X Cachoeira + Una group) to 25% (P. sp4 X Almada group). In relation to other genera in the subfamily, the distance values varied from 9% (Pseudotothyris obtusa x Contas group) to 21% (Otothyris travassosi x Cachoeira/Una group) (Online Resource, ESM 4).
In relation to the species delimitation methods, the RESL algorithm available in BOLD platform discriminated four MOTUs: BOLD:ADI4749 (Almada group), BOLD:ADI4931 (Contas group), BOLD:ADH9476 (Cachoeira + Una group) and BOLD:ADP2528 (Parotocinclus jimi, middle Contas River). The same pattern was obtained by ABGD (p < 0.05), GMYC, bPTP (JOB ID: 48393) and mPTP analyses (Fig. 4).

DNA barcode analysis in Parotocinclus cristatus. Three asterisks on branches represent high support values for all inferences (NJ > 98%, ML > 98%, BI > 0.9). Two asterisks indicate that at least two inferences were highly supported (> 98%, > 0.9). The columns on right summarize the results obtained by distinct species delimitation algorithms. See online version for colored figure
Phylogenetic and divergence time inferences
The topologies of the NJ, ML and BI trees were similar for the COI and Cyt-b (835 pb) markers (Fig. 4; (Online Resource, ESM 6). High bootstrap and posterior probability values supported the same clusters indicated by the species delimitation methods, excepting for the bootstrap values (< 98%) in ML inference based on COI data for the Almada group (Fig. 4). In addition, the BI based on Cyt-b sequences recovered two groups within the samples from Almada River basin, separating the individuals from Lower and Middle/Upper portions (Online Resource, ESM 6). The groups from Contas and Almada River basins were closely related, being placed as the sister group of Cachoeira + Una cluster. On the other hand, the interrelationships among the sampled specimens and other congeneric species and other representatives in Hypoptopomatinae has low support values for both mitochondrial markers. The only exception was the grouping among P. jimi, Parotocinclus sp2, P. cearensis and P. spilosoma observed in BI tree based on COI data (Fig. 4).
As expected for nuclear genes, the ML and BI phylogenetic reconstruction based on the Rhodopsin sequences (473 bp) revealed less accentuated phylogenetic structure (Online Resource, ESM 6). Nonetheless, two major clusters were recovered, represented by samples from Almada + Contas River basins and the populations from Cachoeira + Una basins.
Two groups emerged around 4 million years ago—mya (95% Highest Posterior Densities—HPD: 2.79 ± 4.76) (Early Pliocene) when the Cachoeira + Una group diverged from Contas + Almada cluster (Fig. 5). The subsequent cladogenic event was estimated around 2.15 mya (95% HPD: 1.47 ± 2.87) during Late Pliocene, leading to the separation between groups from Contas and Almada River basins. The divergence between the groups from Lower and Middle/Upper portions of Almada basin has taken place about 0.63 Ma (95% HPD: 0.41 ± 0.89) (Pleistocene).

Timetree for Parotocinclus cristatus based on COI dataset (the posterior probability values are shown below the nodes). The numbers in brackets and the blue bars indicate the 95% Highest Posterior Densities (HPD). See online version for colored figure
Discussion
Cryptic diversity in Parotocinclus cristatus
The taxonomic status and the phylogenetic relationships within Hypoptopomatinae are particularly confusing as evidenced by several reports of new descriptions, redescriptions and reallocations of genera and tribes in which putative diagnostic features have proved to be polymorphic or shared among other taxa (e.g. Roxo et al. 2015, 2017b, 2019; Ramos et al. 2016; Lehmann et al. 2018). Moreover, many Neotropical fish species can be more widely distributed than previously recognized (Garavello 1977; Schaefer 2003; Camelier and Zanata, 2014), thus jeopardizing the definition of their actual range. Thus, the biased estimates of richness (Linnean shortfall) and species ranges (Wallacean shortfall) are critical to conservation efforts and environmental policies since threatened species might remain ignored (Bini et al. 2006; Casciotta et al. 2013; Hortal et al. 2015). Currently, DNA-based approaches have been successfully used to resolve both shortfalls in Neotropical ichthyofauna and to infer their evolutionary history, with emphasis in DNA barcode using COI sequences (e.g. Roxo et al. 2017a; Barreto et al. 2018; Souza et al. 2018).
Accordingly, the genetic divergence among the populations of P. cristatus based on COI indicated a complex pattern of cryptic diversity. The mean intraspecific distance within P. cristatus (4.4%) was ~ 3.4 times lower than that observed among congeneric species (14.8%) (Online Resource, ESM 4). On the other hand, three population groups presented reduced intraspecific divergence (0 to 0.4%) but remarkable high genetic differentiation among each other (5 to 9%), as follows: (1) samples from Cachoeira and Una River basins, (2) the specimens from Almada River basin, and (3) the population of P. cristatus from Contas River (Online Resource, ESM 4). Indeed, taking the genetic distance among and within these groups into account, the barcode gap is above the tenfold threshold established for discriminating species in barcoding studies (Hebert et al. 2003). Similarly, the genetic divergence among these groups is much higher than the minimum value of 2%, widely used to indicate interspecific differences in fishes (e.g. Ward et al. 2009). Therefore, the genetic differentiation among groups from Cachoeira + Una, Almada, and Contas hydrographic basins is compatible with the existence of, at least, three MOTUS (see Floyd et al. 2002), characterizing P. cristatus as a species complex.
Corroborating this suggestion, the phylogenetic inferences based on mitochondrial genes and all species delimitation algorithms recovered the same number of independent taxonomic units within P. cristatus (Fig. 4) (Online Resource, ESM 6). Tang et al. (2014) recommend combining Poisson Tree Process (such as bPTP and mPTP) and GMYC methods in order to increase the reliability of species identification, as carried out in this study. In addition, we included the BIN analysis, also considered a highly informative approach to discriminate MOTUs inasmuch as this method is free of putative biases caused by incomplete or poorly resolved phylogenetic inferences (Ratnasingham and Hebert 2007), as observed in the controversial and species-rich group of Hypoptopomatinae catfishes (Roxo et al. 2019).
The only exception to the abovementioned pattern refers to the tree topology and the haplotype network based on Rhodopsin sequences which grouped the MOTUs from Contas and Almada River basins (Fig. 2a). This result might be related to the relatively low evolutionary rate of this nuclear gene, thus failing in discriminating between closely related species (Behrens-Chapuis et al. 2015). On the other hand, the conspicuous and highly supported genetic differentiation observed in a more conservative DNA marker for the Cachoeira + Una cluster in relation to the others reinforces the divergence of the former as a quite distinctive cluster (Fig. 2a) (Online Resource, ESM 6).
Furthermore, the three clades within P. cristatus presented deep divergence in relation to congeneric species, including P. jimi from Contas River basin, with genetic distances up to 25% (Parotocinclus sp. 4). These values are comparable or even higher than those observed among distinct genera (Online Resource, ESM 4). For instance, Epactionotus bilineatus and Hisonotus depressicauda presented a genetic divergence of 9%, similar to that observed among some Parotocinclus species, revealing the confusing systematic relationships of Hypoptopomatinae and the paraphyletic status of Parotocinclus (Gauger and Buckup, 2005; Cramer et al. 2011; Roxo et al. 2019). These issues are likely to be related to the underrepresentation of genetic data as well as to the large number of “obscure taxa” awaiting identification at species-level in this fish group (Page 2016), as also demonstrated in the present study.
Therefore, the actual richness in dwarf plecos remains largely unresolved, thus hindering their phylogenetic reconstruction (Roxo et al. 2017a). Consequently, undescribed taxa from poorly known regions combined with increased threats to local biodiversity, as observed in the presently studied areas, are potentially drawn to extinction before being properly recognized (Niemiller et al. 2013). This scenario is particularly alarming for the coastal basins from eastern Brazil, since Roxo et al. (2014) proposed that unique evolutionary lineages of dwarf plecos are common along these drainages, favored by the phylopatric behavior of this group of fishes. Likewise, the presence of conspicuous MOTUs within P. cristatus from nearby coastal rivers in northeastern Brazil reiterates the hypothesis proposed by these authors.
Phylogeographic and conservation inferences
The AMOVA based on mtDNA markers confirmed that most of genetic variation in P. cristatus are explained by the differentiation among the clusters from Almada, Contas and Cachoeira + Una hydrographic basins (Table 2), providing additional support to the barcode and phylogenetic inferences. On the other hand, under a phylogeographic approach, the mitochondrial makers revealed four groups (Fig. 2, 3) since the populations from Upper/Middle and Lower Almada were clearly discriminated. This pattern was particularly evident in the results based on Cyt-b sequences, showing the high sensitivity of this marker to discriminate species or unique lineages (Leonardo et al. 2016; Velasco et al. 2016). The high haplotype diversity detected in Cyt-b gene combined with the high number of sampled specimens in Upper/Middle Almada portions could also explain the only significant signal of demographic expansion observed in this group (Table 1; Online Resource, ESM 5). Both features could overestimate the population size (Thomaz et al. 2015).
In addition, the fixation index values (FST) and gene flow (Nm) together with the high and significant correlation between genetic and geographic distances also supported the differentiation of lineages within Almada River basin (Fig. 3). Apparently, the distances among collection sites might have influenced the divergence levels in this basin. The sampled areas between Upper and Middle Almada portions are relatively close geographically from each other (~ 11.5 km) thereby favoring the gene flow among specimens and a less accentuated population structure, while the site sampled in Lower Almada is at least 38.71 km apart from the other populations along this river system (Fig. 1). In general, these data fit the stepping-stone model of genetic differentiation in which migration rates increase among nearby subpopulations, while more isolated groups would accumulate high genetic differences (Slatkin 1987). Such divergence within a single basin is important to direct conservation policies which should focus on conserving local populations. Unfortunately, only a small region in Lower Almada (called “Lagoa Encantada”) has been officially protected but lacks effective management and monitoring (Gomes et al. 2012). In fact, protected areas in Brazil are not designed to assure the conservation of freshwaters (Azevedo-Santos et al. 2018).
Again, the Cachoeira + Una clade presented the largest divergence in relation to the other groups, even considering the analysis using nuclear DNA (Rhodopsin) fragments (Online Resource, ESM 6), regarded as less efficient markers in detecting structure than the mtDNA sequences (Zink and Barrowclough, 2008). Such remarkable differentiation might be explained by the long period of isolation between the Cachoeira + Una populations from the others, estimated in about 4 mya (Early Pliocene), resulting in higher accumulation of mutations after vicariance. Analogously, the closer relationship between Contas and Almada groups could be related to a more recent divergence (2.15 mya), while the population split between Lower and Upper/Middle Almada dated back to 800 thousand years ago, during Pleistocene (Fig. 5).
In fact, the warm climate in Early Pliocene increased the sea level (Weitzman et al. 1988; Camelier et al. 2018). Thus, formerly connected fluvial systems (Thomaz and Knowles 2018) could favor their dispersal and colonization, boosted by the environmental plasticity of Parotocinclus species (Cetra et al. 2009) prior this period. As the sea level increased, these would have become isolated ever since leading to the deep genetic differentiation between Cachoeira + Una and Almada + Contas groups.
On the other hand, the genetic similarity between populations from Cachoeira and Una River basins (Fig. 1) contrasts with the great geographic distance (43.1 km in straight line and even greater if we assume the river pathway) among collection sites and to the fact they belong to distinct hydrographic systems. Nonetheless, successive events of headwater capture (inferred by the presence of several river elbows and proximity between tributaries, highlighted in Online Resource, ESM 7) and the fact that both drainages share a common geological formation (“Barreiras”) across most of their extension (Nacif 2000) could account for the close relationship between local ichthyofauna. Additionally, the Una river basin encompasses a less rugged relief situated at lower altitudes than Cachoeira basin (DePaula et al. 2012). Such difference in landscape is particularly suitable to headwater capture (Christofoletti 1975; Bishop 1995) from the high-altitude basins (e.g. Cachoeira) to those at low altitudes (e.g. Una).
Likewise, the genetic similarities between samples from Almada and Contas basins when compared to the other groups should be associated with the neotectonic activities in the Brazilian Atlantic coast (Saadi et al. 2002). A recent report elegantly demonstrated that relatively recent events of river capture should play a major role in the biogeographic patterns of the ichthyofauna from coastal basins in northeastern Brazil because of the narrow extension of the continental shelf along this region (Thomaz and Knowles 2018). This hypothesis is also attested by the close evolutionary relationships observed for distinct fish groups from Almada and Lower Contas such as Characidae (Barreto et al. 2018) and Cichlidae (Souza et al. 2018). Moreover, the photointepretation of Almada River basin reveals a strong pattern of asymmetric straightness, i.e., most tributaries are found in the left margin along the northern portion and they present a high number of anomalies such as accentuated meandering and several sharp turns (elbows) (Online Resource, ESM 7). These features are compatible with intense geological activity (Christofoletti 1975; Bishop 1995) that could have increased river capture events from Contas into Almada River basin when compared to the nearby southern basins.
The present results revealed a more complex biogeographic pattern than that previously suggested by parsimony analysis of endemicity which placed the ichthyofauna from the Cachoeira and Almada River basins as closely related (Camelier and Zanata 2014), showing the importance of molecular techniques and geological data to infer the evolutionary history of Neotropical fishes. In this sense, caution is advised when the evolutionary relationships of regional ichthyofauna is based only on morphological features and geographic distance among basins, particularly in controversial taxa, such as Hypoptopomatinae and many other Neotropical fish groups. Finally, the high levels of endemism and hidden diversity in Parotocinclus from eastern coastal basins and insufficient knowledge justifies categorizing these drainages as priority areas for freshwater biodiversity conservation before species communities and phylogenetic diversity are eventually lost.
Availability of data and material
The genetic data are available in BOLD Systems (project “Phylogeography of Parotocinclus cristatus—PCA” as part of the FISHBOL campaign) and Supplementary Material.
References
Anjos MS, Bitencourt JA, Nunes LA et al (2020) Species delimitation based on integrative approach suggests reallocation of genus in Hypostomini catfish (Siluriformes, Loricariidae). Hydrobiologia 847:563–578. https://doi.org/10.1007/s10750-019-04121-z
Armbruster JW (2004) Phylogenetic relationships of the suckermouth armoured catfishes (Loricariidae) with emphasis on the Hypostominae and the Ancistrinae. Zool J Linn Soc 141:1–80. https://doi.org/10.1111/j.1096-3642.2004.00109.x
Azevedo-Santos VM, Frederico RG, Fagundes CK et al (2018) Protected areas: a focus on Brazilian freshwater biodiversity. Divers Distrib 25:442–448. https://doi.org/10.1111/ddi.12871
Bandelt HJ, Forster P, Rohl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48. https://doi.org/10.1093/oxfordjournals.molbev.a026036
Barreto SB, Silva AT, Batalha-Filho H, Affonso PRAM, Zanata AM (2018) Integrative approach reveals a new species of Nematocharax (Teleostei: Characidae). J Fish Biol. https://doi.org/10.1111/jfb.13834
Behrens-Chapuis S, Herder F, Esmaeili HR et al (2015) Adding nuclear rhodopsin data where mitochondrial COI indicates discrepancies—can this marker help to explain conflicts in cyprinids? DNA Barcodes 3:187–199. https://doi.org/10.1515/dna-2015-0020
Bini LM, Diniz-Filho JA, Rangel TFLVB et al (2006) Challenging Wallacean and Linnean shortfalls: knowledge gradients and conservation planning in a biodiversity hotspot. Divers Distrib 12:475–482. https://doi.org/10.1111/j.1366-9516.2006.00286.x
Bishop P (1995) Drainage rearrangement by river capture, beheading and diversion. Prog Phys Geogr 19:449–473. https://doi.org/10.1177/030913339501900402
Bitencourt JA, Affonso PRAM, Giuliano-Caetano L et al (2011) Identification of distinct evolutionary units in allopatric populations of Hypostomus cf. wuchereri Günther, 1864 (Siluriformes: Loricariidae): karyotypic evidence. Neotrop Ichthyol 9:317–324. https://doi.org/10.1590/s1679-62252011000200008
Bizerril CRSF (1994) Análise taxonômica e biogeográfica da ictiofauna de água doce do leste brasileiro. Acta Biol Leopold 16:51–80
Blair C, Bryson RW Jr (2017) Cryptic diversity and discordance in single-locus species delimitation methods within horned lizards (Phrynosomatidae: Phrynosoma). Mol Ecol Resour 17:1168–1182. https://doi.org/10.1111/1755-0998.12658
Blessing JJ, Marshall JC, Balcombe SR (2010) Humane killing of fishes for scientific research: a comparison of two methods. J Fish Biol 76:2571–2577. https://doi.org/10.1111/j.1095-8649.2010.02633.x
Britski HA, Garavello JC (2009) Redescription of Parotocinclus bahiensis (Miranda-Ribeiro, 1918) (Pisces, Ostariophysi, Loricariidae). Zootaxa 2143:59–67
Camelier P, Zanata AM (2014) Biogeography of freshwater fishes from the Northeastern Mata Atlântica freshwater ecoregion: distribution, endemism, and area relationships. Neotrop Ichthyol 12:683–698. https://doi.org/10.1590/1982-0224-20130228
Camelier P, Menezes NA, Costa-Silva GJ et al (2018) Molecular and morphological data of the freshwater fish Glandulocauda melanopleura (Characiformes: Characidae) provide evidences of river captures and local differentiation in the Brazilian Atlantic Forest. PLoS ONE 13:e0194247. https://doi.org/10.1371/journal.pone.0194247
Casciotta J, Almirón A, Sánchez S et al (2013) The genus Gymnotus (Gymnotiformes: Gymnotidae) in Argentina. How bad taxonomy results in poor regulations and no conservation. J Appl Ichthyol 29:208–212. https://doi.org/10.1111/jai.12028
Castro RMC (1999) Evolução da ictiofauna de riachos sul-americanos: padrões gerais e possíveis processos causais. In: Caramaschi EP, Mazzoni R, Peres-Neto PR (eds) Ecologia de Peixes de Riachos, 6rd edn. Rio de Janeiro, RJ, pp 1–22
Eschmeyer WN, Fricke R, Van Der Laan R (eds) (2019) Catalog of fishes: Loricariidae. Publishing Eschmeyer's Catalog of Fishes. http://researcharchive.calacademy.org/research/ichthyology/catalog/fishcatmain.asp. Accessed 21 July 2019
Cetra M, Ferreira FC, Carmassi AL (2009) Caracterização das assembleias de peixes de riachos de cabeceira no período chuvoso na bacia do rio Cachoeira (SE da Bahia, NE do Brasil). Biota Neotrop 29:107–115. https://doi.org/10.1590/S1676-06032009000200010
Cetra M, Sarmento-Soares LM, Martins-Pinheiro RF (2010) Peixes de riachos e novas Unidades de Conservação no sul da Bahia. Pan-Am J Aquat Sci 5:11–21
Christofoletti A (1975) Capturas fluviais. In: Enciclopédia Mirador Internacional, 5rd edn, São Paulo, SP, pp 2.049–2.05
Corander J, Marttinen P, Sirén J et al (2008) Enhanced Bayesian modelling in BAPS software for learning genetic structures of populations. BMC Bioinform 9:539. https://doi.org/10.1186/1471-2105-9-539
Cramer CA, Bonatto SL, Reis RE (2011) Molecular phylogeny of the Neoplecostominae and Hypoptopomatinae (Siluriformes: Loricariidae) using multiple genes. Mol Phylogenet Evol 59:43–52. https://doi.org/10.1016/j.ympev.2011.01.002
Darriba D, Taboada GL, Doallo R et al (2012) JModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772–772. https://doi.org/10.1038/nmeth.2109
DePaula FCF, Silva DML, Souza CM (2012) Tipologias Hidroquímicas das Bacias Hidrográficas do Leste da Bahia. Rev Virtual Quim 4:365–373
Dias MS, Oberdorff T, Hugueny B et al (2014) Global imprint of historical connectivity on freshwater fish biodiversity. Ecol Lett 17:1130–1140. https://doi.org/10.1111/ele.12319
Dupanloup I, Schneider S, Excoffier L (2002) A simulated annealing approach to define the genetic structure of populations. Mol Ecol 11:2571–2581. https://doi.org/10.1046/j.1365-294x.2002.01650.x
Excoffier L, Lischer HE (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Floyd R, Abebe E, Papert A et al (2002) Molecular barcodes for soil nematode identification. Mol Ecol 11:839–850. https://doi.org/10.1046/j.1365-294x.2002.01485.x
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
Fujisawa T, Barraclough TG (2013) Delimiting species using single-locus data and the Generalized Mixed Yule Coalescent approach: a revised method and evaluation on simulated data sets. Syst Biol 62:707–724. https://doi.org/10.1093/sysbio/syt033
Garavello JC (1977) Systematics and geographical distribution of the genus Parotocinclus Eigenmann & Eigenmann, 1889 (Ostariophysi, Loricariidae). Arq Zool 28:1–37
Garavello JC (1988) Three new species of Parotocinclus Eigenmann & Eigenmann, 1889 with comments on their geographical distribution (Pisces, Loricariidae). Naturalia 13:117–128
Gauger MF, Buckup PA (2005) Two new species of Hypoptopomatinae from the rio Paraíba do Sul basin, with comments on the monophyly of Parotocinclus and the Otothyrini (Siluriformes: Loricariidae). Neotrop Ichthyol 3:509–518. https://doi.org/10.1590/s1679-62252005000400008
Gery J (1969) The fresh-water fishes of South America. Monogr Biol 19:828–848
Gomes RL, Moraes ME, Moreau AM et al (2012) Aspectos físico-ambientais e de uso e ocupação do solo da bacia hidrográfica do rio Almada-BA. Bol Geogr 30:45–57. https://doi.org/10.4025/bolgeogr.v30i2.16423
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hebert PD (2004) Ten species in one: DNA Barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc Natl Acad Sci USA 101(41):14812
Hebert PDN, Cywinska A, Ball SL et al (2003) Biological identifications through DNA barcodes. Proc R Soc Lond [Biol] 270:313–321
Heled J, Drummond AJ (2008) Bayesian inference of population size history from multiple loci. BMC Evol Biol 8:289. https://doi.org/10.1186/1471-2148-8-289
Hortal J, De Bello F, Diniz-Filho JA et al (2015) Seven shortfalls that beset large-scale knowledge of biodiversity. Annu Rev Ecol Evol Syst 46:523–549. https://doi.org/10.1146/annurev-ecolsys-112414-054400
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Ivanova NV, Zemlak TS, Hanner RH et al (2007) Universal primer cocktails for fish DNA barcoding. Mol Ecol Notes 7:544–548. https://doi.org/10.1111/j.1471-8286.2007.01748.x
Jensen J, Bohonak A, Kelley S (2005) Isolation by distance, web service. BMC Genet 6:13. https://doi.org/10.1186/1471-2156-6-13
Kapli P, Lutteropp S, Zhang J et al (2017) Multi-rate poisson tree processes for single-locus species delimitation under maximum likelihood and Markov chain Monte Carlo. Bioinformatics 33:1630–1638. https://doi.org/10.1093/bioinformatics/btx025
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120. https://doi.org/10.1007/bf01731581
Kumar S, Stecher G, Li M et al (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Lehmann AP, Lima FCT, Reis RE (2018) Parotocinclus yaka, a new species of armored catfish (Loricariidae: Hypoptopomatinae), from the Amazon basin in Brazil. Zootaxa 4521:584. https://doi.org/10.11646/zootaxa.4521.4.7
Leonardo R, Nunes RS, Monteiro ML (2016) Molecular testing on sardines and rulings on the authenticity and nutritional value of marketed fishes: an experience report in the state of Rio de Janeiro, Brazil. Food Control 60:394–400. https://doi.org/10.1016/j.foodcont.2015.08.004
Lundberg JG, Marshall LG, Guerrero J et al (1998) The stage for neotropical fish diversification: a history of tropical South American rivers. In: Malabarba LR, Reis RE, Vari RP et al (eds) Phylogeny and classification of neotropical fishes. EDUPUCRS, Porto Alegre, pp 13–48
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220
Medrado AS, Ribeiro MS, Affonso PRAM et al (2012) Cytogenetic divergence in two sympatric fish species of the genus Astyanax Baird and Girard, 1854 (Characiformes, Characidae) from northeastern Brazil. Genet Mol Biol 35:797–801. https://doi.org/10.1590/s1415-47572012005000075
Medrado AS, Affonso PRAM, Carneiro PL et al (2015) Allopatric divergence in Astyanax aff. fasciatus Cuvier, 1819 (Characidae, Incertae sedis) inferred from DNA mapping and chromosomes. Zool Anz 257:119–129. https://doi.org/10.1016/j.jcz.2015.05.005
Menezes NA, Weitzman SH, Oyakawa OT et al (2007) Peixes de água doce da Mata Atlântica: Lista preliminar das espécies e comentários sobre conservação de peixes de água doce neotropicais. Museu de Zoologia da Universidade de São Paulo, São Paulo
Miller MA, Pfeiffer W, Schwartz T (2010) Creating the CIPRES Science Gateway for inference of large phylogenetic trees. 2010 Gateway Computing Environments Workshop (GCE). https://doi.org/10.1109/gce.2010.5676129
Montoya-Burgos JI (2003) Historical biogeography of the catfish genus Hypostomus (Siluriformes: Loricariidae), with implications on the diversification of Neotropical ichthyofauna. Mol Ecol 12:1855–1867. https://doi.org/10.1046/j.1365-294x.2003.01857.x
Nacif PGS (2000) Ambientes naturais da bacia hidrográfica do rio Cachoeira, com ênfase nos domínios pedológicos. PhD Thesis, Universidade Federal de Viçosa, Viçosa, Minas Gerais, Brazil
Niemiller ML, Graening GO, Fenolio DB et al (2013) Doomed before they are described? The need for conservation assessments of cryptic species complexes using an amblyopsid cavefish (Amblyopsidae: Typhlichthys) as a case study. Biodivers Conserv 22:1799. https://doi.org/10.1007/s10531-013-0514-4
Nogueira C, Buckup PA, Menezes NA et al (2010) Restricted-range fishes and the conservation of Brazilian freshwaters. PLoS ONE 5:e11390. https://doi.org/10.1371/journal.pone.0011390
Oliveira IA, Argolo LA, Bitencourt JD et al (2016) Cryptic chromosomal diversity in the complex “Geophagus” brasiliensis (Perciformes, Cichlidae). Zebrafish 13:33–44. https://doi.org/10.1089/zeb.2015.1169
Ornelas-García C, Domínguez-Domínguez O, Doadrio I (2008) Evolutionary history of the fish genus Astyanax Baird & Girard (1854) (Actinopterygii, Characidae) in Mesoamerica reveals multiple morphological homoplasies. BMC Evol Biol 8:340. https://doi.org/10.1186/1471-2148-8-340
Page RD (2016) DNA barcoding and taxonomy: dark taxa and dark texts. Philos Trans R Soc B 371:20150334. https://doi.org/10.1098/rstb.2015.0334
Paithankar KR, Prasad KS (1991) Precipitation of DNA by polyethylene glycol and ethanol. Nucleic Acids Res 19:1346. https://doi.org/10.1093/nar/19.6.1346
Perdices A, Bermingham E, Montilla A et al (2002) Evolutionary history of the genus Rhamdia (Teleostei: Pimelodidae) in Central America. Mol Phylogenet Evol 25:172–189. https://doi.org/10.1016/s1055-7903(02)00224-5
Pereira LHG, Hanner R, Foresti F, Oliveira C (2013) Can DNA barcoding accurately discriminate megadiverse Neotropical freshwater fish fauna? BMC Genet. https://doi.org/10.1186/1471-2156-14-20
Puillandre N, Lambert A, Brouillet S et al (2012) ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol Ecol 21:1864–1877. https://doi.org/10.1111/j.1365-294x.2011.05239.x
Rambaut A, Drummond AJ, Xie D et al (2018) Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst Biol 67:901–904. https://doi.org/10.1093/sysbio/syy032
Ramos TP, Lehmann AP, Barros-Neto LF et al (2016) Redescription of the endangered hypoptopomatine catfish Parotocinclus spilurus (Fowler, 1941) (Siluriformes: Loricariidae) from the upper rio Jaguaribe basin, northeastern Brazil. Neotrop Ichthyol 14:e150098. https://doi.org/10.1590/1982-0224-20150098
Ramos-Onsins SE, Rozas J (2002) Statistical properties of new neutrality tests against population growth. Mol Biol Evol 19:2092–2100. https://doi.org/10.1093/oxfordjournals.molbev.a004034
Ratnasingham S, Hebert PD (2007) BOLD: the Barcode of life data system (http://www.barcodinglife.org). Mol Ecol Notes 7:355–364. https://doi.org/10.1111/j.1471-8286.2007.01678.x
Ratnasingham S, Hebert PD (2013) A DNA-based registry for all animal species: the barcode index number (BIN) system. PLoS ONE 8:e66213. https://doi.org/10.1371/journal.pone.0066213
Reis RE, Albert JS, Di Dario F et al (2016) Fish biodiversity and conservation in South America. J Fish Biol 89:12–47. https://doi.org/10.1111/jfb.13016
Ribeiro AC (2006) Tectonic history and the biogeography of the freshwater fishes from the coastal drainages of eastern Brazil: an example of faunal evolution associated with a divergent continental margin. Neotrop Ichthyol 4:225–246. https://doi.org/10.1590/s1679-62252006000200009
Ribeiro AC, Lima FCT, Menezes NA (2016) Biogeografia dos Peixes de Água Doce da América do Sul. In: Carvalho CJB, Almeida EAB (eds) Biogeografia da América do Sul: análise de tempo, espaço e forma. Roca, Rio de Janeiro, pp 245–258
Ribeiro AC, Riccomini C, Leite JAD (2018) Origin of the largest South American transcontinental water divide. Sci Rep 8:17144. https://doi.org/10.1038/s41598-018-35554-6
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225. https://doi.org/10.1111/j.1558-5646.1989.tb04220.x
Roxo FF, Albert JS, Silva GSC et al (2014) Molecular phylogeny and biogeographic history of the armored neotropical Catfish Subfamilies Hypoptopomatinae, Neoplecostominae and Otothyrinae (Siluriformes: Loricariidae). PLoS ONE 9:e105564. https://doi.org/10.1371/journal.pone.0105564
Roxo FF, Silva GSC, Ochoa LE et al (2015) Description of a new genus and three new species of Otothyrinae (Siluriformes, Loricariidae). ZooKeys 534:103–134. https://doi.org/10.3897/zookeys.534.6169
Roxo FF, Lujan NK, Tagliacollo VA et al (2017b) Shift from slow- to fast-water habitats accelerates lineage and phenotype evolution in a clade of Neotropical suckermouth catfishes (Loricariidae: Hypoptopomatinae). PLoS ONE 12:e0178240. https://doi.org/10.1371/journal.pone.0178240
Roxo FF, Melo BF, Silva GSC et al (2017a) New species of Parotocinclus (Siluriformes: Loricariidae) from coastal drainages of Rio de Janeiro, southeastern Brazil. Zootaxa 4232:260. https://doi.org/10.11646/zootaxa.4232.2.9
Roxo FF, Ochoa LE, Sabaj MH et al (2019) Phylogenomic reappraisal of the Neotropical catfish family Loricariidae (Teleostei: Siluriformes) using ultraconserved elements. Mol Phylogenet Evol 135:148–165. https://doi.org/10.1016/j.ympev.2019.02.017
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC et al (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34:3299–3302. https://doi.org/10.1093/molbev/msx248
Saadi A, Machette, MN, Haller, KM et al (2002) Map and database of quaternary faults and lineaments in Brazil. Publishing from U.S. Geological Survey Open-File Report 02-230, Version 1.0. http://pubs.usgs.gov/of/2002/ofr-02-230/. Accessed 20 Mar 2019
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425. https://doi.org/10.1093/oxfordjournals.molbev.a040454
Sarmento-Soares LM, Lehmann AP, Martins-Pinheiro RF (2009) Parotocinclus arandai, a new species of hypoptopomatine catfish (Siluriformes: Loricariidae) from the upper rios Jucuruçu and Buranhém, States of Bahia and Minas Gerais, Brazil. Neotrop Ichthyol 7:191–198. https://doi.org/10.1590/s1679-62252009000200009
Schaefer SA (2003) Subfamily Hypoptopomatinae (Armored catfishes). In: Reis RE, Kullander SO, Ferraris CJ (eds) Check list of the freshwater fishes of South and Central America. EDIPUCRS, Porto Alegre, Rio Grande do Sul, pp 321–329
Severi W, El-deir ACA, Felix RTS et al (2010) Composição e abundância da ictiofauna na área de influencia dos reservatórios de Pedra e Funil, bacia do Rio de Contas, Bahia. In: Moura NA, Araújo EL, Bittencourt-Oliveira MC et al (eds) Reservatórios do Nordeste do Brasil: biodiversidade, ecologia e manejo. NUPEEA, Bauru, pp 541–572
Sevilla RG, Diez A, Norén M et al (2007) Primers and polymerase chain reaction conditions for DNA barcoding teleost fish based on the mitochondrial cytochrome b and nuclear rhodopsin genes. Mol Ecol Notes 7:730–734. https://doi.org/10.1111/j.1471-8286.2007.01863.x
Slatkin M (1987) Gene flow and the geographic structure of natural populations. Science 236:787–792. https://doi.org/10.1126/science.3576198
Souza CR, Affonso PRAM, Bitencourt JÁ et al (2018) Species validation and cryptic diversity in the Geophagus brasiliensis Quoy & Gaimard, 1824 complex (Teleostei, Cichlidae) from Brazilian coastal basins as revealed by DNA analyses. Hydrobiologia 809:309–321. https://doi.org/10.1007/s10750-017-3482-y
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. https://doi.org/10.1093/bioinformatics/btu033
Strecker U, Faúndez VH, Wilkens H (2004) Phylogeography of surface and cave Astyanax (Teleostei) from Central and North America based on cytochrome b sequence data. Mol Phylogenet Evol 33:469–481. https://doi.org/10.1016/j.ympev.2004.07.001
Suchard MA, Lemey P, Baele G et al (2018) Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol 4:vey016. https://doi.org/10.1093/ve/vey016
Tajima F, Misawa K, Innan H (1998) The amount and pattern of DNA polymorphism under the neutral mutation hypothesis. Genetics 102–103:103–107. https://doi.org/10.1007/978-94-011-5210-5_10
Tang C, Humphreys AM, Fontaneto D et al (2014) Effects of phylogenetic reconstruction method on the robustness of species delimitation using single-locus data. Methods Ecol Evol 5:1086–1094. https://doi.org/10.1111/2041-210X.122461
Thomaz AT, Knowles LL (2018) Flowing into the unknown: inferred paleodrainages for studying the ichthyofauna of Brazilian coastal rivers. Neotrop Ichthyol 16(16):e180019. https://doi.org/10.1590/1982-0224-20180019
Thomaz AT, Malabarba LR, Bonatto SL et al (2015) Testing the effect of palaeodrainages versus habitat stability on genetic divergence in riverine systems: study of a Neotropical fish of the Brazilian coastal Atlantic Forest. J. Biogeogr 42:2389–2401. https://doi.org/10.1111/jbi.12597
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680. https://doi.org/10.1093/nar/22.22.4673
Trindade ME, Cetra M, Jucá-Chagas R (2010) Ictiofauna do Ribeirão Limoeiro, Bacia do Rio Cachoeira, BA. Biota Neotrop 4:111–117. https://doi.org/10.1590/s1676-06032010000400015
Velasco A, Aldrey A, Pérez-Martín RI et al (2016) Assessment of the labelling accuracy of spanish semipreserved anchovies products by FINS (forensically informative nucleotide sequencing). Heliyon 2:e00124. https://doi.org/10.1016/j.heliyon.2016.e00124
Ward RD, Hanner R, Hebert PD (2009) The campaign to DNA barcode all fishes, FISH-BOL. J Fish Biol 74:329–356. https://doi.org/10.1111/j.1095-8649.2008.02080.x
Weitzman SH, Menezes NA, Weitzman, MJ (1988) Phylogenetic biogeography of the Glandulocaudini (Teleostei: Characiformes, Characidae) with comments on the distribution of other freshwater fishes in eastern and southeastern Brazil. In: Vanzolini PE, Heyer WR (Eds) Proceedings of a workshop on neotropical distribution patterns, Academia Brasileira de Ciências, Rio de Janeiro, Rio de Janeiro, pp 225–256
Zanata AM, Camelier P (2009) Astyanax vermilion and Astyanax vermilion: new characid fishes (Ostariophysi: Characiformes) from Northeastern Bahia, Brazil. Neotrop Ichthyol 7:175–184. https://doi.org/10.1590/S1679-62252009000200007
Zhang J, Kapli P, Pavlidis P et al (2013) A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29:2869–2876. https://doi.org/10.1093/bioinformatics/btt499
Zink RM, Barrowclough GF (2008) Mitochondrial DNA under siege in avian phylogeography. Mol Ecol 17:2107–2121. https://doi.org/10.1111/j.1365-294x.2008.03737.x
Acknowledgements
The authors would like to thank FAPESB (RED0009/2013 and BOL0055/2016) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brazil (CAPES) (Finance Code 001) for the financial support and SISBIO for the collection license (# 26752).
Funding
This work was supported by CAPES (financial code 001) and FAPESB (RED009/2013 and BOL0055/2016).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by JLPdS and JdAB. The first draft of the manuscript was written by JLPdS and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Ethical approval
All collection expeditions and experiments were approved by ICMBIO (SISBIO n. 26752) and CEUA/UESB (n. 32/2013).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
de Sousa, J.L.P., de Araújo Bitencourt, J., Sampaio, I. et al. “More than meets the eye”: phylogeographic inferences and remarkable cryptic diversity and in endemic catfish Parotocinclus (Loricariidae: Hypoptopomatinae) from neglected and impacted basins in South America. Conserv Genet 22, 411–425 (2021). https://doi.org/10.1007/s10592-021-01336-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-021-01336-3