
Abstract
Habitat fragmentation may involve a loss of genetic diversity and increments the vulnerability to species persistence. It could be a particular issue when coupled with other negative factors as the predicted climatic changes and the emergence of infectious diseases. In Southern Iberian Peninsula several endemic amphibian species have confined and fragmented distributions, including the Betic midwife toad Alytes dickhilleni. Herein, we present the first range-wide assessment of genetic diversity and structure in this species, using mitochondrial and microsatellite data. A mitochondrial fragment of the ND4 gene was amplified for 65 individuals and a set of 20 microsatellite loci, specifically developed for this species, was genotyped for 490 individuals from several sampling sites distributed across the species entire range. While both markers revealed high genetic diversity, only for microsatellites a marked genetic substructure was apparent. Our results evidence low levels of gene flow, suggesting the persistence of the species in fragmented habitats for several generations and a very limited connectivity between most of mountain ranges. The high diversity within A. dickhilleni populations could help to respond to the emergence of new diseases and to the predicted effects of climatic changes in Southeastern Iberian Peninsula. We hypothesize that the lack of gene flow is due to the absence of available breeding habitats and recommend that future management efforts of A. dickhilleni include the creation and maintenance of aquatic breeding habitats in a way that most of genetic diversity is preserved.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The genetic consequences of population fragmentation depend upon connectivity and gene flow among patches (Keller and Waller 2002) and are more pronounced in species with smaller ranges, lower dispersal potential and/or mainly confined to mountain isolates (Frankham et al. 2009). Habitat fragmentation leads to overall reductions in population size that can influence gene flow, genetic drift, and intraspecific genetic variation (Slatkin 1987). Consequently, reproduction and survival in the short term and, in the long term, the capacity of populations to respond to environmental change will be reduced and thereby increase extinction risk (Evans and Sheldon 2008; Frankham et al. 2009). Habitat loss or fragmentation coupled with global climate change and the emergence of infectious diseases are among the major factors that have been implicated in population declines, and they are suspected to act as drivers of species extinction (Parmesan and Yohe 2003; Opdam and Wascher 2004; Ewers and Didham 2006; Pounds et al. 2006).
Ongoing climate change is shifting species geographical ranges (IPCC 2014) and the predictions for the near future point to significant range shifts towards higher latitudes and/or altitudes (Chen et al. 2011; Kuntner et al. 2014). Range retractions caused by climate change are currently observed and many species are moving towards extinction or are already extinct (Thomas et al. 2006). Furthermore in fragmented populations, movements and shifts in distribution ranges could be constrained by habitat fragmentation or even delayed if spatial cohesion is very low (Opdam and Wascher 2004). The study by Gao and Giorgi (2008) indicates that the Iberian Peninsula central and southern parts might experience a substantial increase in aridity by the end of this century, making it particularly vulnerable to water stress and desertification.
Amphibians are among the most endangered group of vertebrates (Stuart et al. 2004; Beebee and Griffiths 2005; Araújo et al. 2006). The study by Hof et al. (2011) focused on amphibian species emphasizes that declines are likely to accelerate in the 21st Century, because multiple drivers of extinction, as climate changes, diseases and changes in land-use, could expose populations to higher extinction risks more than previous single causal assessments have suggested. Moreover, amphibians are particularly prone to suffer the negative effects of habitat fragmentation due to their biological (e.g., physiological constrains, small size, low vagility) and demographic characteristics, as shown by several studies on anurans (Funk et al. 2005a, b; Johansson et al. 2005) and urodeles (e.g., Spear et al. 2005; Purrenhage et al. 2009; Emaresi et al. 2011).
In particular, the Betic midwife toad Alytes dickhilleni (Arntzen and García-París 1995) meets most of the criteria outlined above: small range, mostly restricted to mountain habitats, poor dispersal abilities, sensitivity to novel diseases and small clutch size that can be related to low local effective population sizes (García-París 2000; García-París and Arntzen 2002; Martínez-Solano et al. 2003; Bosch et al. 2013). It is considered Vulnerable by the IUCN Red List of Threatened Species (http://www.iucnredlist.org/apps/redlist/details/979/0) due to a very fragmented distribution and a decrease in habitat area and quality, mainly for reproduction (García-París and Arntzen 2002). All known populations are threatened (García-París and Arntzen 2002) and it is also included in the EDGE (Evolutionarily Distinct and Globally Endangered) list of the Zoological Society of London (http://www.edgeofexistence.org/amphibians/top_100.php). The fragmented and confined distribution of A. dickhilleni makes this species particularly sensitive to changes predicted to occur in the Southern of Iberian Peninsula. For this reason, information concerning distribution of genetic structure is important for the development of conservation and management plans and could be of general interest for other amphibian species, particularly the ones facing the same conservation challenges.
In this study, we present the first range-wide assessment of patterns of genetic diversity and structure in A. dickhilleni, making use of markers from the mitochondrial and nuclear genomes, in order to emphasize connectivity and demography and provide valuable information for the conservation and management of its populations.
Materials and methods
Sampling and DNA extraction
Alytes dickhilleni samples were obtained across the species’ entire geographical range (Fig. 1A; Table 1). Tissue samples were collected from adult toe tips or larvae tail clips and preserved in ethanol until DNA extraction. Immediately after tissue collection individuals were released in the field. Genomic DNA was extracted using the EasySpin® Genomic DNA Tissue Kit (Qiagen, Hilden, Germany) following the fabricant protocol.

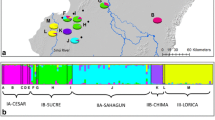
A Distribution of Alytes dickhilleni in Southwestern Iberian Peninsula in a 10 × 10 km squares map (MAGRAMA 2012), with indication of the geographical origin of the samples analyzed. Populations tested for HWE, LD, presence of null alleles and allele dropout, are marked with asterisk. The main mountain ranges are identified as follows: TEJ Sierra de Tejeda, ALM S. de Almijara, ALH S. de Alhama, ALB S. de Albuñuelas, NEV S. Nevada, GAD S. de Gadór, FIL S. de los Fillabres, BAZ S. de Baza, LUC S. de Lucar, HUE S. de Huetor, ARA S. de Arana, CAM S. del Campanario y las Cabras, AC S. de Alta Coloma, MAG S. Mágina, CAZ S. de Cazorla, CAS S. de Castil, SAG S. de la Sagra, VIL S. de las Villas, SEG S. Segura, ALC S. de Alcaraz, TAI S. de Taibilla, NM S. del Nordoeste de Murcia. B Results from individual multilocus genotype clustering analysis. Each individual is represented as a vertical line partitioned into the K (2–4) coloured segments, whose length is proportional to the K colors. Black lines separate individuals from different mountain ranges. Mountain ranges are identified as in Fig. 1. (Color figure online)
Mitochondrial DNA sequencing
A fragment of the NADH dehydrogenase subunit 4 gene (ND4) was amplified combining the forward primer ND4 described by Arévalo et al. (1994) and the reverse primer Aly11727_R (5′-CTA AGA CCA ACG GAT AGA CTG-3′). Polymerase chain reactions (PCR) were performed in a final volume of 10 μL using 5 μL of Phusion Flash High-Fidelity PCR Master Mix (Thermo Scientific) and 0.2 μM of each primer. PCR conditions consisted of a pre-denaturing step of 5 min at 98 °C and 42 cycles of denaturing (30 s at 98 °C), primer annealing (25 s at 55 °C) and extension (60 s at 72 °C), with a final extension of 5 min at 72 °C. PCR products were purified using ExoSAP-IT® PCR clean-up Kit (GE Healthcare, Piscataway, NJ, USA) and sequenced with primers ND4 and the internal primer Aly11168_F (5′-CAA CAC CAA CTA CGA AGC-3′). Cycle sequencing reactions were carried out using the ABI Prism® BigDye® Terminator version 3.1 Cycle Sequencing Kit (Applied Biosystems, Carlsbad, CA, USA) standard protocol. Samples were subsequently sequenced for both strands on an ABI Prism® 3130xl Genetic Analyser Sequencer (Applied Biosystems/HITASHI). The ND4 sequences of A. dickhilleni available in GenBank (EF441309, Gonçalves et al. 2007; and KJ858967, Maia-Carvalho et al. 2014a) were also included in this study. All sequences generated are deposited in GenBank under the accession numbers KP036330-KP036392 (Table 1).
Microsatellite genotyping
Twenty specific microsatellite markers were genotyped for this study: four dinucleotides (AlyA115, AlyB103, AlyB107, Aly108; Albert et al. 2010), three trinucleotides (AlyC1, AlyC132, AlyD2; Albert et al. 2010) and 13 tetranucleotides (Adickμ01 to Adickμ13; Molecular Ecology Resources Primer Development Consortium et al. 2011). The program multiplex manager version 1.0 (Holleley and Geerts 2009) was used as a starting point for optimizing multiplex PCR conditions. Three of the markers could not be amplified in multiplex reactions (Adickμ03, Adickμ06 and Adickμ11). Only three were amplified in triplex (Adickμ07, AlyB108 and AlyC132) and the other thirteen in diplex reactions (see Table S1, supplementary material). PCRs were optimized in a 5 μL of final volume and 2.5 μL of Qiagen® PCR Multiplex Kit Master Mix (Qiagen, Hilden, Germany) for simplexes and 10 μL of final volume and 2.5 μL of Qiagen® PCR Multiplex Kit Master Mix (Qiagen, Hilden, Germany) for diplexes and triplexes. PCR conditions for simplexes consisted of a pre-denaturing step of 15 min at 95 °C; 35 cycles of denaturing (30 s at 95 °C), primer annealing (see Table S1, supplementary material) and extension (45 s at 72 °C); 8 cycles of denaturing (30 s at 95 °C), primer annealing (45 s at 53 °C) and extension (45 s at 72 °C); and a final extension of 60 min at 72 °C. For multiplexes an additional touchdown step with a decrease of 0.5 °C each cycle was added after the initial pre-denaturing step consisting in a set of cycles of denaturing (30 s at 95 °C), primer annealing (see Table S1, supplementary material) and extension (45 s at 72 °C). Microsatellites were amplified with fluorescent dyes (PET, NED, 6-FAM and VIC). PCR products were tested on a 2 % agarose gel electrophoresis and were run on an ABI Prism® 3130xl Genetic Analyser Sequencer (Applied Biosystems/HITASHI). genemapper version 4.0 (Applied Biosystems/HITASHI) was used to perform alleles scoring for each locus.
Analysis of mtDNA data
ND4 gene sequences were checked by eye, edited and then aligned using the program ABI Prism® seqscape version 2.5.0 (Applied Biosystems). Alytes muletensis and A. maurus were used as outgroups (Maia-Carvalho et al. 2014a). The sequences of ND4 for A. muletensis (Accession nr. KJ858969; Gonçalves et al. 2009) and A. maurus (Accession nr. EF441312 and EF441312; Gonçalves et al. 2009) were downloaded from GenBank. A first approach to explore the mtDNA data was done by estimating relationships between haplotypes using the median-joining network algorithm in network version 4.6.1.0 (Bandelt et al. 1999). A second approach was made using a Bayesian coalescent method with a Markov Chain Monte Carlo (MCMC) sampling scheme implemented in beast version 1.7.2 (Drummond and Rambaut 2007) to estimate phylogenetic relationships between haplotypes. The best-fitted substitution model determined through the calculation of Akaike information criterion (AIC) with jmodeltest version 0.1.1 (Guindon and Gascuel 2003; Posada 2008) was the TrN model (Tamura and Nei 1993) without rate variation across sites. We used a Bayesian Skyline model (Drummond et al. 2005) with 10 groups for the coalescent prior and the strict molecular clock method with a normal prior with a mean of 0.0075, corresponding to a pairwise genetic distance of 1.5 % per million years (Crawford 2003; Burns and Crayn 2006) and a standard deviation of 0.0025. Preliminary analyses suggested that it fit the data better than relaxed clock models (data not shown). Five independent runs were made to avoid analysis to be trapped on local optima. All analyses started with randomly generated trees and ran four Metropolis coupling MCMC for 25 million generations, with sampling at intervals of 1,000 generations that produced 25,000 sampled trees. To estimate the time to the most recent common ancestor (TMRCA) for detected haplotypes we used as calibration point the well-known association between the split of three Alytes lineages (A. dickhilleni, A. muletensis and A. maurus) included in the Baleaphryne group and the end of the Messinian Salinity Crisis (Martínez-Solano et al. 2003). Same analysis was applied to the haplotypes from Southwestern supported group. After checking for stationary and convergence of the chains and the run length with the software tracer version 1.5 (Rambaut and Drummond 2009), the first 6,250 trees produced before reaching stationary were discarded as burn-in. The other 18,750 trees produced in each of the independent analyses were used to estimate the final Bayesian tree. Only posterior probability values above 0.95 were considered as indicating that clades were significantly supported.
Several genetic diversity parameters were estimated using the program DnaSP version 5.10 (Librado and Rozas 2009): number of haplotypes (h), number of polymorphic sites (S) and haplotype (Hd) and nucleotide diversity (π). We also tested the association between the estimates of interpopulation genetic and geographical distances (isolation by distance—IBD), using Mantel tests (Mantel 1967) implemented in the program alleles in space (Miller 2005). The statistical significance of the values was obtained by 1,000 randomization steps.
To assess population demography, three neutrality tests were performed using DnaSP: Tajima’s D (Tajima 1989), Fu’s Fs (Fu 1997) and Ramos-Onsins & Rozas’s R2 (Ramos-Onsins and Rozas 2002). In addition, distribution of pairwise differences (mismatch distribution) was also calculated with R2. The significances of Fs and R2 parameters were calculated using coalescent simulations based on theta value and no recombination, with a confidence interval of 95 % and 50,000 replications. To depict the change of the effective population size since the time to the most recent common ancestor (TMRCA) of the sampled mitochondrial haplotypes, the demographic history was reconstructed through a Bayesian skyline plot (BSP) using the software tracer version 1.5 (Rambaut and Drummond 2009). Effective population size was defined as θ (N e t, where N e is the effective population size and t is the generation time) and time as years.
Analysis of microsatellite data
Diversity and genetic structure
Standard indices of genetic diversity, i.e., total number of alleles (Na), allelic richness (R), observed (Ho) and expected heterozygosities (He) for each locus, were estimated with fstat version 2.9.3.2 (Goudet 2002). To determine possible genotyping errors, null alleles and allelic dropout, four populations with suitable number of samples (Fuente Boliches, 19; Poveda, 39; Colomera, 44; Cortijo de Lancas, 65—Table 1, Fig. 1 A) were examined with microchecker version 2.2.3 (van Oosterhout et al. 2004). Linkage disequilibrium between all pairs of loci and Hardy–Weinberg equilibrium were tested in the same populations using fstat version 2.9.3.2 (Goudet 2002). P values for 5 % nominal level in both analyses were adjusted for multiple comparisons (Rice 1989). The program freena (Chapuis and Estoup 2007) was used to estimate unbiased Fst values (Weir 1996) in the presence of null alleles, with 50,000 bootstrap resampling. Fst values with bias induced by the presence of null alleles were also calculated to compare with unbiased values, both for all dataset and for each pair of population clusters.
The distribution of the genetic variation across the entire dataset was investigated with a Factorial Correspondence Analysis (FCA), as implemented in genetix version 4.05 (Belkhir et al. 2004), and the population structure using the Bayesian multilocus clustering algorithm implemented in the software structure version 2.1 (Pritchard et al. 2000), assuming admixture, without using prior information on origin locality of individuals. We used an admixture model with correlated allele frequencies for K values from 2 to 6, and performed 5 runs for each population cluster (K) value with 1,000,000 MCMC interactions and a burn-in period of 100,000. To infer the optimal value of K the log probabilities of the data (LnP(K); Pritchard et al. 2000) estimated with structure were plotted against K values. The ΔK (Evanno et al. 2005) values were also estimated and plotted against K with structure harvester (Earl and von Holddt 2011). The geographical consistency of the clusters and the K max for which K max + 1 does not refine the clusters were then checked. The software clumpp version 1.1.2 (Jakobsson and Rosenberg 2007) was used to find the optimal alignments of the replicate cluster analyses for the best K value of structure.
To evaluate the relation between all individuals genotyped for microsatellite loci we constructed a phylogenetic tree of individuals. The software microsatellite analyser (MSA) version 4.05 (Dieringer and Schlštterer 2003) was used to calculate the genetic distance between the individuals. We used the Cavalli-Sforza and Edwards chord distance to calculate the distances (Cavalli-Sforza and Edwards 1967), since we have a considerable number of individuals. One thousand genetic distance matrices were replicated. Phylogenetic trees were constructed for each of the 1,000 replicate datasets using the program fitch with global rearrangements and the input sequence order randomized. The program consense was used to create a majority-rule consensus tree. Both programs are implemented in the computer package phylip version 3.69 (Felsenstein 2010). The final tree was drawn and edited using FigTree version 1.3.1 (http://tree.bio.ed.ac.uk/software/figtree/).
F-statistics (Weir and Cockerham 1984) and their significance were calculated as well as pairwise Fst values between clusters. To assess genetic differentiation we also analyzed our data set with smogd version 1.2.5 (Crawford 2010) to estimate Jost’s D (Jost 2008) and its confidence intervals (CI). Additionally, we used the program genepop version 4.0.1 (Raymond and Rousset 1995) to perform Mantel tests (Mantel 1967) to test the Isolation By Distance (IBD) hypothesis with microsatellite data for the entire dataset, for all pairwise comparisons between groups and within each different group identified in the structure runs.
Migration rates
To detect migration between structure detected groups we used the Bayesian approach implemented in bayesass version 3.0.1 (Wilson and Rannala 2003) to calculate migration rates and direction of migration that occurred recently (m), i.e., within the last few generations (Wilson and Rannala 2003). Three independent MCMC runs with different starting seed numbers were performed for 10 million generations and sampled at intervals of 1,000. The first one million generations were used as burn-in after checking for convergence with preliminary runs.
Changes in population size
Two methods implemented in bottleneck version 1.2.02 (Piry et al. 1999) were used to detect changes in effective population sizes. These approaches were applied to every population cluster detected with structure. The first method is based on the detection of heterozygosity excess. As suggested by Piry et al. (1999), TPM was used as the most suitable mutation model to the current microsatellite data set with a variation of 12 and a proportion of SSM in TPM of 95 %. The results obtained separately for each locus were combined using the Wilcoxon test based on 10,000 replications. This test is more sensitive for detecting bottlenecks occurring over approximately the last 2-4N e generations. The second method used is a qualitative descriptor of allele frequency distribution. This test is more appropriate for detecting more recent population declines, mainly over the last few dozen generations (Cornuet and Luikart 1996, Luikart et al. 1998). The main advantage of this method is that no reference population or data on historical levels of genetic variation is needed to determine if a population has been recently bottlenecked (Luikart et al. 1998).
Additionally, we calculated the mean ratio of the number of alleles to range in allele size (M) with the software m_p_val (Garza and Williamson 2001). During a bottleneck rare alleles are quickly removed from the population reducing the number of observed alleles faster than the size range of those alleles, resulting in a reduced M-ratio. M values were compared to critical M values (M c ) determined with the program critical_m (Garza and Williamson 2001), below which it can be assumed that an observed ratio is from a population that has experienced a significant reduction in size. To calculate Mc we need three TPM parameters: p s (the fraction of mutations that are larger than single steps), Δg (the mean size of non one-step changes) and \(\theta \;( = 4N_{e} \mu )\) before the bottleneck. As we did not have species-specific information we varied Δg and θ which are more influential than p s (Garza and Williamson 2001). First we varied the θ from 1 to 50, which we assumed to be more dependent from effective population size variation, keeping the default value of Δg (3.5). For populations where no bottlenecks were detected we pushed the bottleneck signature by decreasing Δg to 2.8—mean value determined from a literature survey by Garza and Williamson (2001).
Results
Mitochondrial DNA data
A fragment of the mitochondrial ND4 gene with 776 base pairs was successfully amplified for a total of 65 individuals from 38 sampling localities distributed across the species’ distribution range (Table 1; Fig. 1A). A total of 31 polymorphic sites (S), including 15 parsimony-informative, were detected across the 65 sequences obtained, yielding 23 haplotypes (h) and revealing a relative high haplotype and nucleotide diversity (Table 2). A star-like median-joining network (Fig. 2) reveals that the most common haplotype (Ha1) occurs in 15 individuals distributed across species range, except the most southwest and northeast extremes, and 14 haplotypes occur only once. Some geographical concordance in haplotype distribution can be detected: three haplotypes are restricted to the Southwest of the distribution range (Ha19-21), four are confined to the Northeastern distribution range (Ha10, 14, 22, 23) and four haplotypes (Ha5-7, 13) occur only in a central distribution (Fig. 2; Table 1).

Median-joining network for ND4 haplotypes detected in A. dickhilleni. Each gray circle represents a different haplotype with size proportional to its relative frequency. Black dots along branches represent nucleotide changes, the white dot represents an insertion and black triangles represent unsampled haplotypes
Results of the beast analyses were consistent across runs and Effective Sample Sizes (ESSs) were greater than 200 for all parameters in the model in all runs. Only two well-supported haplotypes groups with Bayesian Posterior Probabilities (BPPs) above 0.95 are recovered (Fig. 3): (i) one, groups the three haplotypes (Ha19-21) from the Southwestern region of distribution (from Sierras Tejeda, Almijara, Alhama, and Albuñuelas to the west extreme of Sierra Nevada); and (ii) the other, groups the two most Northeastern haplotypes (Ha22, 23) from the same sampling site (Puerto de las Crucetillas). These two well-supported groups are allocated in the opposite extremes of the distribution range. The estimated TMRCA for A. dickhilleni ND4 haplotypes is 364,600 years (95 % HPD interval 156,300–603,100) dating back to Pleistocene, while for the supported group from SW distribution is 299,700 (95 % HPD interval 85,000–545,100).

Bayesian inferred haplotype tree from partial Alytes dickhilleni ND4 gene sequences. Bayesian posterior probabilities higher than 0.95 are near nodes and at tips of the branches are haplotype codes (codes as in Fig. 2)
All neutrality tests estimated for the dataset are congruent (Fig. 4), detecting significant departures from neutral expectations and indicating the occurrence of recent demographic expansion. The distribution of expected and observed frequencies for pairwise number of differences calculated with R2 also presents an unimodal distribution under a scenario of demographic fluctuation (Fig. 4). Furthermore the BSP analysis is congruent with the neutrality tests showing an increment in effective population sizes until about 50-kyr ago where it reaches a stationary state (Fig. 5). No evidences for an IBD scenario were observed for mtDNA (data not shown).

Observed distributions (bars) of pairwise differences among individual sequences across distribution range and the expected Poison distribution under demographic expansion model (line) and demographic parameters estimated. R2, Ramos-Onsins & Rozas’s R2; Fs, Fu’s Fs; D, Tajima’s D; *significant values at P < 0.05; **significant values at P < 0.01

Log-linear Bayesian skyline plot representing the mitochondrial demographic history of A. dickhilleni. The horizontal axis is the number of years in the past and is estimated based on the rate of 0.0075 substitutions per site per year. The vertical axis is equal to the product of the effective population size by the generation time in years. The solid line represents the mean θ and the dashed lines delineate the 95 % HPD limits
Microsatellite data
Diversity and genetic structure
A total of 490 individuals from 76 sampling sites were genotyped at 20 polymorphic microsatellite loci (Table S2, supplementary material). A total of 741 alleles were observed with numbers of alleles per locus ranging from 5 (A115) to 83 (Adickµ11) with an average of 26.97 alleles per locus. Observed heterozygosity (Ho) among loci was variable ranging from 0.318 (D2) to 0.863 (Adickµ07) with an average of 0.648 over all samples (Table S2, supplementary material).
No evidences for scoring errors due to stuttering or large allele dropout were detected after microcheker analysis. Null alleles may be present at four loci (Adickµ02, Adickµ13, AlyC132 and AlyD2) in Colomera (44), at one locus (Adickµ04) in Fuente Boliches (19) and another (Adickµ11) in Poveda (39) and at two loci (Adickµ06 and Adickµ12) in Cortijo de Lancas (65) as suggested by the general excess of homozygotes for most allele size classes. In the absence of population structure, the presence of null alleles does not unbias the Fst estimates but in populations with significant differentiation (as in our study; see below), Fst is overestimated (Chapuis and Estoup 2007). The global uncorrected Fst value for the presence of null alleles in our dataset (Fst = 0.205) is similar to corrected Fst (0.203). Neither significant linkage disequilibrium nor departures from Hardy–Weinberg equilibrium were detected after correction for multiple comparisons in any analyzed population.
The results of the Bayesian clustering analysis performed with the program structure regarding the optimal number of clusters that best explains the distribution of genetic diversity based on the log probability of data (LnP(K); Pritchard et al. 2000) plotted against K are unclear. To address this problem the ΔK method described in Evanno et al. (2005) was applied to our data and according to this method the K value given by structure that best represents the maximum population structure is four. These four ancestral population clusters for microsatellite variation agree with the Kmax from which Kmax + 1 does not refine clusters and has a high geographical consistency. Following these criteria four population clusters will be considered for further analyses and discussion. The individual assignment probability to a certain cluster is generally very high and the clusters are clearly defined (Fig. 1B). Southwest group (SW, yellow in Fig. 1) clusters individuals from Sierras de Tejeda, Almijara, Alhama, Albuñuelas and West of Sierra Nevada. The Eastern group (E, light green in Fig. 1) comprises populations from Sierra de Baza, East of Sierra Nevada, Sierra de Gádor, Sierra de los Filabres and Sierra de Lucar. Finally, populations from Sierras de Huétor, Arana, del Campanario, Alta Coloma and Mágina (west group, W, dark green in Fig. 1) are split from populations of Sierras de Castril, la Sagra, Cazorla, Segura, las Villas, Alcaraz, Taibilla and West and Central Sierras of Murcia (Northeast group, NE, blue in Fig. 1). Results from FCA were concordant with Bayesian clustering analysis showing the same four clusters, corresponding to the a priori defined population groups (Fig. 6).

Factorial correspondence analysis (FCA) of genetic variation in the 20 microsatellite loci analyzed for Alytes dickhilleni. (Color figure online)
The results of UPGMA tree constructed with the 20 microsatellites are congruent with the Bayesian tree for mtDNA. The individuals from the SW cluster defined by structure are grouped as a monophyletic group, while the other three groups are paraphyletic (Fig. 7).

Neighbour-joining phylogenetic tree of all 490 A. dickhilleni genotyped individuals for the 20 microsatellite markers. Individual colors are according to Fig. 1. (Color figure online)
For each population group the genetic diversity parameters calculated are in Table S2 (supplementary material). All population groups exhibited considerable levels of expected heterozygosity and allelic richness, ranging from 0.513 in group E to 0.596 in group NE and from 17.362 in group SW to 24.289 in group NE, respectively.
Pairwise Fst estimates range from 0.063 to 0.162 and are all significant (Table 3). Most pairwise Fst values are high or moderately high, except between groups SW and NE (0.069) or W and NE (0.063). The comparisons between group SW and all others present the higher Fst values except with NE group. With Jost’s D, the differentiation estimates are considerable higher than with Fst. The comparisons between group SW and all others present the higher D values. None of these estimates have CI’s overlapping zero. Furthermore, we don’t have evidence for IBD neither within each identified cluster nor between clusters (data not shown).
Migration rates
The results from Bayesian individual assignment tests suggest restricted gene flow between population groups. All the runs to estimate contemporary migration rates (m; within the last few generations) consistently show reduced levels of gene flow (Fig. 8). All the tested migration rates were lower than 0.78 % with a 95 % confidence interval ranging between 0.003 and 2.286 % demonstrating a limited gene flow among population groups.

Bayesian estimates of migration rates among structure detected groups (m values). Polygon sizes reflect relative group sizes. Arrows represent the direction of gene flow among population groups
Changes in population size
With bottleneck we found no significant heterozygosity excess in any of the four groups, and thus the null hypothesis of mutation-drift equilibrium cannot be rejected (Table 4). Also allele frequency distributions for all groups are concordant with Wilcoxon tests, showing a normal L-shaped distribution, with allele frequencies in the lowest class (<0.1) ranging from 0.686 in group NE to 0.911 in group W, showing no reductions in effective population size in any population group (Table 4). In contrast, with M-ratio we detected signals of decrease in effective population size for some groups (Table 4). For SW group all M values are below the M c independently of the θ showing a bottleneck signature. For the other groups altering θ had impact in the strength of the bottleneck signal. In group E M < M c only with θ = 1 and for group W with θ = 5, 10 and 50. Finally for group NE, M is always higher then M c . For those who M > M c , reducing Δg increases the possibility of a significant M, which was verified for group E, W and for θ = 1 in group NE. Thus M-ratio detects at least some signal of bottlenecked populations.
Discussion
Alytes dickhilleni is a relatively recent differentiated species when compared with other congeneric species, whose the split of its sister taxon A. maurus might be associated with the opening of the Strait of Gibraltar (Maia-Carvalho et al. 2014a) at the end of the Messinian Salinity Crisis, 5.3 Myr (Krijgsman et al. 1999). This work is the most complete genetic study to date of this endemic species and allowed to assess the importance of several populations or population groups for its persistence and evolution.
We found relative high diversity for both mtDNA and microsatellites. These values are well within previously reported genetic variation in amphibians, namely within those obtained for congeneric species with wider distributions ranges and deeper divergence times such as A. cisternasii and A. obstetricans (see Table 2; Gonçalves et al. 2009; Maia-Carvalho et al. 2014b). The geographical distribution of the different population groups within A. dickhilleni, defined by both mtDNA and microsatellites, shows some interesting biogeographical features. The mitochondrial analysis only identified one monophyletic group of populations from SW region with statistical support in the Bayesian inferred tree. This result was also congruent with the results of UPGMA tree constructed with the 20 microsatellites. Moreover, the comparison between populations from SW region and all others presents the higher estimations of differentiation parameters for both markers. Our coalescent estimates of the TMRCA for all haplotypes in A. dickhilleni range approximately from 160,000 to 600,000 years ago, with a median value of 364,600 years ago suggesting a Pleistocene origin for the ancestor of the mtDNA groups. These are good evidences supporting the hypothesis that this species persisted in the southwestern limit of its distribution range during the last glacial period, whereas populations occurring northernmost are probably the result of recent expansions, as shown by unimodal distribution of mismatch analysis of mtDNA data and BSP.
The results of the Bayesian clustering identified four clusters with a great correspondence with geographic distribution of fragmented populations (or mountain groups). Genetic differentiation is very marked, mainly between isolated mountain ranges, meaning that fragmentation should play an important role as a cause of population structure detected with microsatellites. The group NE is the most extensive and seems to have a relatively continuous distribution. It extends from Sierras de Cazorla/Castril until Sierra de Alcaraz and also Northwest/Central Murcia province where the distribution is again more fragmented. These are mostly humid mountains where the gene flow between populations is more likely to occur than in drier areas, as in Sierras de los Filabres or de Gadór (Bosch and González-Miras 2012). The population structure detected within groups matches with restricted geographical locations as mountains or even several single populations, meaning that even at a finer scale there is a lack of gene flow and the differentiation is considerable, detonating the importance of low dispersal abilities and the complex orography as drivers of population structure.
For several amphibian species with marked mtDNA structure and high differentiation the persistence in separated glacial refugia is pointed to be the cause in several regions (Alexandrino et al. 2000; Monsen and Blouin 2003; Canestrelli et al. 2007). For A. dickhilleni we do not have a marked mtDNA structure, but we do have high levels of overall genetic diversity, a marked genetic structure disclosed with microsatellites and levels of differentiation between population groups pointing to very limited gene flow. These are good evidences supporting the idea that this species persists at least for several generations in restricted areas or even in the localities associated to reproduction, enough to gather a differentiation that is detected with microsatellites.
Our results show that migration levels between genetically distinct population groups are small. The estimates of gene flow are consistently low across tested groups (m ≤ 0.78 %). This is not surprising since the extant A. dickhilleni populations have a very fragmented distribution across mountainous areas (García-París and Arntzen 2002) making dispersal between populations a hard challenge. This limited dispersal is probably due to the Betic midwife toad dependence of permanent water points for reproduction, preventing migratory movements (Arntzen and García-París 1995; García-París 2000). Patterns of moderate to high genetic differentiation and diversity and reduced gene flow appear to be a general pattern among amphibians in fragmented mountain habitats (see for example Monsen and Blouin 2003; Milá et al. 2010). The results obtained for the Mallorcan midwife toad A. muletensis (Kraaijeveld-Smit et al. 2005) are particularly comparable with the results presented herein. Both species have a fragmented distribution confined to mountain habitats (García-París and Arntzen 2002; Román 2002) and are subjected to similar environmental pressures. A. muletensis population diversity (H e = 0.38–0.71) is slightly lower than A. dickhilleni and differentiation (Fst = 0.53–0.12) is a bit higher which can be explained by smaller and more fragmented populations (Kraaijeveld-Smit et al. 2005). Even for species that are not restricted to mountain habitats, dispersal is more limited and genetic diversity is higher in high altitude populations (Funk et al. 2005a; Martínez-Solano et al. 2005; Giordano et al. 2007; Martínez-Solano and González 2008). Likewise, other organisms with low dispersal abilities inhabiting fragmented habitats show these patterns of genetic variation. Chiucchi and Gibbs (2010) and Smith et al. (2009) found quite similar results based on microsatellite data for the eastern massasauga rattlesnake Sistrurus c. catenatus (Northeast of United States) and for the pygmy bluetongue skink Tiliqua adelaidensis (north of Adelaide, South Australia), respectively, two species inhabiting fragmented habitats for a long-term, characterized by low migration rates and high genetic variation.
Microsatellites seem to maintain rare alleles in frequencies expected for stable populations in mutation-drift equilibrium and no significant heterozygotic excess. In contrast M-ratio detected consistent bottleneck signatures in SW group for all pre-bottleneck θ values. For the other groups M-ratio also detects decrease in effective population sizes but only for some pre-bottleneck θ or Δg values. It could be interpreted as a weak, if any, bottleneck signal. However we should interpret these results with caution, as we do not have species specific on μ and Δg and taking into account the paraphyletic origin of each group (Fig. 7) the bottleneck signals can be masked. Our results could suggest a more historical basis for bottlenecks, mainly for E, W and NE as distributions of alleles frequencies and heterozygosity excess tend to be erased in approximately 0.2–4N e generations (Luikart and Cornuet 1998). A review about microsatellite-based bottleneck (Piry et al. 1999) tests indicates that (i) they do not detect bottlenecks in vertebrate populations known to have experienced declines, largely as a result of limited sample sizes and (ii) the proportion of multi-step mutations is, on average, underestimated, resulting in higher probability of detect bottlenecks in stable populations than expected based on the nominal significance level (Peery et al. 2012). This second limitation is not verified for our dataset. In other hand low M-ratios can persist for several hundreds or thousands of generations (Garza and Williamson 2001). Moreover the BPS shows us an increment of effective population size in the last 360-kyr that stabilized around 50-kyr. These results, combined with low gene flow and high genetic structure, support the hypothesis that Betic midwife toad populations persisted in fragmented habitats for a long-term. Several studies on amphibian populations have demonstrated similar results (Kraaijeveld-Smit et al. 2005; Martínez-Solano and González 2008). On the other hand several amphibian populations have experienced strong bottleneck events due to recent habitat fragmentation (Andersen et al. 2004), recent demographic expansion (Johansson et al. 2006) or show very low levels of genetic diversity strongly suggesting a recent decrease in effective population size (Noël et al. 2007).
Evolutionary and conservation implications
The loss of genetic diversity and gene flow restriction in fragmented amphibian populations involve a high degree of vulnerability (Beebee 2005; Funk et al. 2005b). However for the Betic midwife toad the high genetic diversity and the marked population structure detected with microsatellites, the lack of considerable gene flow among population groups, and the non-detection of bottleneck events suggest the persistence in fragmented habitats for several generations. These findings have important implications for the conservation of this threatened species, suggesting that the high genetic diversity detected may prevent the detrimental effects of inbreeding and genetic drift. Rather ecological factors may play a larger role in the determination of long-term population survival. This species requires permanent or almost permanent water places for a prolonged larval development (Martínez-Solano et al. 2003). These habitats are scarce and mainly restricted to mountains where adults seem to be constrained, which is the main cause for population fragmentation (Bosch and González-Miras 2012). The modification of natural breeding habitats for human usage caused an important dependence for artificial water places (e.g., fountains, reservoirs, water troughs for livestock, wells) and this dependence turns populations highly vulnerable to changes in human handling or even abandonment of such places, which is pointed to be the main threat to A. dickhilleni persistence (García-París and Arntzen 2002; Bosch and González-Miras 2012).
All groups detected within the Betic midwife toad distribution range have high genetic diversity and the extinction of any of it involves a loss of considerable genetic diversity. Populations with high diversity have potentially higher levels of standing genetic variation, which has an important role in rapid adaptation to new environmental conditions (Barrett and Schluter 2008). In this way, each of the four detected groups should be preserved in order to maintain genetic diversity, since these high diversity levels could be important to A. dickhilleni persistence, for example in the context of rapid environmental changes and/or emergence of new diseases. Rapid evolution will be necessary for the survival under drastic environmental changes (Palumbi 2001). Therefore high diversity within A. dickhilleni populations could help to respond to the predicted effect of climatic changes in Southeastern Iberian Peninsula (Gao and Giorgi 2008; Rodríguez-Puebla et al. 2010), as the decrease of the hydroperiod of adequate water points to A. dickhilleni reproduction. The recently detected chytridiomycosis, caused by the chytrid fungus Batrachochytrium dendrobatidis, in natural populations of this species (Bosch et al. 2013) is another risk factor. The South of Spain presents medium/high vulnerability for the expansion of this pathogenic fungus (Rödder et al. 2009) that could be particularly driven by the predicted climatic changes (Pounds et al. 2006). Within each population group several reproduction sites should be managed as conservation sites so that most of genetic diversity is covered and the creation of new breeding sites will help to promote or maintain connectivity. It is especially important across SW and NE groups, where populations still seem to keep at least some degree of gene flow, across Sierra de Huetor and Sierra Alta Coloma until Sierra Mágina, that are genetically identic, and within the other studied mountain ranges. These genetic features should be taking into account to establish conservation management actions at both local (e.g., creation of new reproduction sites) and regional scales (e.g., captivity breeding, translocation of individuals or habitat restoration) in order to preserve the high genetic diversity.
Finally, further research efforts should be focused on a landscape genetic approach to test the relative influence of ecological and environmental constraints on population connectivity and gene flow (see review in Manel and Holderegger 2013); and bioclimatic modulation to predict the potential effects of future climate change on population connectivity and genetic diversity to anticipate impacts and guide conservation strategies (Wasserman et al. 2013).
References
Albert EM, Arroyo JM, Godoy JA (2010) Isolation and characterization of microsatellite loci for the endangered Midwife Betic toad Alytes dickhilleni (Discoglossidae). Cons Genet Resour 3:251–253
Alexandrino J, Froufe E, Arntzen JW, Ferrand N (2000) Genetic subdivision, glacial refugia and postglacial recolonization in the golden-striped salamander, Chioglossa lusitanica (Amphibia: Urodela). Mol Ecol 9:771–781
Andersen LW, Fog K, Damgaard C (2004) Habitat fragmentation causes bottlenecks and inbreeding in the European tree frog (Hyla arborea). Proc R Soc Lond B 271:1293–1302
Araújo MB, Thuiller W, Pearson RG (2006) Climate warming and the decline of amphibians and reptiles in Europe. J Biogeogr 33:1712–1728
Arévalo E, Davis SK, Sites JW Jr (1994) Mitochondrial DNA sequence divergence and phylogenetic relationships among eight chromosome races of the Sceloporus grammicus complex (Phrynosomatidae) in central Mexico. Syst Biol 43:387–418
Arntzen JW, García-París M (1995) Morphological and allozyme studies of midwife toads (genus Alytes) including the description of two new taxa from Spain. Contrib Zool 65:5–34
Bandelt HJ, Forster P, Rohl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48
Barrett RDH, Schluter D (2008) Adaptation from standing genetic variation. Trends Ecol Evol 23:38–44
Beebee TJC (2005) Conservation genetics of amphibians. Heredity 95:423–427
Beebee TJC, Griffiths RA (2005) The amphibian decline crisis: a watershed for conservation biology? Biol Conserv 125:271–285
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F (2004) genetix 4.05, Population genetics software for Windows TM. Université de Montpellier II. Montpellier
Bosch J, González-Miras E (2012) Seguimiento de Alytes dickhilleni: informe final. Monografías SARE. Asociación Herpetológica Española, Ministerio de Agricultura, Alimentación y Medio Ambiente. Madrid
Bosch J, García-Alonso D, Fernández-Beaskoetxea S, Fisher MC, Garner TWJ (2013) Evidence for the introduction of lethal chytridiomycosis affecting wild betic midwife toads (Alytes dickhilleni). EcoHealth 10:82–89
Burns EL, Crayn DM (2006) Phylogenetics and evolution of bell frogs (Litoria aurea species-group, Anura: Hylidae) based on mitochondrial ND4 sequences. Mol Phylogenet Evol 39:573–579
Canestrelli D, Verardi A, Nascetti G (2007) Genetic differentiation and history of populations of the Italian treefrog Hyla intermedia: lack of concordance between mitochondrial and nuclear markers. Genetica 130:241–255
Cavalli-Sforza LL, Edwards AWF (1967) Phylogenetic analysis: models and estimation procedures. Am J Hum Genet 19:233–257
Chapuis M-P, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631
Chen C, Hill JK, Ohlemüller R, Roy DB, Thomas CD (2011) Rapid range shifts of species associated with high levels of climate warming. Science 333:1024–1026
Chiucchi JE, Gibbs HL (2010) Similarity of contemporary and historical gene flow among highly fragmented populations of an endangered rattlesnake. Mol Ecol 19:5345–5358
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Crawford AJ (2003) Relative rates of nucleotide substitution in frogs. J Mol Evol 57:636–641
Crawford NG (2010) smogd: software for the measurement of genetic diversity. Mol Ecol Resour 10:556–557
Dieringer D, Schlštterer C (2003) microsatellite analiser (MSA): a platform independent analysis tool for large microsatellite data sets. Mol Ecol Notes 3:167–169
Drummond AJ, Rambaut A (2007) beast: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214–221
Drummond AJ, Rambaut A, Shapiro B, Pybus OG (2005) Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol 22:1185–1192
Earl DA, von Holddt BM (2011) structure harvester: a website and program for visualizing Structure output and implement the Evanno method. Cons Genet Resour 4:359–361
Emaresi G, Pellet J, Dubey S, Hirzel AH, Fumagalli L (2011) Landscape genetics of the Alpine newt (Mesotriton alpestris) inferred from a strip-based approach. Conserv Genet 12:41–50
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620
Evans SR, Sheldon BC (2008) Interspecific patterns of genetic diversity in birds: correlations with extinction risk. Conserv Biol 22:1016–1025
Ewers RM, Didham RK (2006) Confounding factors in the detection of species responses to habitat fragmentation. Biol Rev 81:117–142
Felsenstein J (2010) phylip (Phylogeny Inference Package) version 3.69, University of Washington, Seattle
Frankham R, Ballou JD, Briscoe DA (2009) Introduction to conservation genetics, 2nd edn. Cambridge University Press, Cambridge
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
Funk WC, Blouin MS, Corn PS, Maxell BA, Pilliod DS, Amish S, Allendorf F (2005a) Population structure of Columbia spotted frogs (Rana luteiventris) is strongly affected by the landscape. Mol Ecol 14:483–496
Funk WC, Greene AE, Corn PS, Allendorf FW (2005b) High dispersal in a frog species suggests that it is vulnerable to habitat fragmentation. Biol Lett 1:13–16
Gao X, Giorgi F (2008) Increased aridity in the Mediterranean region under greenhouse gas forcing estimated from high resolution simulations with a regional climate model. Global Planet Change 62:195–209
García-París M (2000) Alytes (Alytes) dickhilleni. In: Ramos MA et al (eds) Fauna Ibérica, vol 24., Museo Nacional de Ciencias NaturalesCSIC, Madrid, pp 288–293
García-París M, Arntzen JW (2002) Alytes dickhilleni (Arntzen and García-París 1995). In: Pleguezuelos JM, Márquez R, Lizana M (eds) Atlas y Libro Rojo de los Anfibios y Reptiles de España, Dirección General de Conservación de la Naturaleza, Asociación Herpetológica Española, Madrid, pp 76–78
Garza JC, Williamson EG (2001) Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10:305–318
Giordano AR, Ridenhour BJ, Storfer A (2007) The influence of altitude and topography on genetic structure in the long-toed salamander (Ambystoma macrodactulym). Mol Ecol 16:1625–1637
Gonçalves H, Martínez-Solano I, Ferrand N, García-París M (2007) Conflicting phylogenetic signal of nuclear vs mitochondrial DNA markers in midwife toads (Anura, Discoglossidae, Alytes): deep coalescence or ancestral hybridization? Mol Phylogenet Evol 44:494–500
Gonçalves H, Martínez-Solano I, Pereira R, Carvalho B, García-París M, Ferrand N (2009) High levels of population subdivision in a morphologically conserved mediterranean toad (Alytes cisternasii) result from recent, multiple refugia: evidence from mtDNA, microsatellites and nuclear genealogies. Mol Ecol 18:5143–5160
Goudet J (2002) fstat, a program to estimate and test gene diversities and fixation indices (version 2.9.3.2). Available from http://www2.unil.ch/popgen/softwares/fstat.htm
Guindon S, Gascuel O (2003) A simple, fast and accurate method to estimate large phylogenies by maximum-likelihood. Syst Biol 52:696–704
Hof C, Araújo MB, Jetz W, Rahbek C (2011) Additive threats from pathogens, climate and land-use change for global amphibian diversity. Nature 480:516–519
Holleley CE, Geerts PG (2009) Multiplex manager 1.0: a crossplatform computer program that plans and optimizes multiplex PCR. Biotechniques 46:511–517
IPCC (2014) Summary for policymakers. In: Climate change 2014: impacts, adaptation, and vulnerability. Part A: global and sectoral aspects. Contribution of working group II to the fifth assessment report of the intergovernmental panel on climate change (eds Field CB et al) Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, pp. 1–32.
Jakobsson M, Rosenberg NA (2007) clumpp: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Johansson M, Primmer CR, Sahlsten J, Merilä J (2005) The influence of landscape structure on occurrence, abundance and genetic diversity of the common frog, Rana temporaria. Glob Change Biol 11:1664–1679
Johansson M, Primmer CR, Merilä J (2006) History vs. current demography: explaining the genetic population structure of the common frog (Rana temporaria). Mol Ecol 15:975–983
Jost L (2008) Gst and its relatives do not measure differentiation. Mol Ecol 17:4015–4026
Keller LF, Waller DM (2002) Inbreeding effects in wild populations. Trends Ecol Evol 17:230–241
Kraaijeveld-Smit FJL, Beebee TJC, Griffiths RA, Moore RD, Schley L (2005) Low gene flow but high genetic diversity in the threatened Mallorcan midwife toad Alytes muletensis. Mol Ecol 14:3303–3315
Krijgsman W, Hilgen FJ, Raffi I, Sierro FJ, Wilson DS (1999) Chronology, causes and progression of the Messinian salinity crisis. Nature 400:652–655
Kuntner M, Năpăruş M, Li D, Coddington JA (2014) Phylogeny predicts future habitat shifts due to climate change. PLoS ONE 9(6):e98907. doi:10.1371/journal.pone.0098907
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Luikart G, Cornuet JM (1998) Empirical evaluation of a test for identifying recently bottlenecked populations from allele frequency data. Conserv Biol 12:228–237
Luikart G, Allendorf FW, Cornuet JM, Sherwin WB (1998) Distortion of allele frequency distributions provides a test for recent population bottlenecks. J Hered 89:238–247
MAGRAMA (2012) Inventario Español de Especies Terrestres. Inventario Español del Patrimonio Natural y de la Biodiversidad. Ministerio de Agricultura, Alimentación y Medio Ambiente. http://www.magrama.es/es/biodiversidad/temas/inventarios-nacionales/inventario-especies-terrestres/inventario-nacional-de-biodiversidad/default.aspx
Maia-Carvalho B, Gonçalves H, Ferrand N, Martínez-Solano I (2014a) Multilocus assessment of phylogenetic relationships in Alytes (Anura, Alytidae). Mol Phylogenet Evol DOI: 10.1016/j.ympev.2014.05.033
Maia-Carvalho B, Gonçalves H, Martínez-Solano I, Gutiérrez-Rodríguez J, Lopes S, Ferrand N, Sequeira F (2014b) Intraspecific genetic variation in the common midwife toad (Alytes obstetricans): subspecies assignment using mitochondrial and microsatellite markers. J Zool Syst Evol Res 52:170–175
Manel S, Holderegger R (2013) Ten years of landscape genetics. Trends Ecol Evol 28:614–621
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220
Martínez-Solano I, González EG (2008) Patterns of gene flow and source-sink dynamics in high altitude populations of the common toad Bufo bufo (Anura: Bufonidae). Biol J Linn Soc 95:824–839
Martínez-Solano I, París M, Izquierdo E, García-París M (2003) Larval growth plasticity in wild populations of the betic midwife toad, Alytes dickhilleni (Anura: Discogloddidae). Herpetol J 13:89–94
Martínez-Solano I, Rey I, García-París M (2005) The impact of historical and recent factors on genetic variability in a mountain frog: the case of Rana iberica (Anura: Ranidae). Anim Conserv 8:431–441
Milá B, Carranza S, Guillaume O, Clobert J (2010) Marked genetic structuring and extreme dispersal limitation in Pyrenean brook newt Calotriton asper (Amphibia: Salamandridae) revealed by genome- wide AFLP but not mtDNA. Mol Ecol 19:108–120
Miller M (2005) Alleles In Space (AIS): computer software for the joint analysis of interindividual spatial and genetic information. J Hered 96:722–724
Molecular Ecology Resources Primer Development Consortium et al (2011) Permanent genetic resources added to molecular ecology resources database december 2010–2031 January 2011: isolation and characterization of 13 highly polymorphic microsatellite loci in the Betic midwife toad Alytes dickhilleni. Mol Ecol Resour 11:586–589
Monsen KJ, Blouin M (2003) Genetic structure in a montane ranid frog: restricted gene flow and nuclear–mitochondrial discordance. Mol Ecol 12:3275–3286
Noël S, Ouellet M, Galois P, Lapointe F-L (2007) Impact of urban fragmentation on the genetic structure of the eastern red-backed salamander. Conserv Genet 8:599–606
Opdam P, Wascher D (2004) Climate change meets habitat fragmentation: linking landscape and biogeographical scale levels in research and conservation. Biol Conserv 117:285–297
Palumbi SR (2001) Humans as the world’s greatest evolutionary force. Science 293:1786–1790
Parmesan C, Yohe G (2003) A globally coherent fingerprint of climate change impacts across natural systems. Nature 421:37–42
Peery MZ, Kirby R, Reid BN, Stoelting R, Doucet-Bëer E, Robinson S, Vásquez-Carrillo C, Pauli JN, Palsbøll PJ (2012) Reliability of genetic bottleneck tests for detecting recent population declines. Mol Ecol 21: 3403–3418
Piry S, Luikart G, Cornuet JM (1999) bottleneck: a computer program for detecting recent reduction in the effective population size using allele frequency data. J Hered 90:502–503
Posada D (2008) jmodeltest: phylogenetic model averaging. Mol Biol Evol 25:1253–1256
Pounds JA, Bustamente MR, Coloma LA, Consuegra JA, Fogden MPL et al (2006) Widespread amphibian extinctions from epidemic disease driven by global warming. Nature 439:161–167
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Purrenhage JL, Niewiarowski PH, Moore FB-G (2009) Population structure of spotted salamanders (Ambystoma maculatum) in a fragmented landscape. Mol Ecol 18:235–247
Rambaut A, Drummond AJ (2009) tracer version 1.5, Available from http://beast.bio.ed.ac.uk/Tracer, Accessed at May 2012
Ramos-Onsins SE, Rozas J (2002) Statistical properties of new neutrality tests against population growth. Mol Biol Evol 19:2092–2100
Raymond M, Rousset F (1995) genepop (Version 1.2): a population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Rödder D, Kielgast J, Bielby J, Schmidtlein S, Bosch J, Garner TJW, Veith M, Walker S, Fisher MC, Lötters S (2009) Global amphibian extinction risk assessment for the panzootic chytrid fungus. Diversity 1: 52–66
Rodríguez-Puebla C, Encinas AH, García-Casado LA, Nieto S (2010) Trends in warm days and cold nights over the Iberian Peninsula: relationships to large-scale variables. Clim Change 100:667–684
Román A (2002) Alytes muletensis (Sanchiz y Adrover, 1979). Atlas y Libro Rojo de los Anfibios y Reptiles de España (eds Pleguezuelos JM, Márquez R, Lizana M), Dirección General de Conservación de la Naturaleza, Asociación Herpetológica Española, Madrid, pp. 79–81
Slatkin M (1987) Gene flow and the geographic structure of natural populations. Science 236:787–792
Smith AL, Gardner MG, Fenner AL, Bull CM (2009) Restricted gene flow in the endangered pygmy bluetongue lizard (Tiliqua adelaidensis) in a fragmented agricultural landscape. Wildl Res 36:466–478
Spear SF, Peterson CR, Matocq MD, Storfer A (2005) Landscape genetics of the blotched tiger salamander (Ambystoma tigrinum melanostictum). Mol Ecol 14:2553–2564
Stuart SN, Chanson JS, Cox NA, Young BE, Rodrigues ASL, Fischman DL, Waller RW (2004) Status and trends of amphibian declines and extinctions worldwide. Science 306:1783–1785
Tajima F (1989) Statistical-method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10:512–526
Thomas CD, Franco AMA, Hill JK (2006) Range retractions and extinction in the face of climate warming. Trends Ecol Evol 21:415–416
van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Wasserman TN, Cushman SA, Littell JS, Shirk AJ, Landguth EL (2013) Population connectivity and genetic diversity of American marten (Martes americana) in the United States northern Rocky Mountains in a climate change context. Conserv Genet 14:529–541
Weir BS (1996) Genetic data analysis II. Sinauer Associates, Sunderland, Mass
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of populations structure. Evolution 38:1358–1370
Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163:1177–1191
Acknowledgments
For the help during sample collection we thank David Garcia and Alberto Escolano. For technical support during lab work we thank Susana Lopes and Bruno Carvalho. We thank to Fernando Sequeira and Iñigo Martínez-Solano for their valuable comments on the earlier version of this manuscript. This work was supported through Project “Genomics and Evolutionary Biology” cofinanced by North Portugal Regional Operational Programme 2007/2013 (ON.2—O Novo Norte), under the National Strategic Reference Framework (NSRF), through the European Regional Development Fund (ERDF); the Program Operacional Factores de Competitividade (COMPETE), and by national funds from Fundação para a Ciência e a Tecnologia (FCT), through the research Project PTDC/BIA-BEC/099915/2008 to HG, and CGL 2008-04814-C02/BOS from Spanish Ministerio de Ciencia y Innovación to JFB. Partial funding for field work was provided by Ministerio de Ciencia e Innovación, Spain, project TATANKA CGL2011-25062 (P.I. R Márquez). GD and HG are supported by a PhD grant (SFRH/BD89750/2012) and a postdoctoral Grant (SFRH/BPD/26555/2006), respectively, from FCT.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dias, G., Beltrán, J.F., Tejedo, M. et al. Limited gene flow and high genetic diversity in the threatened Betic midwife toad (Alytes dickhilleni): evolutionary and conservation implications. Conserv Genet 16, 459–476 (2015). https://doi.org/10.1007/s10592-014-0672-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-014-0672-2