
Abstract
Metastasis is the major cause of morbidity and mortality in cancer patients. An understanding of the genes that regulate metastasis and development of therapies to target these genes is needed urgently. Since members of the LIM kinase (LIMK) family are key regulators of the actin cytoskeleton and are involved in cell motility and invasion, LIMK is considered to be a good therapeutic target for metastatic disease. Here we investigated the consequences of LIMK inhibition on growth and metastasis of human and mouse mammary tumors. LIMK activity was reduced in tumor cells by expression of dominant-negative LIMK1, by RNA interference or with a selective LIMK inhibitor. The extent of phosphorylation of the LIMK substrate, cofilin, of proliferation and invasion in 2D and 3D culture and of tumor growth and metastasis in mice were assessed. Inhibition of LIMK activity efficiently reduced the pro-invasive properties of tumor cells in vitro. Tumors expressing dominant-negative LIMK1 grew more slowly and were less metastatic in mice. However, systemic administration of a LIMK inhibitor did not reduce either primary tumor growth or spontaneous metastasis. Surprisingly, metastasis to the liver was increased after administration of the inhibitor. These data raise a concern about the use of systemic LIMK inhibitors for the treatment of metastatic breast cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Metastasis is a major cause of morbidity and mortality and an understanding of the genes that regulate metastasis and development of therapies to target these genes are needed urgently. Regulation of the actin cytoskeleton is an important aspect of metastasis, since one of the prerequisites for tumor cells to leave the primary site and invade other tissues is increased motility driven by changes in actin polymerization and depolymerization [1, 2]. The regulation of actin treadmilling in assembly of dynamic protruding regions of the cell occurs through the action of accessory proteins, one of which is actin depolymerizing factor (ADF)/cofilin that, together with its regulators, influences cell motility and invasion [3, 4].
The family of the ADF that includes cofilin, is a major regulator of the actin cytoskeleton [5]. Binding of ADF/cofilin to actin filaments (F-actin) results in the severing and depolymerization by sequestering actin monomers [6]. Severing of filamentous actin (F-actin) creates free barbed ends and its depolymerization generates a pool of actin monomers that are used by the Arp2/3 complex to generate actin filaments that are necessary for cell motility [7]. Cofilin activity is regulated through its phosphorylation on Ser3 mainly by LIM kinase1&2 (LIMK1&2) [8–10]. When phosphorylated on Ser3, cofilin loses its ability to bind to and depolymerize F-actin, resulting in increased F-actin levels [11]. Positive regulators of cofilin activity include phosphatases such as members of the slingshot phosphatase family (SSH) [12] and chronophin [13].
The activity of LIMK is regulated mainly by Rho-GTPases (Rho, Rac and Cdc42) through their downstream kinases ROCK, PAK1 and PAK4 [14–16] that phosphorylate LIMK1&2 on a threonine residue (T508/T505, respectively) [17, 18]. The two LIMK proteins have identical genomic structure and share 50 % overall identity, with 70 % identity in the kinase domain [19, 20]. They both phosphorylate cofilin, however their cellular localization is distinct [21].
Conflicting data exist on the role of LIMK1 in regulating tumor cell motility and invasiveness. LIMK1 is expressed at high levels in a variety of tumors [22, 23] and cell lines [24]. Increased expression of LIMK1 enhances the invasiveness of MCF7 breast cancer cells and normal prostate cells in vitro and in mice [22, 24]. Down-regulation of LIMK1 activity within the tumor cells by dominant-negative LIMK1 reduces the invasiveness of MDA-MB-231 and MDA-MB-435 breast cancer lines [24, 25]. Inhibition of LIMK1&2 by RNA interference or pharmacological agents blocks the invasive capacity of MDA-MB-231 cells [26]. Furthermore, down-regulation of LIMK1, SSH1L or cofilin in the highly invasive MM1 hepatoma line decreases their motility and invasiveness [27].
In contrast, expression of the kinase domain of LIMK1 that totally inhibits cofilin activity in MTLn3 rat mammary carcinoma cells was found to suppress EGF-stimulated motility and invasion in vitro [28, 29]. Furthermore, enhanced LIMK1 expression decreases the number of intravasated MTLn3 cells, decreases lung metastasis and increases survival of rats bearing MTLn3 mammary tumors. MTLn3 tumors expressing kinase-dead LIMK1 give rise to increased lung metastases and reduced overall survival [29]. However, LIMK levels in tumor cells growing in the lung after intravenous injection remained high, indicating that elevated LIMK is permissive for growth in the lung but not for spontaneous metastasis in this particular cell line [29]. Since the inhibitory effects of elevated LIMK1 on motility and invasion are reversed by increased expression of cofilin, [29], it is apparent that the balance of the LIMK1/cofilin ratio determines the cellular response and that small changes in LIMK activity could either increase or decrease invasiveness, depending on the phosphorylation state of cofilin at that point in time [26].
To further clarify the role of LIMK1 in migration and invasion and to assess the consequences of LIMK inhibition on tumor growth and metastasis, we used a syngeneic mammary tumor model to measure the impact of down-regulation of LIMK1 activity specifically within the tumor cells. These experiments clearly show that inhibition of LIMK activity either by expression of dominant-negative LIMK1, by siRNA or by pharmacological inhibition with BMS3 [30] effectively reduces the pro-invasive properties of breast cancer cells, both mouse and human, in vitro. Although targeted reduction of LIMK1 activity in the tumor cells did inhibit tumor growth in mice, systemic administration of a LIMK inhibitor to tumor-bearing mice did not alter primary tumor growth and actually increased metastasis to liver. None of the assays performed could explain the lack of inhibition of metastasis, or the unexpected enhancement of liver metastasis.
Materials and methods
Reagents
Antibodies were used at the indicated dilutions: rat anti-LIMK1 [31] (1:3,000) and anti-LIMK2 mAbs [21] (1:1,000), anti-cofilin (Cytoskeleton, Cat#ACFL02 1:5,000), anti-p-cofilin (Cell Signaling Technology, Cat# 3311S, 1:3,000), anti-actin (Santa Cruz Biotechnology, Cat# SC-1616 HRP, 1:5,000). BMS3 [30] was dissolved in DMSO at 10 mM before dilution in DMEM to the final concentration.
Cell lines
The 4T1.2 line [32] was maintained in α-MEM/5 % FCS with antibiotics. The viral packaging line Phoenix–ECO (Dr. G. Nolan, Stanford University), was maintained in DMEM/10 % FCS with antibiotics. MDA-MB-231 cells (authenticated by CellBank Australia in 2011 by STR profiling) were maintained in DMEM/10 % FCS with antibiotics.
Mice
Female Balb/c and Balb/c SCID mice 6–8 weeks old were purchased from the Animal Resources Centre, Perth or Walter and Eliza Hall Institute (WEHI), Melbourne, Australia. Female NOD/SCID/IL2-Rgamma chain null (NSG) mice (5–6 weeks) were purchased from WEHI. All experiments were completed following institutional animal ethics approval.
Expression of DN-LIMK1 and LIMK1 in 4T1.2 cells
A flag-tagged dominant-negative kinase-dead construct for LIMK1 (DN-LIMK1) [8] was cloned into both the pBABE-puromycin and MSCV-IRES-Cherry retroviral vectors. The mCherry fluorescent protein construct was provided by Dr. R. Tsien, UCSD. Viral supernatants generated by transient transfection into the Phoenix–ECO packaging line were used to infect 4T1.2 cells followed by selection for puromycin resistance (pBABE) or flow cytometry for cherry fluorescence (MSCV-cherry). Similarly, full length Flag-LIMK1 was cloned into pBABE-puro. A pool of single cell clones expressing similar levels of Flag-tag was generated from the pBABE-puro cells whilst a population of cherry fluorescent positive cells was obtained from the MSCV-cherry vector cells.
Inhibition of LIMK1/LIMK2 by siRNA
MDA-MB-231 cells were transfected with siRNA targeting LIMK1 or both LIMK1&2 or with a non-targeting siRNA (250 pmol, Dharmacon), using Lipofectamine™ 2000 (Invitrogen), according to the manufacturer’s instructions. The siRNA sequences are:
LIMK1: TGGCAAGCGTGGACTTTCA; LIMK2: CTATCAAGGTGACACACAA. Down-regulation of LIMK1&2 was confirmed by immunoblotting.
Immunoblotting
4T1.2 or MDA-MB-231 cells were incubated for 18 h in BMS3 prior to preparation of lysates for gel electrophoresis and western blotting using primary antibodies as specified above and IR-Dye800 conjugated anti-rabbit IgG (Rockland, Gilbertsville, PA, Cat#611-132-122) and goat anti-mouse 680 nm (Molecular Probes Cat#A21059) with a Li-Cor Odyssey infrared imager or by ECL detection. Tumors were snap frozen upon removal from the mouse and crushed while frozen for preparation of lysates, to maintain protein phosphorylation.
Chemotactic migration and invasion assays
Gelatin-coated Transwell inserts seeded with 2 × 105 4T1.2 cells in DMEM/0.1 % BSA were transferred to wells containing α-MEM/2 % FCS. After 6 h at 37 °C, cells migrating through the 8 μm membrane pores were fixed with neutral buffered formalin, stained with DAPI and scored by microscopy. The 4T1.2 invasion assay involved the same protocol except that the cells were seeded in Matrigel and incubated at 37 °C for 24 h. Five fields at 400× magnification were scored for each transwell membrane.
Proliferation assays
Cells were plated into 96 well plates at 500 cells/well. At 24 h intervals, cells were fixed in 10 % trichloroacetic acid, rinsed and stained with 0.4 % sulforhodamineB (SRB) dissolved in 1 % acetic acid. Protein-bound dye was released with 10 mM Tris base, and the absorbance was measured at 550 nm. Five replicate wells were used per time point. To assess the effect of BMS3, cells (5 × 103) were plated in a 96 well plate and incubated for 4 days in the presence of 1 % DMSO (control) or 1, 5 or 10 μM BMS3.
For analysis of proliferation in three-dimensional (3D) cultures, cells (4 × 103) were plated into 96 well plates pre-coated with 50 μl of Cultrex Basement Membrane extract (BD#354230) and treated for 4 days with 1 % DMSO or 1 or 5 μM BMS3. Proliferation was measured by addition of 20 μl of the Cell Titer96® Aqueous One Solution kit (Promega; Madison, WI) and the absorbance at 490 nm measured 3 h later. Each sample was plated in triplicate and the experiment was repeated twice. Proliferation was calculated as average values for each experiment. The histogram represents the average proliferation where the controls were adjusted to 100.
Morphological analysis of 3D cultures
Cells were grown in growth factor reduced Cultrex Basement Membrane extract (BD#354230) as described previously [33]. In brief, an eight-chamber glass slide (Lab-TEK II, Naperville, IL) was coated with 50 μl Cultrex. Cells (5 × 103 well) resuspended in DMEM low glucose medium supplemented with 2 % FCS and 2 % Cultrex were plated onto the coated chamber slides and treated with BMS3 dissolved in DMSO. The final DMSO concentration was 0.1 %. The medium was replaced every 4 days. Cell morphology was monitored by phase contrast microscopy (Olympus) before staining with DAPI to visualize nuclei and rhodamine phalloidin to visualize F-actin for confocal microscopy analysis.
B- and T- cell proliferation assays
Six Balb/c mice were injected intraperitoneally (IP) with 1 mg BrdU. From the following day, they received 0.8 mg/ml BrdU in their drinking water. Three mice received a daily IP injection of 20 mg/kg BMS3 while the other three received the vehicle (10 % DMSO/0.5 % solutol in saline). The mice were culled at day 7, their spleens removed and single cell suspensions prepared. For B cell proliferation, cells were stained with anti-B220 and anti-BrdU and for T cell proliferation cells were stained with anti-CD4, anti-CD8 and anti-BrdU followed by FACS analysis as described previously [34].
For in vitro B and T cell proliferation assays, mice were injected IP with either 20 mg/kg BMS3/day or vehicle. After 7 days the mice were culled, spleens removed and single cell suspensions prepared. Cells (1 × 107/ml) were labelled with 2.5 mM CFSE for 10 min at 37 °C before being washed twice and resuspended in RPMI and 10 % FCS and seeded into 24 well plates. For B cell proliferation, cells were incubated with 2 μg/ml LPS in the presence or absence of 10 μM BMS3 and for T cell proliferation, cells were incubated with 2 μg/ml Con A in the presence or absence of 10 μM BMS3. After 48 h incubation at 37 °C, cell proliferation was analysed by FACS as described previously [34].
Tumor growth and metastatic burden in mice
Female Balb/c mice (15–20 per group) were injected into the fourth mammary gland with 105 cells. Primary tumor growth was monitored and mice were either culled all on the same day or as a group when the average primary tumor size reached 1,500 mm3. Lungs and spines were removed for measurement of relative tumor burden (RTB) by quantitative real time PCR (q-PCR) as described previously [35]. RTB was quantified by Taqman genomic real time PCR of the neomycin resistance gene or mCherry fluorescent protein DNA expressed only by the tumor cells, using the vimentin gene as a measure of total cellularity of the organ. The PCR reactions were multiplexed, each probe having a different fluorescent dye, to eliminate sampling and pipetting errors. Tumor burden was calculated from the difference in cycle number at which the products of the two reactions (neomycin resistance or mCherry and vimentin genes) reach a preset threshold.
From extra mice, some lung and liver samples were taken for histology. These were fixed in 10 % buffered formalin and embedded in paraffin. To detect tumor nodules, 5 μM sections were stained with hematoxylin and eosin (H&E).
Determination of the pharmacokinetics of BMS3
Mice (2 per time point) were given a single intraperitoneal injection of BMS3 at 20 mg/kg and culled 0.5, 1, 3, 7.5 or 24 h later. Plasma was recovered from each mouse by cardiac puncture and BMS3 was quantified using LC–MS. Aliquots of plasma samples (50 μl) were treated with acetonitrile (120 μl), mixed and centrifuged before the supernatant was removed and analysed by LC–MS using a Waters Micromass Quattro Premier mass spectrometer coupled to a Waters Acquity UPLC separations system. The mobile phase consisted of an acetonitrile–water gradient (containing 0.05 % formic acid) and a Phenomenex Polar RP 4 μm (50 × 1 mm) column was used. Plasma samples were quantified against calibration standards prepared in blank plasma from mice. The calibration was conducted in the range of 1–10,000 ng/ml (R 2 > 0.999), and the lower limit of quantitation (LLQ) for BMS3 was 1 ng/ml.
Treatment of tumor bearing mice with the LIMK inhibitor BMS3
Female Balb/c or NSG mice (15 per group) were injected into the fourth mammary gland with 1 × 105 4T1.2 cells. SCID mice (5 per group) were injected into the fourth mammary gland with 1 × 106 MDA-MB-231 cells. Mice were administered 20 mg/kg of the BMS3 inhibitor in 10 % DMSO/0.5 % solutol in saline or with vehicle alone, by daily IP injection. Balb/c mice were culled on day 34, NSG mice on day 30 and SCID mice on day 29. To assess extravasation of tumor cells into lung, Balb/c mice (7 per group) were treated for 4 days with either BMS3 or vehicle prior to intravenous inoculation of 1 × 106 4T1.2 tumor cells. After 10 min precisely, each mouse was culled, lungs were excised, rinsed in PBS and snap frozen. The tumor burden in lungs was assessed by q-PCR using mCherry DNA as the reporter gene.
Statistical analysis
Data that were normally distributed and with similar variances were analysed by unpaired t test or Anova with post hoc comparisons with Tukey’s multiple comparison test. Otherwise, data were analysed by a non-parametric test (Mann–Whitney U test) as indicated in the Figure legends.
Results
Inhibition of LIMK1 activity suppresses proliferation, migration and invasion
To establish its relevance to tumor growth and metastasis, LIMK1 activity was reduced in the highly metastatic 4T1.2 mammary tumor line [32, 35] by expression of a DN-LIMK1 and was increased by expression of exogenous LIMK1. Pools of six clones with similar expression of Flag-tagged LIMK1 proteins were used. Compared to a pool of clones containing the pBABE vector control, expression of Flag-LIMK1 resulted in increased phospho-cofilin levels, while expression of DN-LIMK1 reduced p-cofilin (Fig. 1a). Expression of DN-LIMK1 significantly reduced chemotactic migration (Fig. 1b) and decreased proliferation in 2D culture (Fig. 1c). A 30 % reduction in the ability of LIMK1 to phosphorylate cofilin led to a 50 % reduction in both proliferation and migration, under the conditions used here. Since inhibition of LIMK1 is likely to have indirect downstream consequences, it is not surprising that the effects on proliferation and migration are different than the direct effect on the phosphorylation of the LIMK1 substrate, cofilin.

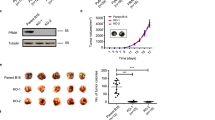
Effect of DN-LIMK1 expression on 4T1.2 cells and tumors. a Western blot analysis of cell lysates from 4T1.2-pBABE, 4T1.2-pBABE-LIMK1 and 4T1.2-pBABE-DN-LIMK1 cells probed for LIMK1, cofilin, p-cofilin and actin. The numbers below indicate the p-cofilin/cofilin ratio. b Chemotactic migration (after 6 h) and invasion (after 24 h) of 4T1.2-pBABE and 4T1.2-pBABE-LIMK1 pooled clones towards 2 % FCS. Results are the average ± SEM of two experiments of duplicate samples expressed as a percentage of the migration or invasion of 4T1.2-pBABE. c Growth of 4T1.2-pBABE and 4T1.2-pBABE-DN-LIMK1 pools in 2D and 3D cultures using the SRB assay. Data represent the cumulative growth over 4 or 6 days for 2D and 3D cultures, respectively. d Growth of 4T1.2-pBABE and 4T1.2-pBABE-DN-LIMK1 mammary tumors in Balb/c mice. Tumors were harvested when the average volume for the group reached 1,500 mm3 (15 mice per group). e Metastatic burden in lungs of mice bearing 4T1.2-pBABE or 4T1.2-DN-LIMK1 tumors as determined by q-PCR at day 26 for 4T1.2-pBABE and day 33 for 4T1.2-pBABE-DN-LIMK1. f Histology of an H&E stained lung section showing presence of metastatic nodules (M) surrounded by normal lung tissue (L). Bar = 100 μM. The statistical analysis was performed by unpaired t test
There is mounting evidence that growth of breast cancer cells in 3D matrices more closely resembles their growth in vivo [36]. Recently, a role for myosin light chain kinase (MLCK) in the proliferation and invasion of MDA-MB-231 cells in 3D cell culture was reported [33]. Since LIMKs, like MLCK, are important for the formation of actin stress fibers, we assessed the effects of reduced LIMK1 activity on invasion and proliferation of 4T1.2 cells in a 3D matrix. Cells expressing DN-LIMK1 were less invasive (Fig. 1b) and had reduced proliferation in 3D culture (Fig. 1c).
Inhibition of LIMK1 activity in tumor cells reduces primary tumor growth and metastasis
Primary 4T1.2-DN-LIMK1 tumors grew more slowly than the control tumors, resulting in a significant reduction in final tumor volume and weight at harvest (p < 0.001) (data not shown). In a separate experiment, mice were culled when the average primary tumor volume reached 1,500 mm3. Since the 4T1.2-DN-LIMK1 primary tumors grew more slowly, they were harvested 1 week later (Fig. 1d). Spontaneous metastasis to lung in mice bearing 4T1.2-DN-LIMK1 tumors was on average lower, but the difference was not statistically significant (Fig. 1e). To demonstrate the presence of metastatic disease, an H&E stained section of lung from a mouse bearing a vector control tumor is shown in Fig. 1f.
This experiment was repeated with new populations of 4T1.2 cells expressing mCherry and either DN-LIMK1 or the MSCV vector. mCherry-expressing cells were selected by flow cytometry, resulting in a population of cells rather than a pool of clones. 4T1.2-MSCV-DN-LIMK1 tumors again grew more slowly compared to control tumors (Fig. 2a). The weights of the primary tumors (Fig. 2b) were significantly reduced in the 4T1.2-DN-LIMK1 tumor-bearing mice. The primary tumor weights confirmed the reduced tumor volumes measured in these mice. A significant reduction in metastatic tumor burden in the lung was demonstrated by a weight decrease (Fig. 2c) and by q-PCR (Fig. 2d). Taken together, these experiments demonstrate that reduced LIMK1 activity in 4T1.2 tumor cells suppresses tumor growth and metastasis.

Effect of DN-LIMK1 in 4T1.2 cherry expressing cell populations on tumor growth and metastasis. A population of 4T1.2-cherry cells expressing DN-LIMK1 or vector was injected into the mammary glands of two groups of 15 Balb/c mice. All mice were culled on day 28. a Growth rates of the 4T1.2-DN-LIMK1 and 4T1.2-MSCV primary mammary tumors. b Final tumor weights. c Lung weights and d metastatic burden in lung as determined by q-PCR. The statistical analysis was performed by unpaired t test
The effects of pharmacological inhibition of LIMK activity on growth and invasion of 4T1.2 cells
Given the impact of DN-LIMK1 on tumor growth and metastasis, we next assessed the effect of LIMK inhibition using BMS3, a small molecule that selectively inhibits the kinase activity of LIMK1&2 with equal potency at the nanomolar level [30]. BMS3 has been tested for activity against 40 other kinases, showing activity only against LIMK and AMPKA1, with low activity against FGFR1 [32]. BMS3 exposure reduced p-cofilin levels in 4T1.2 cells (Fig. 3a) and at 5 μM, inhibited 2D (Fig. 3b) and 3D (Fig. 3c) proliferation of 4T1.2-pBABE and 4T1.2-pBABE-LIMK1 cells. Consistent with the reduced proliferation of 4T1.2-DN-LIMK1 cells (Fig. 1b, c), expression of Flag-LIMK1 increased proliferation (Fig. 3b, c). The inhibition of growth in 3D culture is demonstrated in Supplementary Fig. 1.

Effect of BMS3 on proliferation of 4T1.2 cells in 2D and 3D culture. a Inhibition of LIMK1 activity in 4T1.2 cells by incubation for 22 h with 0–10 μM BMS3 followed by immunoblotting with anti-p-cofilin and anti-tubulin antibodies. The numbers below represent the p-cofilin:tubulin ratio, where the level in untreated cells is adjusted to one. b and c Comparison of proliferation in 2D and 3D culture of 4T1.2-pBABE and 4T1.2-pBABE-LIMK1 pooled clones in the absence or presence of 1 or 5 μM BMS3. b Cumulative proliferation of cells in 2D culture over 4 days and c in 3D culture over 6 days. Each sample was plated in triplicate and the experiment was repeated three times. The bars represent mean ± SEM. * indicates p value of <0.005 as determined by Anova with post hoc comparisons with Tukey’s multiple comparison test
The effect of LIMK inhibition by BMS3 or by siRNA on p-cofilin levels and proliferation of human breast cancer cells
To verify that LIMK is also important in human breast cancer, the consequences of inhibition of LIMK activity by BMS3 or LIMK down-regulation by siRNA were assessed using the human MDA-MB-231 line. Cells were treated with varying concentrations of BMS3 (Fig. 4a) or with specific LIMK1 and LIMK2 siRNAs. Introduction of LIMK1 or LIMK2 siRNA resulted in 30 or 40 % reduction of p-cofilin, respectively, while a combination of both siRNAs reduced p-cofilin levels by 66 % after 4 days in 2D culture (Fig. 4b). The combined LIMK1/2 siRNAs or 10 μM BMS3 had a similar inhibitory impact on proliferation in both 2D and 3D cultures (Fig. 4c, d). Inhibition of LIMK by BMS3 caused changes in morphology and prevented local invasion of MDA-MB-231 grown in 3D culture (Supplementary Fig. 2a). After 10 days in culture in the absence of BMS3, the colonies had lost their round shape and began to invade the matrix (arrow). In the presence of 1 μM BMS3, the colonies remained a regular shape and their size was reduced significantly by day 10. Colony size was further decreased with increasing BMS3 concentration, reflecting the reduced proliferation shown in Fig. 4d. Similar results were obtained for 4T1.2 cells (Supplementary Fig. 2b).

Effect of LIMK inhibition on MDA-MB-231 cells. a Immunoblot analysis of cofilin and p-cofilin in MDA-MB-231 cells incubated for 16 h with 0–5 μM BMS3. Tubulin was used as a loading control. b Immunoblot analysis of LIMK1, LIMK2 and p-cofilin levels following incubation of MDA-MB-231 cells with LIMK1 and/or LIMK2 siRNA for 6 days. The numbers below represent the p-cofilin:cofilin ratio. c Cumulative proliferation over 4 days in 2D or d over 6 days in 3D culture of MDA-MB-231 cells. BMS3 was used at 10 μM. Data are the mean ± SEM of three experiments, each with four replicates. **p < 0.0001, *p > 0.05 as determined by unpaired t test
Effect of BMS3 on tumor growth and metastasis in mice
Since down-regulation of LIMK1 activity by expression of DN-LIMK1 reduced 4T1.2 tumor growth and metastasis and BMS3 had similar effects in vitro on both 4T1.2 and MDA-MB-231 cells, we examined the ability of systemic treatment with the LIMK inhibitor BMS3 to block tumor growth and/or metastasis. For 4T1.2 tumor growth, we used both immune competent Balb/c mice and the severely immunocompromised NSG mice that lack a functional immune system and are deficient in cytokine signalling. Use of NSG mice enabled us to examine tumor growth and metastasis where any potential impairment of immune responses by the inhibitor would not impact on tumor growth. MDA-MB-231 tumors were grown in immune deficient SCID mice.
First, we measured the level of BMS3 in plasma of mice at various times after a single intraperitoneal injection of 20 mg/kg of BMS3 (Supplementary Fig. 3a). The concentration of BMS3 decreased with time but was still detectable at 24 h. The half-life of BMS3 was estimated to be 5.6 h. When used systemically, BMS3 inhibited p-cofilin by ~60 % in orthotopic MDA-MB-231 tumors in mice treated daily with 20 mg/kg BMS3 (Supplementary Figs. 3b, c), but with only a slight lag in tumor growth (Supplementary Fig. 3d). Thus, we can demonstrate that the inhibitor is active on its target in primary tumors, even 30 h after the last BMS3 treatment. A higher dose of 40 mg/kg had a similar impact on p-cofilin levels in tumors, leading us to select 20 mg/kg as the preferred dose for the in vivo experiments.
Two groups of 20 Balb/c or NSG mice bearing mammary 4T1.2 tumors received daily intraperitoneal injections of 20 mg/Kg BMS3 or vehicle, commencing the day after tumor cell inoculation. BMS3 had no effect on primary tumor growth, although the tumors grew faster in the NSG mice (Fig. 5a). No changes in final primary tumor weight between BMS3 and vehicle treated groups were found (data not shown). Spleen weights at harvest were not significantly different in Balb/c mice but were lower in BMS3 treated NSG mice. The smaller spleen weights in NSG mice compared to Balb/c mice reflect their inability to mount an immune response (Fig. 5b). Lung weights were higher in NSG mice, reflecting their higher metastatic burden, but the weights were not altered by inhibitor treatment in either cohort of mice (Fig. 5c). Quantitation of tumor burden in lung confirmed the organ weight measurements. Both cohorts of the inhibitor treated mice showed a non-significant trend towards greater metastatic burden. Consistent with the total lung weights, NSG mice had a substantially larger tumor burden in the lung compared to Balb/c mice (Fig. 5d). Metastatic burden in the spine was unaltered by BMS3 in both cohorts (Fig. 5e). Visible liver metastatic burden was greater in both cohorts of the BMS3-treated mice and this was confirmed by q-PCR, with significant increases in liver tumor burden (Fig. 5f). H&E images of metastatic nodules in liver from the control and treated mice are shown in Supplementary Fig. 4. In the vehicle treated mouse, a small nodule adjacent to a blood vessel is shown. BMS3 treated mice had more tumor nodules, which tended to be larger in size (Supplementary Fig. 4).

Effect of BMS3 on tumor growth. Forty Balb/c and forty NSG mice were inoculated with 4T1.2 cells (1 × 105) into the 4th mammary gland. The next day, twenty from each group commenced daily IP injections of BMS3 (20 mg/kg) whilst the other twenty mice received vehicle. NSG mice were culled on day 30 and Balb/c mice on day 34. a Primary tumor growth rates. b Spleen weights. c Lung weights at harvest. Metastatic burden in lung (d), spine (e) and liver (f) as determined by q-PCR. For panels d–f, black and grey symbols represent vehicle or inhibitor treated mice, respectively. Statistical analysis of the results in b and c was completed using unpaired t test while that of d-f used the Mann–Whitney test
In a separate experiment using Balb/c mice, we again observed no differences in 4T1.2 primary tumor growth (Supplementary Fig. 5a). Spleen and lung weights were increased significantly in BMS3-treated mice (Supplementary Fig. 5b, c) but quantitation of tumor burden did not result in a significant increase in metastasis to either lung or spine (Supplementary Fig. 5d, e). However, visual inspection revealed an increased number of metastases in other sites, especially liver and diaphragm, as well as metastases in organs not normally involved, including spleen, cecum and stomach (Supplementary Fig. 5f). In summary, although the BMS3 inhibitor reduced the level of p-cofilin in primary tumors, it was not effective in reducing tumor burden or spontaneous 4T1.2 metastasis, despite its ability to inhibit growth and invasion of 4T1.2 and MDA-MB-231 cells in 2D and 3D cultures.
The results obtained in immunocompromised NSG mice did not support the hypothesis that the inhibitor affected the immune system. However, to explore this further in mice treated with BMS3, we noted that B cell proliferation was inhibited by nearly 50 % but there was no effect on T cell proliferation (Fig. 6a, b). Incubation of B cells in vitro with BMS3 inhibited their LPS-stimulated proliferation by 50 % regardless of whether the cells were harvested from mice treated with BMS3 or DMSO (vehicle) (Fig. 6c). A significant reduction (23 %) of in vitro proliferation was evident when B cells from BMS3 treated mice were stimulated with LPS alone without further exposure to the inhibitor (Fig. 6c, middle columns). For the in vitro T cell proliferation assays, cells isolated from mice treated with BMS3 or DMSO were incubated with ConA for 48 h in the presence or absence of BMS3. The inhibitor reduced in vitro proliferation by 50 %, regardless of whether or not the cells were derived from mice treated with BMS3 (Fig. 6d). No difference in T cell proliferation was observed when cells harvested from inhibitor or vehicle treated mice were stimulated with ConA alone (Fig. 6d, middle columns), confirming results obtained from the in vivo proliferation assay (Fig. 6b). Thus, inhibition of LIMK activity in immune competent mice by treatment with BMS3 potentially impairs the capacity of B cells to mount an immune response against the 4T1.2 tumor. However, this impairment does not explain the increased liver metastasis in the NSG mice.

BMS3 inhibits the proliferation of B cells but not T cells in mice and in vitro. a and b Mice (3/group) were treated for 7 days with BMS3 (20 mg/kg/day) or vehicle, with administration of 1 mg of BrdU or DMSO by IP injection on day 1. The BrdU injected mice were given a continuous supply of BrdU (0.8 mg/ml) in drinking water. Mice were culled on day 7 and B and T cells isolated from spleens. a Percentage of proliferating B cells as determined by FACS analysis with anti-BrdU and anti-B220 antibodies. b T cell proliferation as determined by FACS analysis with anti-CD4, anti-CD8 and anti-BrdU antibodies. c and d Mice (3/group) were treated for 7 days as described above but without BrdU. Spleen cells were labelled with CFSE (2.5 mM) before culturing. B and T cell proliferation was stimulated with LPS (2 μg/ml) or ConA (2 μg/ml), respectively, in the presence or absence of BMS3 (10 μM). After 48 h, the extent of proliferation was measured by FACS using the antibodies described above and the number of cell divisions assessed from loss of CFSE fluorescence. c B cell proliferation. d T cell proliferation. Statistical analysis of a and b was completed using the Mann–Whitney test while c and d were analysed by Anova with post hoc comparisons with Tukey’s multiple comparison test
Another possible explanation for the failure of BMS3 to block invasion and metastasis in mice is damage to vascular endothelium by inhibition of LIMK activity, leading to enhanced vessel permeability. We therefore assessed the ability of tumor cells to extravasate into lung tissue of mice treated with vehicle or BMS3 for 4 days prior to intravenous injection of 106 cells. Precisely 10 min later, the mice were culled, the lungs removed and the number of tumor cells scored by q-PCR of the mCherry reporter DNA. No difference in the initial number of tumor cells extravasated into the lung between the vehicle and BMS3 treated mice was observed (Supplementary Fig. 6).
Recently, we have found that levels of LIMK1, LIMK2 and p-cofilin are increased when PMC42 breast cancer cells transition into mesenchymal cells following EGF stimulation (unpublished observation), indicating that LIMKs may have a role in epithelial to mesenchymal transition (EMT). We therefore reasoned that inhibition of LIMK could induce mesenchymal to epithelial transition (MET), which is known to correlate with increased growth of secondary tumors [37]. Enhanced MET could therefore explain the elevated level of metastasis by accelerating growth of micrometastases. While incubation of epithelial PMC42 cells with EGF or FGF resulted in EMT and LIMK activation, incubation of the mesenchymal cells with BMS3 reduced LIMK activity, but did not cause an MET (data not shown). Thus, the increase in metastasis following inhibition of LIMK cannot be explained by enhancement of MET in disseminated tumor cells.
Discussion
Given the role of LIMK1 in cancer cell motility and invasion, and evidence indicating that LIMK1 levels are increased in breast and prostate cancer metastasis [22], it was reasonable to expect that inhibition of its activity might reduce or prevent metastasis. Since published data on the role of LIMK are somewhat contradictory, as discussed in the Introduction, the aim of this study was to confirm or refute a role for LIMK in metastasis. This issue is of great importance as several pharmaceutical and biotechnology companies have identified inhibitors of LIMK activity for the treatment of human diseases, including cancer and glaucoma [30, 38].
In agreement with the hypothesis that LIMK1 drives metastasis, a reduction of LIMK1 activity specifically within the highly invasive 4T1.2 tumor cells using a dominant-negative construct resulted in reduced proliferation, chemotactic migration and invasion as well as reduced primary tumor growth and spontaneous metastasis to lung. Previously we demonstrated that increased LIMK1 activity in MDA-MB-231 cells, which naturally express high levels of LIMK1 protein, increases invasion in vitro and experimental bone metastasis in mice, while DN-LIMK1 has the opposite effect [24]. Here we have shown an inhibition of proliferation and invasion in MDA-MB-231 cells following reduced expression of LIMK1&2 using siRNA or reduced activity using a pharmacological agent. Interestingly, the down-regulation of LIMK1/2 expression has a more profound effect on proliferation of MDA-MB-231 cells than inhibition of the kinase activity using the inhibitor, indicating that LIMK may have additional functions unrelated to its kinase activity.
However, in animal studies, unexpected results were obtained with the LIMK selective inhibitor. Instead of the anticipated reduction in primary tumor growth and metastasis, there was no change in primary tumor growth and no reduction in metastasis to several sites. In fact, we found increased metastasis to liver when using BMS3, raising the possibility that an off-target effect of BMS3 on the liver may have led to increased metastasis to this organ. Since similar results were obtained using immune-competent Balb/c mice and the profoundly immune-suppressed NSG mice, it is unlikely that the increase in metastasis to liver can be ascribed to an impairment of the immune system by the inhibitor, despite our demonstration of reduced B cell proliferation by the inhibitor. We could also discount enhanced extravasation of tumor cells into the lung and changes to EMT and MET as other possible mechanisms to explain our data.
We demonstrated by western blotting for p-cofilin that BMS3 was effective in inhibiting LIMK activity in the primary tumors. Although it is likely that LIMK activity was also inhibited in the metastases, it would have been valuable to show this directly by measuring p-cofilin levels in the liver metastases. It is always possible that the cells that did escape from the primary tumor, despite inhibition of LIMK activity, were able to grow in sites less well accessed by BMS3. Alternatively, these disseminated tumor cells may have evolved resistance to BMS3. Unfortunately, we were not able to achieve satisfactory measurement of p-cofilin in metastases by immunohistochemistry in the formalin fixed tissues. It is clear from our experiments that the inhibitor is active on its target in primary tumors, even 30 h after the last BMS3 treatment, strongly indicating that the dose was adequate. However, we cannot discount the possibility that a higher dose and/or a different dosing schedule could have resulted in more potent target inhibition and therefore a better therapeutic benefit.
Several reports demonstrating increased levels of p-LIMK1/2 and p-cofilin during mitosis indicate that LIMK might have a role in cell cycle regulation [39]. For example, reduced LIMK1 expression in HeLa cells resulted in a delay in the onset of anaphase [40]. Furthermore, suppression of LIMK1 expression in prostate cancer cells resulted in cell cycle arrest at G2/M and retardation of cell growth [22, 41]. The involvement of LIMK2 in the cell cycle was shown by increased cyclin A levels in response to ROCK activation via LIMK2 [42]. However, while we found that expression of DN-LIMK1 did reduce proliferation in vitro and tumor growth in vivo, the use of a systemic LIMK inhibitor did not reduce tumor growth in mice.
It is well established that growth of cells in 3D basement membrane cultures resembles their in vivo growth more so than in 2D culture [36]. We therefore adapted the 3D growth system [33] to study the effects of LIMK inhibition on cell growth and invasion. Inhibition of MLCK in MDA-MB-231 and 4T1 cells prevents stress fiber formation as well as proliferation in 3D culture [33]. Since LIMKs are also important in the regulation of stress fiber formation, we studied the effects of LIMK inhibition on growth of these cells in 3D culture. Incubation of both cell lines with increasing concentrations of the BMS3 reduced colony size as well as invasion into the matrix in a concentration dependent manner, while elevated expression of LIMK1 in 4T1.2 cells increased their proliferation in 2D and 3D culture.
It is unclear why our results differ from those of Wang et al. [29] who demonstrated that increased expression of LIMK1 in MTLn3 cells did not alter proliferation but decreased invasion in vitro. As it was proposed that the level of p-cofilin determines the ability of cells to metastasise [43] it is possible that the contradictory results can be explained by the use of different cell lines and/or by differences in the ratio of LIMK to cofilin. Recently, Scott et al. [26] reported that while BMS3 had no effect on proliferation and motility of MDA-MB-231 cells in 2D culture, it inhibited their invasion in 3D culture.
To our knowledge this is the first work addressing the role of LIMK small molecule inhibitors in metastasis. Recently, Prudent et al. [44] demonstrated the role of a new LIMK inhibitor Pyr1 in the inhibition of LIMK activity in tumor cells and in mice. In an in vivo model of leukemia L1210-bearing mice, they demonstrated that vehicle treated mice died by day 70, whereas all the Pyr1-treated mice survived. However, this paper used a leukemia line and did not address the role of Pyr1 in metastasis. Pyr1, unlike BMS3 [32], enhances microtubule formation and it is this activity that inhibited L1210 cell and tumor growth.
In summary, our data demonstrate that caution should be exercised when considering the application of LIMK inhibitors for therapy of invasive cancer. It is important to report that the genetic reduction of tumor-localized LIMK has a very different response to pharmacological inhibition of LIMK in the whole animal. It is also important to use syngeneic tumor models so that any impact on the immune system can be assessed. We have shown that specific targeting of LIMK activity within tumor cells reduces metastasis, even in aggressive 4T1.2 tumors. However, systemic use of LIMK inhibitors is not effective, possibly due to perturbation of normal tissue and cellular processes that might otherwise be acting to suppress tumor growth or metastasis. This may be due to specific action of these inhibitors on LIMK activity in normal tissues or to off-target activities of these inhibitors.
Abbreviations
- LIMK:
-
LIM kinase
- ADF:
-
Actin depolymerizing factor
- F-actin:
-
Filamentous action
- SSH:
-
Slingshot
- siRNA:
-
Small interfering RNA
References
Olson MF, Sahai E (2009) The actin cytoskeleton in cancer cell motility. Clin Exp Metastasis 26:273–287
Pollard TD, Borisy GG (2003) Cellular motility driven by assembly and disassembly of actin filaments. Cell 112:453–465
Wang W, Eddy R, Condeelis J (2007) The cofilin pathway in breast cancer invasion and metastasis. Nat Rev Cancer 7:429–440
Sahai E (2007) Illuminating the metastatic process. Nat Rev Cancer 7:737–749
Bamburg JR (1999) Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu Rev Cell Dev Biol 15:185–230
Bamburg JR, Bernstein BW (2008) ADF/cofilin. Curr Biol 18:R273–R275
Yamaguchi H, Condeelis J (2007) Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta 1773:642–652
Arber S, Barbayannis FA, Hanser H et al (1998) Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 393:805–809
Yang N, Higuchi O, Ohashi K et al (1998) Cofilin phosphorylation by LIM-kinase1 and its role in Rac-mediated actin reorganization. Nature 393:809–812
Sumi T, Matsumoto K, Takai Y et al (1999) Cofilin phosphorylation and actin cytoskeletal dynamics regulated by rho- and Cdc42-activated LIM-kinase 2. J Cell Biol 147:1519–1532
Bernard O (2007) LIM kinases, regulators of actin dynamics. Int J Biochem Cell Biol 39:1071–1076
Niwa R, Nagata-Ohashi K, Takeichi M et al (2002) Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell 108:233–246
Gohla A, Birkenfeld J, Bokoch GM (2005) Chronophin, a novel HAD-type serine protein phosphatase, regulates cofilin-dependent actin dynamics. Nat Cell Biol 7:21–29
Maekawa M, Ishizaki T, Boku S et al (1999) Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 285:895–898
Edwards DC, Sanders LC, Bokoch GM et al (1999) Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol 1:253–259
Dan C, Kelly A, Bernard O et al (2001) Cytoskeletal changes regulated by the PAK4 serine/threonine kinase are mediated by LIM kinase 1 and cofilin. J Biol Chem 276:32115–32121
Ohashi K, Nagata K, Maekawa M et al (2000) Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J Biol Chem 275:3577–3582
Sumi T, Matsumoto K, Nakamura T (2001) Specific activation of LIM kinase 2 via phosphorylation of threonine 505 by ROCK, a Rho-dependent protein kinase. J Biol Chem 276:670–676
Ikebe C, Ohashi K, Fujimori T et al (1997) Mouse LIM-kinase 2 gene: cDNA cloning, genomic organization, and tissue-specific expression of two alternatively initiated transcripts. Genomics 46:504–508
Nunoue K, Ohashi K, Okano I et al (1995) LIMK-1 and LIMK-2, two members of a LIM motif-containing protein kinase family. Oncogene 11:701–710
Acevedo K, Moussi N, Li R et al (2006) LIM kinase 2 is widely expressed in all tissues. J Histochem Cytochem 54:487–501
Davila M, Frost AR, Grizzle WE et al (2003) LIM kinase 1 is essential for the invasive growth of prostate epithelial cells: implications in prostate cancer. J Biol Chem 278:36868–36875
Saxena M, Singh S, Negi MP et al (2010) Expression profiling of G2/M phase regulatory proteins in normal, premalignant and malignant uterine cervix and their correlation with survival of patients. J Cancer Res Ther 6:167–171
Yoshioka K, Foletta V, Bernard O et al (2003) A role for LIM kinase in cancer invasion. Proc Natl Acad Sci USA 100:7247–7252
Bagheri-Yarmand R, Mazumdar A, Sahin AA et al (2006) LIM kinase 1 increases tumor metastasis of human breast cancer cells via regulation of the urokinase-type plasminogen activator system. Int J Cancer 118:2703–2710
Scott RW, Hooper S, Crighton D et al (2010) LIM kinases are required for invasive path generation by tumor and tumor-associated stromal cells. J Cell Biol 191:169–185
Horita Y, Ohashi K, Mukai M et al (2008) Suppression of the invasive capacity of rat ascites hepatoma cells by knockdown of Slingshot or LIM kinase. J Biol Chem 283:6013–6021
Zebda N, Bernard O, Bailly M et al (2000) Phosphorylation of ADF/cofilin abolishes EGF-induced actin nucleation at the leading edge and subsequent lamellipod extension. J Cell Biol 151:1119–1128
Wang W, Mouneimne G, Sidani M et al (2006) The activity status of cofilin is directly related to invasion, intravasation, and metastasis of mammary tumors. J Cell Biol 173:395–404
Ross-Macdonald P, de Silva H, Guo Q et al (2008) Identification of a nonkinase target mediating cytotoxicity of novel kinase inhibitors. Mol Cancer Ther 7:3490–3498
Foletta VC, Moussi N, Sarmiere PD et al (2004) LIM kinase 1, a key regulator of actin dynamics, is widely expressed in embryonic and adult tissues. Exp Cell Res 294:392–405
Lelekakis M, Moseley JM, Martin TJ et al (1999) A novel orthotopic model of breast cancer metastasis to bone. Clin Exp Metastasis 17:163–170
Barkan D, Kleinman H, Simmons JL et al (2008) Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res 68:6241–6250
Lyons AB (2000) Analysing cell division in vivo and in vitro using flow cytometric measurement of CFSE dye dilution. J Immunol Methods 243:147–154
Eckhardt BL, Parker BS, van Laar RK et al (2005) Genomic analysis of a spontaneous model of breast cancer metastasis to bone reveals a role for the extracellular matrix. Mol Cancer Res 3:1–13
Shaw KR, Wrobel CN, Brugge JS (2004) Use of three-dimensional basement membrane cultures to model oncogene-induced changes in mammary epithelial morphogenesis. J Mammary Gland Biol Neoplasia 9:297–310
Korpal M, Ell BJ, Buffa FM et al (2011) Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med 17:1101–1108
Harrison BA, Whitlock NA, Voronkov MV et al (2009) Novel class of LIM-kinase 2 inhibitors for the treatment of ocular hypertension and associated glaucoma. J Med Chem 52:6515–6518
Amano T, Kaji N, Ohashi K et al (2002) Mitosis-specific activation of LIM motif-containing protein kinase and roles of cofilin phosphorylation and dephosphorylation in mitosis. J Biol Chem 277:22093–22102
Kaji N, Muramoto A, Mizuno K (2008) LIM kinase-mediated cofilin phosphorylation during mitosis is required for precise spindle positioning. J Biol Chem 283:4983–4992
Davila M, Jhala D, Ghosh D et al (2007) Expression of LIM kinase 1 is associated with reversible G1/S phase arrest, chromosomal instability and prostate cancer. Mol Cancer 6:40
Croft DR, Olson MF (2006) The Rho GTPase effector ROCK regulates cyclin A, cyclin D1, and p27Kip1 levels by distinct mechanisms. Mol Cell Biol 26:4612–4627
Oser M, Condeelis J (2009) The cofilin activity cycle in lamellipodia and invadopodia. J Cell Biochem 108:1252–1262
Prudent R, Vassal-Stermann E, Nguyen CH et al (2012) Pharmacological inhibition of LIM kinase stabilizes microtubules and inhibits neoplastic growth. Cancer Res 72(17):4429–4439
Acknowledgments
We thank Christina Restall for technical assistance and advice, Dr Bala Murthy for advice on FACS analysis, Dr. Roger Tsien for provision of the cherry fluorescent protein vector, Alison Gregg and Julia Morizzi for analysis of BMS3 levels in mice, Dr. Siddhartha Deb for pathology advice and Dr. Duncan Campbell for assistance with the statistical analysis. This work was supported by grants from the NIH (R21CA098229) and from the NHMRC of Australia, Fellowship support from NHMRC (OB) and from NBCF (Australia) (RLA). The authors acknowledge financial support from the Cancer Therapeutics CRC, established and supported under the Australian Government’s Cooperative Research Centre Program.
Conflict of interest
No potential conflicts of interest were disclosed.
Author information
Authors and Affiliations
Corresponding authors
Additional information
R. Li and J. Doherty contributed equally to this study. R.L. Anderson and O. Bernard are equal senior authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Li, R., Doherty, J., Antonipillai, J. et al. LIM kinase inhibition reduces breast cancer growth and invasiveness but systemic inhibition does not reduce metastasis in mice. Clin Exp Metastasis 30, 483–495 (2013). https://doi.org/10.1007/s10585-012-9553-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-012-9553-6