
Abstract
B chromosomes are dispensable elements observed in many eukaryotic species, including the African cichlid Astatotilapia latifasciata, which might have one or two B chromosomes. Although there have been many studies focused on the biology of these chromosomes, questions about the evolution, maintenance, and potential effects of these chromosomes remain. Here, we identified a variant form of the hnRNP Q-like gene inserted into the B chromosome of A. latifasciata that is characterized by a high copy number and intron-less structure. The absence of introns and presence of transposable elements with a reverse transcriptase domain flanking hnRNP Q-like sequences suggest that this gene was retroinserted into the B chromosome. RNA-Seq analysis did not show that the B variant retroinserted copies are transcriptionally active. However, RT-qPCR results showed variations in the canonical hnRNP Q-like copy expression levels among exons, tissues, sex, and B presence/absence. Although the patterns of transcription are not well understood, the exons of the B retrocopies were overexpressed, and a bias for female B+ expression was also observed. These results suggest that retroinsertion is an additional and important mechanism contributing to B chromosome formation. Furthermore, these findings indicate a bias towards female differential expression of B chromosome sequences, suggesting that B chromosomes and sex determination are somehow associated in cichlids.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
B chromosomes, also known as supernumerary, accessory, or extra chromosomes, are dispensable elements observed in many eukaryotic species, such as animals, plants, and fungi. The transmission of B chromosomes does not follow Mendelian laws of inheritance, and the number of B chromosomes accumulate according to drive mechanisms during cell division (Burt and Trivers 2006). Evidence suggests that many supernumerary chromosomes originated from an autosomal set (A complement) of chromosomes but followed their own and independent evolutionary history (reviewed by Beukeboom 1994).
B chromosomes have been described in many fish species, including representatives of the cichlid family (Perciformes), which is the richest family of vertebrates and has more than 3000 species distributed in Central and South America, Africa, Madagascar, and South India. Cichlids represent a diverse fauna and model organism for genetic and evolutionary studies; species from the Great Lakes of East Africa underwent rapid sympatric speciation (Kocher 2004). There are several genomic resources (whole genomes sequenced, transcriptomes, miRNomes, genetic maps, and chromosome maps) available for cichlids (Mazzuchelli et al. 2011; Brawand et al. 2014), and many of these species are reproductively viable in the laboratory.
Among the African cichlids, B chromosomes were first described in Astatotilapia latifasciata from Lake Nawampasa, a satellite lake of Lake Kyoga (part of the Lake Victoria system) and in Metriaclima lombardoi from Lake Malawi in East Africa (Poletto et al. 2010a, 2010b). Subsequently, B chromosomes were identified in 12 species of African cichlids of Lake Victoria (Yoshida et al. 2011; Kuroiwa et al. 2014) and more than six species of Lake Malawi (Clark et al. 2017). The cichlid fish A. latifasciata harbors zero, one, or two B chromosomes and was previously studied through classical and molecular cytogenetics studies. In addition, the supernumerary chromosomes of this species might have evolved from a small chromosome fragment from the A complement followed by the invasion of many families of repetitive DNAs (Poletto et al. 2010a; Fantinatti et al. 2011). In addition to studies at the cytogenetic level, large-scale genomic analyses by next generation sequencing (NGS) of whole genomes of A. latifasciata both with and without the B chromosome (s), as well as a microdissected B chromosome were conducted (Valente et al. 2014). This study showed that the B chromosome of A. latifasciata contains thousands of sequences duplicated from essentially every chromosome in the ancestral karyotype, including transposable elements and genes. Although most genes on the B chromosome are fragmented, a few genes are largely intact. Among the genes detected on the B chromosome of A. latifasciata, the heterogeneous nuclear ribonucleoprotein (hnRNP) Q-like (heterogeneous nuclear ribonucleoprotein Q-like) gene is highlighted because of its retrogene characteristics (Valente et al. 2014).
The hnRNP Q-like gene has three protein isoforms called hnRNP Q1–Q3, resulting from the alternative splicing of one gene copy (Quaresma et al. 2009). The hnRNP Q-like gene belongs to a hnRNP family comprising approximately 20 proteins (hnRNPs A-U) involved in fundamental processes such as DNA transcription, messenger RNA (mRNA) splicing, export, degradation, and translation (Huelga et al. 2012). For example, hnRNP Q participates in the mammalian circadian clock for the activation of Period (Per) and Cryptochrome (Cry) genes. hnRNP Q also interacts with 5’UTR of mCry mRNA, suppressing its translation and reducing mCRY1 protein levels (Lim et al. 2016). hnRNP Q binds to the UTR of mPer3 mRNA and promotes mRNA translation and accelerates mRNA decay (Kim et al. 2011). The hnRNP Q-like gene also has retrogene features in several other animals, including Macaca mulatta, Homo sapiens, Rattus norvegicus, Nomascus leucogenys, Pongo abelii, Pan troglodytes, Oryctolagus cuniculus, Sus scrofa, Vicugna pacos, and Dasypus novemcinctus (Kabza et al. 2014).
Characterizing the presence of a retrogene-like sequence on the B chromosome may provide important information on the chromosome structural composition. Understanding B chromosome transcriptional profiles can help to evaluate their impact on cell biology. In A. latifasciata, B chromosomes can influence the cell biology in a complex manner, potentially favoring their self-maintenance and self-perpetuation (Valente et al. 2017). Thus, we investigated the hnRNP Q-like genomic organization and transcriptional profiles in tissues of A. latifasciata. This analysis provided evidence of hnRNP Q-like gene retroinsertion and duplication in the B chromosomes and transcriptional profile variations associated with the presence/absence of the B chromosome. Furthermore, these findings also indicated that B chromosomes and sex determination are somehow associated in A. latifasciata, similar to other cichlids (Yoshida et al. 2011; Clark et al. 2017).
Materials and methods
Samples
Animal samples were obtained from the fish facility of the Integrative Genomics Laboratory, Sao Paulo State University, Botucatu, Brazil. The experimental research on animals was employed in the present study according to the ethical principles of animal research adopted by the Brazilian College of Animal Experimentation and approved through the Institute of Biosciences/UNESP—Sao Paulo State University ethics committee on the use of animals (protocol no. 486-2013). Tissues for DNA and RNA extraction were sampled from males and females with one or two B chromosomes (here named B+) or without B chromosomes (0B; here named B−). The B presence/absence in the samples was determined using polymerase chain reaction (PCR) with specific primers for B chromosome identification (Fantinatti and Martins 2016). The presence of one or two B chromosomes was identified through cytogenetic analysis.
DNA samples from B+ (1B) and B− individuals were extracted from fin clips using a phenol-chloroform method (Sambrook and Russel 2001) and a DNeasy Blood & Tissue kit (Qiagen, Hilden, Germany). RNA samples were obtained from the muscle, brain, and heart of B+ (1B) and B− male and female individuals. Total RNA extraction was conducted using TRIzol reagent (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions. RNA quantification was performed using the NanoVue Spectrophotometer (GE Healthcare Life Sciences, Chicago, USA), and the integrity (RNA integrity number—RIN) was evaluated using a 2100 Bioanalyzer Instrument (Agilent Technologies, Santa Clara, USA).
Genomic analyses
One set of Illumina HiSeq1000 sequencing data analyzed in the present study (whole 2B and 0B genomes) was previously generated for the African cichlid A. latifasciata (Valente et al. 2014) (Table 1). An additional set of reads was obtained for four individuals including B+ and B− A. latifasciata males and females (Table 1). The total read datasets were aligned against the Metriaclima zebra cichlid using Bowtie2 (Langmead and Salzberg 2012), based on a “very sensitive” preset for the pair-end reads. The alignments are available in Sacibase (sacibase.ibb.unesp.br). Alignments against the M. zebra reference enabled a comparison of exon copy number alterations in the hnRNP Q-like gene through the sequenced genomes. In the present study, we focused on a specific region of M. zebra scaffold 3 containing the hnRNP Q-like gene, which is homologous to a retrogene-like sequence observed in A. latifasciata (Valente et al. 2014).
The variant copies of the hnRNP Q-like gene in B+ genomes were identified in a comparison of 0B and 2B read alignments and visualized using Tablet Genome Browser (1.12.12.05) (Milne et al. 2010). To quantify nucleotide variations in exonic regions, we generated an index (called delta or Δ) for each exon. The visual analysis of B+ and B− genomes using Tablet Software was conducted to determine the presence or absence of point mutations, which received a value of 1 or 0, respectively. To differentiate true mutations from sequencing errors, we considered only mutations present in more than five reads. The total number of point mutations, divided by the exon length, resulted in a normalized value per exon. This normalized value was divided by the mean read coverage of each corresponding genome, resulting in a specific value for each exon, summarized by the formula:
MR1 was subsequently used to calculate the delta values, which represent nucleotide diversity between B+ and B− genomes, per exon:
Regions with higher variability (higher delta) and lower variability (lower delta) were selected as the most representative for further analysis.
Bedtools (Quinlan 2014) was used to extract the total genome coverage and per exon coverage from all A. latifasciata alignments. To normalize the exon mean coverage values, the coverage ratios between the alignments were calculated using one male 0B sample (M1-0B) as reference. For each exon, the coverage value was divided by each ratio. To calculate proportions, the mean coverage values from each exon were divided by the M1-0B mean coverage. The analysis was performed using a custom Python script.
We recovered a canonical copy of hnRNP Q-like via the assembly of extracted reads from the high coverage region of the gene (scaffold 3: position 9,112,721..9,126,380 in the reference genome) using SAMtools (Li et al. 2009). These reads were subjected to de novo assembly using the Velvet assembler (Zerbino and Birney 2008) with a k-mer of 69 determined using Kmergenie software (Chikhi and Medvedev 2013). Furthermore, we performed assemblies to recover the hnRNP Q-like retrocopies in all read datasets. For this analysis, an artificial retrocopy was generated using the M. zebra gene sequence without intron sequences (manually removed). This artificial retrocopy was used as a reference to align all sets of reads (0B, 1B, and 2B) using Bowtie2 (“very sensitive” parameters). The reads aligned against the artificial retrocopy were isolated and assembled using the Velvet assembler (k-mer of 67, 121, and 73 for 0B, 1B, and 2B, respectively). The canonical and retrocopies obtained in the assemblies and the reference genome were aligned using Muscle (Edgar 2004).
To recover single nucleotide variants, the reads from all six datasets were aligned against the canonical assembled hnRNP Q-like gene with Bowtie2 (“very sensitive” parameters). Alignments were pre-processed using GATK (McKenna et al. 2010) and Picard tools (http://broadinstitute.github.io/picard). Variant calling was performed with SAMtools 1.2 and filtered for qualities above Phred 30. All analyses used default parameters. The gene assembly, alignments, and related variant calling files (VCF) are available in Sacibase.
Flanking regions (FR) of the hnRNP Q-like gene of M. zebra reference were queried to search for transposable elements (TEs) against the Repbase database (Jurka et al. 2005) at the Genetic Information Research Institute (Giri) (http://www.girinst.org/repbase/). A search for open reading frames (ORFs) was performed using Geneious Pro 4.8.5 (Drummond et al. 2009) to identify the potential protein coding regions of the FRs. ORFs corresponding to sequences of TEs were submitted to the Pfam database (Finn et al. 2010) to obtain more information on the TEs.
Several segments of the hnRNP Q-like gene were amplified using PCR in B+ (1B) and B− males and females with primers designed in representative regions of exons 1 to 11 (Table 2). The amplicons were cloned into the pGEM-T vector system (Promega, Madison, USA), and the clones were sequenced using an ABI Prism 3100 DNA sequencer (Perkin-Elmer, Waltham, USA) with ABI Prism Big Dye Terminator Cycle Sequencing Ready Reaction kits (Perkin-Elmer).
To evaluate the relative copy number of the hnRNP Q-like gene, genomic quantitative PCR (genomic-qPCR) was performed with genomic DNA from B+ (1B) and B− males and females (24 samples in total: nine samples of B+ males, eight B+ females, three B− males, and four B− females). The regions amplified corresponded to exons 2 (primers Ex2F and Ex2R) and 10 (primers Ex10F and Ex10R) (Table 2). The GDR (gene dose ratio) value was obtained using relative quantification (ΔCt); the relative gene copy number was calculated as 2−ΔCt, where ΔCt = Cttarget − Ctreference (Bel et al. 2011). The single-copy autosomal gene hypoxanthine phosphoribosyltransferase (Hprt) was used as the reference. The qPCR was performed using the StepOne Real-Time PCR System (Life Technologies, Carlsbad, CA, USA). The target and reference genes were simultaneously analyzed in duplicate. The program included 1 cycle at 95 °C for 10 min, followed by 45 cycles at 95 °C for 15 s and 60 °C for 1 min. The primer efficiency was calculated using the LingRegPCR program (Ramakers et al. 2003).
Furthermore, primers for the exon-exon junctions (EEJ) were designed (Table 2) to confirm the absence of introns in the retrocopies and apply over three samples of each B+ males, B+ females, B− males, and B− females.
Transcript analysis
The transcriptional levels of the hnRNP Q-like copies were evaluated using RT-qPCR and RNA sequencing (RNA-seq) analysis of the brain, muscle, and heart of B+ (1B) and B− males and females (16 samples of the brain, 24 samples of the muscle, and 23 samples of the heart). RNA extraction was conducted using TRIzol reagent (Life Technologies, USA). Only samples with RIN greater than seven were used and treated with DNase I (Thermo Fisher, Waltham, USA). The reverse transcription reaction of total mRNA was performed using the High Capacity Kit RNA-to-cDNA Master Mix (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions. The regions corresponding to exons 2 and 10 were amplified using the same primers used for genomic-qPCR (primer sets Ex2F/Ex2R and Ex10F/Ex10R—Table 2) analyses of genomic DNA. The UBCE (ubiquitin-conjugating enzyme) gene was used as an endogenous control. The qPCR was performed using the StepOne™ Real-Time PCR System (Life Technologies, Carlsbad, CA, USA). The target and reference genes were simultaneously analyzed in duplicate. The program included 1 cycle at 95 °C for 10 min, followed by 40 cycles at 95 °C for 15 s, and 55 °C for 1 min. Primer efficiency was calculated using the LingRegPCR program (Ramakers et al. 2003). Normalization of the data was performed using the Q-Gene program (Muller et al. 2002; Simon 2003). Data obtained from qPCR were analyzed by comparing the variables sex and B chromosome presence/absence using a generalized linear model (GLM). A statistical model was used to adjust the averages considering a Gamma distribution with SAS software. The significance level cutoff was a p value lower than 0.05.
Differential expression analysis was performed with the reference transcriptomes of the brain, muscle, and gonads assembled using triplicates of male and female samples with B+ and B− genomes. Sequencing generated approximately 30 million reads per sample using a Hi-seq Illumina platform 2000 (Marques DF, Conte MA, Fantinatti BEA, Nakajima RT, Valente GT, Kocher TD, Martins C (2017) Effects of a B chromosome on gene transcription in the cichlid fish Astatotilapia latifasciata. In preparation). Expression was calculated using the FPKM (fragments per kilobase of exon per million fragments mapped) obtained using RSEM v1.2.21 (Li et al. 2010).
A transcript from the hnRNP Q-like gene was identified from de novo transcriptome assembly (Marques DF, Conte MA, Fantinatti BEA, Nakajima RT, Valente GT, Kocher TD, Martins C (2017) Effects of a B chromosome on gene transcription in the cichlid fish Astatotilapia latifasciata. In preparation). Alignments from 36 samples against the reference gene transcript were used to extract single nucleotide variations (SNVs). Protocols for variant calling were similar to a previous analysis. Gmap (Wu and Watanabe 2005) was used to map hnRNP Q-like transcripts to the gene assembly to match transcriptome and genomic SNVs.
Evolutionary analysis of hnRNP Q-like
The hnRNP Q-like copies from 0B, 1B, and 2B A. latifasciata karyomorphs were recovered using the Illumina data. Reads were obtained from whole genome datasets of one representative of each of the 0B, 1B, and 2B genomes. The B consensus copy was constructed based on the hnRNP Q-like retrocopies retrieved from the 2B genome. The hnRNP Q-like genes from fishes, mammals, Archelosauria (birds and turtle), frog, coelacanth, and lamprey were retrieved from the National Center for Biotechnology Information (NCBI) (www.ncbi.nlm.nih.gov) and Ensembl (http://www.ensembl.org/index.html). Mammalian hnRNP Q-like retrocopies were downloaded from RetrogeneDB (http://retrogenedb.amu.edu.pl/) (Kabza et al. 2014).
The sequences were aligned according to the following seven steps according to taxonomic groups: first step, cichlids; second step, other fishes (except coelacanth); third step, mammals; fourth step, Archelosauria; fifth step, tetrapods (includes mammals and Archelosauria alignments plus Xenopus tropicalis and Latimeria chalumnae genes); sixth step, ray-fined fishes; and seventh step, vertebrates (includes tetrapods and ray-fined fish alignments plus lamprey gene). For the first to fourth alignments, Clustal Omega (default) aligner (http://www.ebi.ac.uk/Tools/msa/clustalo/) was used, and for the other alignments, ClustalW (cost matrix IUB, gap open cost 15 and gap extend cost 6.66) was used. The first to sixth alignments were edited using the Gblocks server using the three first Gblocks options for the first, second, and sixth alignments; the second and third options for the third alignment; and the first and second options for the fourth to fifth alignments (http://molevol.cmima.csic.es/castresana/Gblocks_server.html). The seventh alignment was manually edited. Moreover, the edited alignments were submitted to the estimation of proportion sites (using a neighbor-joining algorithm) and saturation test using the Xia algorithm implemented in DAMBE5 (Xia 2013). The re-alignment steps (fifth to seventh) were performed with the edited alignments.
The best-fit evolutionary model for the vertebrate alignment (Supplementary File 1) was calculated using jModelTest2 on XSEDE (three substitution schemes + f + i + g 4, -t ML, -S BEST, information criteria BIC, -p, -v) in Cipres Science Gateway (Miller et al. 2010). The phylogenetic reconstruction was determined by maximum likelihood using Phyml implemented in Seaview (Gouy et al. 2010). The parameters for phylogeny were a GTR+G model (four rate categories and fixed gamma shape at 0.9310), approximate likelihood ratio test (Shimodaira-Hasegawa-like), starting tree using BioNJ and tree searching operation based on the best of NNI and SPR. The tree was rooted at the lamprey sequence.
Results
Genomic structure of hnRNP Q-like copies
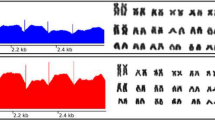
The canonical copy of the hnRNP Q-Like gene is 8992 base pairs in length with 13 exons alternatively spliced into four mRNA isoforms. Genomic alignments of Illumina reads from A. latifasciata identified 13 exons of the hnRNP Q-like gene in scaffold 3 of M. zebra that were over represented by a higher coverage in the B+ genomes (Fig. 1; Supplementary Tables 1 and 2). The coverage of B+ exons varied from 1.7 to 2.8× within the four sequenced 1B samples and approached 6.9× coverage for the 2B genome (Figs. 1 and 2; Supplementary Tables 2, 3 and 4; Supplementary Figure 1). The same analysis was performed using the genome datasets of Pundamilia nyererei and Oreochromis niloticus available at BouillaBase (www.bouillabase.org) and did not confirm higher coverage of exonic regions for the hnRNP Q-like gene as observed in A. latifasciata.

Comparative analysis of the hnRNP Q-like gene structure. a Read coverage of 0B, 1B, and 2B genomes aligned against scaffold 3 of the M. zebra reference genome. Scale on the right indicates the number of read coverage and the coverage ratio. b Structure of the hnRNP Q-like gene with introns (lines), coding DNA sequences (CDS) (blue blocks), and UTRs (red blocks)

Genomic features of the hnRNP Q-like gene. a Read coverage of hnRNP Q-like exons 1 to 13 among 0B (M1_0B, F1_0B), 1B (M2_1B, M3_1B, F2_1B), and 2B (M4_2B) samples normalized to the 0B sample (M1_0B). Colors indicate different individuals. b Read coverage ratio for hnRNP Q-like exons 1 to 13 among 0B, 1B, and 2B samples. The coverage ratio was calculated using the M1_0B sample as a reference. The last row of the table shows the median coverage ratio for all exons. Colors indicate the ratio of one individual vs. the M1-0B reference genome. c GDR estimation using genomic-qPCR for exons 2 and 10 in 0B and 1B genomes. *p < 0.05; **p < 0.01. M male, F female
Nucleotide diversity analyses of hnRNP Q-like gene sequences showed that B+ genomes have higher mutation ratios compared to B− genomes (Fig. 3a; Supplementary Figures 2 and 3). Nucleotide diversity was independently analyzed for each of the hnRNP Q-like exons, focusing on exons 2 and 10, which presented lower and higher delta values, respectively (Supplementary Figure 3). The search for TEs in flanking regions provided evidence of TE relics at a low level of integrity but included reverse transcriptase nucleotide sequences. The TEs belong to class I elements, representing nonLTR and LTR retrotransposons (Table 3; Fig. 3a).

Features of hnRNP Q-like gene. a A recovered 14-Kb contig containing the hnRNP Q-like gene with exon (boxes) and intron structure (line). Mapping of repetitive elements LINE/Dong-R4 and DNA/CMC-EnSpm based on RepeatMasker annotation is indicated. Genome and transcriptome single nucleotide variations (SNV) are also indicated. Genomic SNVs were predominantly obtained from the M4-2B sample, with lower variation compared to B− samples. Few SNVs are present in the transcripts, which indicates no transcription of B+ sequences. Read coverage of six A. latifasciata samples (M1_0B, F1_0B, M2_1B, M3_1B, F2_1B, M4_2B) aligned against the de novo assembly of the hnRNP Q-like gene. The alignment pattern is similar to that observed for A. latifasciata reads against the M. zebra reference in Fig. 1. b De novo assembly of the canonical hnRNP Q-like gene and retrocopies of 1B and 2B A. latifasciata samples compared to the reference gene of M. zebra. Exons are in red and introns in yellow. The lines connecting exons indicate the absence of introns in 1B and 2B retrocopies. c DNA fragments containing the exons 1–6, 5–8, and 8–11 obtained by PCR and sequencing of 1B samples and aligned against the reference hnRNP Q-like gene. The red boxes indicate exons and lines indicate gaps introduced to allow alignment. d Electrophoresis of PCR products for the exon-exon junctions EEJ1/2–EEJ2/3, EEJ6/7–EEJ7/8, and EEJ9/10–EEJ10/11 obtained for B+ and B− males and females. NC negative control. Ladder markers in base pairs are indicated on the left
From the extracted reads aligned to the hnRNP Q-like gene, we obtained the A. latifasciata canonical copy assembly. The same procedure was performed using reads from all samples aligned against the hnRNP Q-like retrogene sequence construct. However, the de novo assembly based on Illumina sequence datasets provided a high number of variable sequences, revealing mutations not recovered by PCR (Fig. 3b). The results from the de novo assembly of the B− genome recovered only one contig corresponding to the entire hnRNP Q-like gene. For the B+ genome, several contigs were obtained, likely reflecting mutations in the hnRNP Q-like copies of the B chromosome (Fig. 3b).
The PCR with primer sets corresponding to exons 1–6, 5–8, and 8–11 (Table 2) only worked in the B+ genome. In the B− genome, PCR with the same set of primers failed to amplify any fragment, likely reflecting the presence of long introns (~3000 bp in the M. zebra genome). The PCR fragment sequencing results revealed the absence of introns in B+ sequences (Fig. 3c). The primers designed over the exon-exon junctions (Table 2) amplified fragments of expected size exclusive of B+ genomes (Fig. 3d).
The relative copy number of the hnRNP Q-like gene was estimated using genomic-qPCR and showed a higher GDR in B+ (1B) individuals compared to B− (0B) individuals. B+ samples have at least double copies of the hnRNP Q-like gene compared to B− samples (Fig. 2c). Exons 2 and 10 showed statistically significant differences in the B chromosome presence between male and females (Supplementary Table 5). Only one B− female sample showed a high copy number of hnRNP Q-like that was similar to the B+ samples (Supplementary Table 6), and this sample was not included in the GDR analysis presented in Fig. 2 and Supplementary Table 5.
The phylogeny of the hnRNP Q-like gene was determined using sequences of several vertebrate species. A total of 62 sequences were used for phylogenetic reconstruction (including mammals, birds, turtle, fishes, amphibians, and lamprey) (Supplementary File 1), and saturation was not evident for all alignments; thus, the final alignment was suitable for phylogenetic inferences. The results showed that the cladogram branches are well supported, even for retrocopy grouping (Supplementary Figure 4).
The hnRNP Q-like transcriptional variation
The search for B+ genome SNVs among transcriptome libraries did not recover any specific variation (Fig. 3; Supplementary Figure 2), showing the complete absence of hnRNP Q-like B copies among the transcribed sequences. The same result was obtained from the analysis of the transcriptomes of other cichlid species. Furthermore, the transcriptome assembly recovered one contig of the hnRNP Q-like gene in the 0B genome. The FPKM results showed no differential expression of the hnRNP Q-like gene in the brain and muscle tissues when comparing the B− and B+ groups. However, differences in the expression level were observed when the gonad tissues of males and females were compared (Supplementary Table 7), representing normal tissue heterogeneity.
RT-qPCR analysis of the expression levels of the hnRNP Q-like gene in the muscle, brain, and heart of B+ and B− individuals revealed significant variations (Fig. 4). Considering the differences among the samples with the same gender-genetic background, B+ females display higher expression of both exons (exons 2 and 10 in the heart and brain, respectively), highlighted in the red box of Fig. 4. Moreover, considering the presence vs. absence of B, exons 2 and 10 were highly expressed in the hearts of B+ males and females (Fig. 4).

Relative transcription levels (RT-qPCR) of exons 2 and 10 of the hnRNP Q-like gene in different tissues of Astatotilapia latifasciata. In a, the horizontal bars in the graphs indicate the median, the box spans the interquartile range, and the whiskers delineate the minimum and maximum of all data. Different colors represent the distinct genotypes investigated: males and females; B− and B+. Statistics: asterisks indicate significant values: *p < 0.05; **p < 0.01. In b, the GLM statistics of qPCR expression levels, comparing sex and B chromosome presence/absence, as indicated in the “Materials and methods” section. In the red box, we highlight male/female expression differences considering B chromosome and sex as variables. Significance was only observed in females
Discussion
The hnRNP Q-like retrogene is exclusive of B+ genomes
The high copy number and higher exon coverage of the hnRNP Q-like gene in B+ genomes is consistent with the idea that such copies were retroinserted into the B chromosome and underwent subsequent duplication events. The coverage ratio variation (1.7–2.8× for 1B genomes and 6.9× for the 2B genome) clearly shows that the copy number of the hnRNP Q-like gene varies among different individuals with higher copy number in the B+ samples. These data indicate that 1B samples can have one (1.7× coverage) to two (2.8× coverage) additional copies of hnRNP Q-like, and the 2B sample can have four copies (6.9× coverage) of the gene. Copy number variations of B located sequences can arise within a single generation as observed in other cichlids (Clark et al. 2017) and were also detected for hnRNP Q-Like retrogene in A. latifasciata. The genomic coverage ratios obtained in the Illumina data were confirmed using genomic-qPCR, revealing variations in the copy number of hnRNP Q-like, with much higher statistically significant values in individuals with B chromosomes. One B− female individual presented a high GDR value for the hnRNP Q-like region as observed among the B+ individuals (discussed later in the “Sex-related variations” section).
Further analysis detected a canonical copy of the hnRNP Q-like gene present in B+ and B− genomes. Additionally, B+ genomes contain variant copies of the hnRNP Q-like gene that may represent retroinserted copies. The existence of such variant copies could reflect the duplication events of the gene after retroinsertion and a higher mutation rate of the B chromosome. Moreover, B+ individuals have much higher copy variation among exons compared to B− individuals, which could reflect a type of independent dynamism after retroinsertion. In fact, the assembly strategy to recover this retrogene from B+ genomes provided more than one contig, suggesting that hnRNP Q-like B copies are fragmented, although a few intact sequences (subjacent exon linkage) are still preserved. It has been demonstrated that duplicated copies of sequences originated early in the B origin subsequently degenerate by accumulating mutations during the evolution of B chromosomes (Banaei-Moghaddam et al. 2013; Valente et al. 2014). Some of the rye B genic sequences have lower similarity to their A-located counterparts, reflecting their faster degeneration or earlier insertion into B chromosomes (Banaei-Moghaddam et al. 2014). B chromosomes of A. latifasciata are a genomic mosaic comprising degenerated sequences derived from most of the A chromosome set (Valente et al. 2014). The high level of copy number variation among B+ samples might be correlated with the relaxed selective pressure over the B chromosome. Variation in copy number of B block sequences among siblings of the same mother of the cichlid M. lombardoi suggests that B mutations can arise in a single generation (Clark et al. 2017).
The absence of introns in the hnRNP Q-like gene present in the B chromosomes indicates its retrogene nature. Retroposition is a process in which mRNAs are reverse-transcribed into DNAs and inserted into a new position in the genome. Retrotransposed copies lack many of their parental gene genetic features, such as introns and regulatory elements (Pan and Zhang 2009). Retrotransposition is an important evolutionary force for the origin of new and potentially functional retrogenes, which are intron-depleted (Huang et al. 2009). Retrocopies of hnRNP Q-like were not detected in the cichlid P. nyererei, which also harbors a B chromosome (Valente et al. 2014), or in any other cichlid genome investigated here. The hnRNP Q-like gene also has retrogene features in several mammals, including M. mulatta, H. sapiens and R. norvegicus (Kabza et al. 2014). Two members of the hnRNP family, hnRNPF and hnRNPH2, are also retrogenes, but represent a special group of retroelements that contain introns (Flabet et al. 2009). Relics of retrotransposable elements were identified in the flanking regions of the hnRNP Q-like B copy, suggesting that TEs have moved the gene from the A complement to the B chromosome. The flanking regions of retrogenes can harbor ancient TE insertions that accumulated mutations and subsequent insertions and rearrangements, reducing the activity and similarity of the element in known families (Wicker et al. 2007).
The phylogenetic tree of the hnRNP Q-like gene revealed that B retrocopies are closely related to the A genome copy. However, one retrocopy from a 1B sample was grouped to the P. nyererei cichlid. Moreover, vertebrates experienced independent rounds of retroinsertion of the nRNP Q-like gene because each retrocopy was grouped with its paralogs in mammals and A. latifasciata.
Transcriptional variation of hnRNP Q-like gene
Extensive tissue-specific transcription variations were observed and associated with the presence/absence of the B chromosome and/or phenotypic sex, although none of these variations presented a common pattern, including differences between exons 2 and 10. Despite the absence of a clear expression pattern, the presence of B increased exon expression (except for one case further discussed). We propose that B hnRNP Q-like exons may be not contiguously linked. This characteristic could be associated with independent duplication events in the sequence after insertion. The insertions could locate the exons close to different promoters/cis-regulatory elements that may reflect the variant expression pattern observed here.
Although no differential expression was observed between B− and B+ genomes using RNA-Seq data, RT-qPCR showed different expression among the analyzed exons, with a female bias. Both techniques can yield different results, particularly when comparing low expressed genes, as demonstrated in a previous genome-wide transcription study (Everaert et al. 2017). The RNA-Seq data obtained in the present study revealed that hnRNP Q-like has fold-change values smaller than 2, and while variations in FC were positive, indicating higher expression in B+ individuals, these alterations were not statistically significant. The fold-change values presented herein were within the range according to Everaert et al. (2017) and may reflect differences between RNA-Seq and RT-qPCR methods.
B-specific mutations were not detected among the transcripts of the hnRNP Q-like gene. These data suggest that the hnRNP Q-like B variant copies are retropseudogenes that lose their transcriptional activity. However, canonical copies of hnRNP Q-like B copies can be transcriptionally active and may influence cell molecular mechanisms. Many DNA fragments from all A chromosomes invaded the A. latifasciata B chromosome, and individual transposition events (including retrogenes) were important for the insertion of those sequences (Valente et al. 2014). Retrotransposed gene copies are often described as pseudogenes that lack regulatory regions and consequently degenerate (Bai et al. 2007). B chromosomes are dispensable and may accumulate mutations. More likely, a duplicated gene undergoes inactivation and pseudogenization, rather than acquiring a new function. Although gene duplication via retrotransposition has long been considered to originate from degenerated non-active copies, a significant number of retrogenes escape elimination and evolve into functional genes (Ciomborowska et al. 2013). There are several examples of functional retrocopies as observed in the Ard1b retrogene, expressed to compensate for the inactivation of the Ard1a gene (Pang et al. 2009). The retrogene Sep5 in Drosophila is expressed during embryogenesis, has functional consequences for the septin complex, and may regulate the complex interactions with other proteins or membranes (O’Neill and Clark 2013). The examination of retrogene distribution in eight mammalian genomes and four non-mammalian genomes identified many functional retrogenes (Pan and Zhang 2009). Ciomborowska et al. (2013) identified 25 “orphan” retrogenes that replaced their progenitors in the human genome, and all of these elements are functional.
Sex-related variations
Although the Illumina data did not reveal genomic variations in hnRNP Q-like B related to sex, genomic-qPCR detected one B− female individual with a high GDR value similar to the values for both B+ males and B+ females. This female genome likely shares genomic blocks with the B chromosome. B chromosomes and sex chromosomes are somehow correlated and represent one of the most astonishing characteristics of B chromosomes (for review, Martins et al. 2011). B chromosomes are female-specific in Lithochromis rubripinnis (cichlids of Lake Victoria, East Africa), and crosses demonstrated that the presence of B leads to a female-biased sex ratio (Yoshida et al. 2011). However, B chromosomes have been associated with both males and females in 12 other cichlid species in Lake Victoria, including A. latifasciata (Poletto et al. 2010a, 2010b; Yoshida et al. 2011; Kuroiwa et al. 2014). B chromosomes were also inferred only in females among seven species in Lake Malawi (East Africa) cichlids (M. lombardoi, M. zebra “Boadzul,” M. zebra “Nkhata Bay,” Metriaclima greshakei, Metriaclima mbenji, Labeotropheus trewavasae, and Melanochromis auratus) (Clark et al. 2017). The correlation between B chromosomes and females may reflect a drive mechanism that acts during female meiosis, suggesting that the B chromosome has a higher fitness in females (Clark et al. 2017).
Here, we observed that the expression in samples with the same gender-genetic background (excluding the possibility of expression variation that is sex-related) was higher in B+ females compared to B− females; however, this differential expression was not observed in both analyzed exons in the three collected tissues. The only case in which B presence did not reflect high expression in all tissues or exons was in the male B+ vs. male B− comparison. The differential expression of hnRNP Q-like transcripts between the testis and ovaries may be associated with the regular effect of such genes on the physiology of the gonads, as observed for other hnRNP genes (Shao et al. 2012). Transcriptional variation linked to B chromosome and sexual phenotype was previously detected for the BncDNA repetitive element in A. latifasciata (Ramos et al. 2017).
Although B and sex chromosome association has been described for various species, their relationship to each other is still poorly understood and needs further investigation. The effects of B chromosomes in sex determination remains unclear, but these results suggest that the female genome and B chromosomes may share similar genomic segments containing the hnRNP Q-like gene.
Conclusion
Retrotransposition contributes to B chromosome constitution. Although B-specific chromosome retrocopies are not transcriptionally active, the canonical hnRNP Q-like B copies, with additional transcripts of the gene, could influence cell functions. Moreover, the exon copy number variation, differential regulation of B chromosome exons, and female B+ differential expression suggest that B retrocopies may have assumed many gene functions during their evolutionary history. Thus, B chromosomes could be relevant to cell biology rather than the classical parasitism related to their perpetuation during cell and individual generations.
Abbreviations
- CDS:
-
Coding DNA sequence
- FPKM:
-
Fragments per kilobase of exon per million fragments mapped
- FR:
-
Flanking regions
- GDR:
-
Gene dose ratio
- GLM:
-
Generalized linear model
- hnRNP Q-like:
-
Heterogeneous nuclear ribonucleoprotein Q-like
- ORFs:
-
Open reading frames
- qPCR:
-
Quantitative PCR
- RIN:
-
RNA integrity number
- RT-qPCR:
-
Reverse transcription quantitative PCR
- SNVs:
-
Single nucleotide variations
- TEs:
-
Transposable elements
- UBCE:
-
Ubiquitin-conjugating enzyme
- UTRs:
-
Untranslated regions
References
Bai Y, Casola C, Feschotte C, Betran E (2007) Comparative genomics reveals a constant rate of origination and convergent acquisition of functional retrogenes in Drosophila. Genome Biol 8:R11
Banaei-Moghaddam AM, Meier K, Karimi-Ashtiyani R, Houben A (2013) Formation and expression of pseudogenes on the B chromosome of rye. Plant Cell 25:2536–2544
Banaei-Moghaddam AM, Martis MM, Macas J et al (2014) Genes on B chromosomes: old questions revisited with new tools. Biochim Biophys Acta 1849:64–70
Bel Y, Ferré J, Escriche B (2011) Quantitative real-time PCR with SYBR green detection to assess gene duplication in insects: study of gene dosage in Drosophila melanogaster (Diptera) and in Ostrinia nubilalis (Lepidoptera). BMC Res Notes 4:84
Beukeboom LW (1994) Bewildering Bs: an impression of the 1st B-chromosome conference. Heredity 73:328–336
Brawand D, Wagner CE, Li YI et al (2014) The genomic substrate for adaptive radiation in African cichlid fish. Nature 513:375–381
Burt A, Trivers R (2006) Genes in conflict: the biology of selfish genetic elements. Belknap Press, Cambridge
Chikhi R, Medvedev P (2013) Informed and automated k-mer size selection for genome assembly. Bioinformatics 30:31–37
Ciomborowska J, Rosikiewicz W, Szklarczyk D, Makałowski W, Makałowska I (2013) “Orphan” retrogenes in the human genome. Mol Biol Evol 30:384–396
Clark FE, Conte MA, Ferreira-Bravo IA, Poletto AP, Martins C, Kocher TD (2017) Dynamic sequence evolution of a sex-associated B chromosome in Lake Malawi cichlid fish. J Hered 108(1):53–62
Drummond AJ, Ashton B, Cheung M et al (2009) Geneious v4.8.5. Available from http://www.geneious.com
Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 9:113
Everaert C, Luypaert M, Maag J et al (2017) Benchmarking of RNA-sequencing analysis workflows using whole-transcriptome RT-qPCR expression data. Sci Rep 7:1559
Fantinatti BEA, Martins C (2016) Development of chromosomal markers based on next-generation sequencing: the B chromosome of the cichlid fish Astatotilapia latifasciata as a model. BMC Genet 17:119
Fantinatti BEA, Mazzuchelli J, Valente GT, Cabral-de-Mello DC, Martins C (2011) Genomic content and new insights on the origin of the B chromosome of the cichlid fish Astatotilapia latifasciata. Genetica 139:1273–1282
Finn RD, Mistry J, Tate J et al (2010) The Pfam protein families database. Nucleic Acids Res 38:D211–D222
Flabet M, Bueno M, Potrzebowski L, Kaessmann H (2009) Evolutionary origin and functions of retrogene introns. Mol Biol Evol 26:2147–2156
Gouy M, Guindon S, Gascuel O (2010) SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27:221–224
Huang C-J, Lin W-Y, Chang C-M, Choo K-B (2009) Transcription of the rat testis-specific Rtdpoz-T1 and -T2 retrogenes during embryo development: co-transcription and frequent exonisation of transposable element sequences. BMC Mol Biol 10:74
Huelga SC, Vu AQ, Arnold JD, Liang TY, Liu PP et al (2012) Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep 1:167–178
Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J (2005) Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110:462–467
Kabza M, Ciomborowska J, Makałowska I (2014) RetrogeneDB—a database of animal retrogenes. Mol Biol Evol 31:1646–1648
Kim DY, Kwak E, Kim SH, Lee KH, Woo KC et al (2011) hnRNP Q mediates a phase-dependent translation-coupled mRNA decay of mouse Period3. Nucleic Acids Res 39:8901–8914
Kocher TD (2004) Adaptive evolution and explosive speciation: the cichlid fish model. Nat Rev Genet 5:288–298
Kuroiwa A, Terai Y, Kobayashi N et al (2014) Construction of chromosome markers from the Lake Victoria cichlid Paralabidochromis chilotes and their application to comparative mapping. Cytogenet Genome Res 142:112–120
Langmead B, Salzberg S (2012) Fast gapped-read alignment with Bowtie 2. Nature Meth 9:357–359
Li H, Handsaker B, Wysoker A et al (2009) 1000 genome project data processing subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079
Li B, Ruotti V, Stewart RM, Thomson JA, Dewey CN (2010) RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 26:493–500
Lim I, Jung Y, Kim DY, Kim KT (2016) HnRNP Q has a suppressive role in the translation of mouse cryptochrome 1. PLoS One 8:e0159018
Martins C, Cabral-de-Mello DC, Valente GT, Mazzuchelli J, Oliveira SG, Pinhal D (2011) Animal genomes under the focus of cytogenetics. Nova Science Publisher, Hauppauge
Mazzuchelli J, Yang F, Kocher TD, Martins C (2011) Comparative cytogenetic mapping of Sox2 and Sox14 in cichlid fishes and inferences on the genomic organization of both genes in vertebrates. Chromosom Res 19:657–667
McKenna A, Hanna M, Banks E et al (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303
Miller MA, Pfeiffer W, Schwartz T (2010) Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Gateway Computing Environments Workshop (GCE), New Orleans, pp 1–8
Milne I, Bayer M, Cardle L, Shaw P, Stephen G, Wright F, Marshall D (2010) Tablet—next generation sequence assembly visualization. Bioinformatics 26:401–402
Muller PY, Janovjak H, Miserez AR, Dobbie Z (2002) Processing of gene expression data generated by quantitative real-time RT-PCR. BioTechniques 32:1372–1374
O’Neill RS, Clark DV (2013) Evolution of three parent genes and their retrogene copies in Drosophila species. Int J Evol Biol 2013:693085
Pan D, Zhang L (2009) Burst of young retrogenes and independent retrogene formation in mammals. PLoS One 4:e5040
Pang AL, Peacock S, Johnson W, Bear DH, Rennert OM, Chan W (2009) Cloning, characterization, and expression analysis of the novel acetyltransferase retrogene Ard1b in the mouse. Biol Reprod 81:302–309
Poletto AB, Ferreira IA, Cabral-de-Mello DC, Nakajima RT, Mazzuchelli J, Ribeiro HB, Venere PC, NirchioM KTD, Martins C (2010a) Chromosome differentiation patterns during cichlid fish evolution. BMC Genet 11:50
Poletto AB, Ferreira IA, Martins C (2010b) The B chromosome of the cichlid fish Haplochromis obliquidens harbors 18S rRNA genes. BMC Genet 11:1
Quaresma AJ, Bressan GC, Gava LM, Lanza DC, Ramos CH, Kobarg J (2009) Human hnRNP Q re-localizes to cytoplasmic granules upon PMA, thapsigargin, arsenite and heat-shock treatments. Exp Cell Res 315:968–980
Quinlan AR (2014) BEDTools: the swiss-army tool for genome feature analysis. In Current protocols in bioinformatics. Curr Protoc Bioinformatics 47:11.12.1–11.12.34
Ramakers C, Ruijter JM, Deprez RH, Moorman AF (2003) Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett 339:62–66
Ramos E, Cardoso AL, Brown J, Marques DF, Fantinatti BEA, Cabral-de-Mello DC, Martins C (2017) The repetitive DNA element BncDNA, enriched in the B chromosome of the cichlid fish Astatotilapia latifasciata, transcribes a potentially noncoding RNA. Chromosoma 126:313–323
Sambrook J, Russel DW (2001) Molecular cloning. A laboratory manual, 3rd ed. Cold Spring
Shao R, Wang X, Weijdegård B, Norström A, Fernandez-Rodriguez J, Brännström M, Billig H (2012) Coordinate regulation of heterogeneous nuclear ribonucleoprotein dynamics by steroid hormones in the human fallopian tube and endometrium in vivo and in vitro. Am J Physiol Endocrinol Metab 302:1269–1282
Simon P (2003) Q-gene: processing quantitative real-time RT-PCR data. Bioinformatics 19:1439–1440
Valente GT, Conte MA, Fantinatti BEA et al (2014) Origin and evolution of B chromosomes in the cichlid fish Astatotilapia latifasciata based on integrated genomic analyses. Mol Biol Evol 31:2061–2072
Valente GT, Nakajima RT, Fantinatti BEA, Marques DF, Almeida RO, Simões RF, Martins C (2017) B chromosomes: from cytogenetics to systems biology. Chromosoma 126:73–81
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P et al (2007) A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8:973–982
Wu TD, Watanabe CK (2005) GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2:1859–1875
Xia X (2013) DAMBE5: a comprehensive software package for data analysis in molecular biology and evolution. Mol Biol Evol 30:1720–1728
Yoshida K, Terai Y, Mizoiri S et al (2011) B chromosomes have a functional effect on female sex determination in Lake Victoria cichlid fishes. PLoS Genet 7:e1002203
Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829
Acknowledgments
This work was financially supported through grants from the São Paulo Research Foundation (FAPESP) (2011/03807-7; 2013/04533-3; 2015/16661-1) and the National Counsel of Technological and Scientific Development (CNPq) (474684/2013-0; 305321/2015-3).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval
The experimental procedure was conducted according to the international guidelines of Sao Paulo State University and approved through the Institutional Animal Care and Use Committee (IACUC) (protocol no. 486-2013-CEEA/IBB/UNESP). The animals were euthanized via immersion in a water bath containing benzocaine at 250 mg/l for 10 min.
Consent for publication
Not applicable.
Availability of data and material
All raw data are available in SaciBase (http://sacibase.ibb.unesp.br/).
Competing interests
The authors declare that they have no competing interests.
Additional information
Responsible Editor: Fengtang Yang.
Rights and permissions
About this article

Cite this article
Carmello, B.O., Coan, R.L.B., Cardoso, A.L. et al. The hnRNP Q-like gene is retroinserted into the B chromosomes of the cichlid fish Astatotilapia latifasciata . Chromosome Res 25, 277–290 (2017). https://doi.org/10.1007/s10577-017-9561-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10577-017-9561-0