
Abstract
The structural maintenance of chromosome (SMC) proteins constitute the cores of three protein complexes involved in chromosome metabolism; cohesin, condensin and the Smc5-Smc6 complex. While the roles of cohesin and condensin in sister chromatid cohesion and chromosome condensation respectively have been described, the cellular function of Smc5-Smc6 is as yet not understood, consequently the less descriptive name. The complex is involved in a variety of DNA repair pathways. It contains activities reminiscent of those described for cohesin and condensin, as well as several DNA helicases and endonucleases. It is required for sister chromatid recombination, and smc5-smc6 mutants suffer from the accumulation of unscheduled recombination intermediates. The complex contains a SUMO-ligase and potentially an ubiquitin-ligase; thus Smc5-Smc6 might presently have a dull name, but it seems destined to be recognized as a key player in the maintenance of chromosome stability. In this review we summarize our present understanding of this enigmatic protein complex.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
SMC proteins
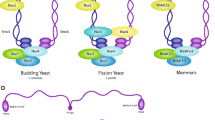
Structural maintenance of chromosome (SMC) proteins are key regulators of the structural and functional organization of chromosomes. Highlighting their important function is the fact that their origin precedes that of histones (Nasmyth and Haering 2005). While bacterial genomes contain a single SMC gene whose product forms a homodimer, in eukaryotes there are at least six different SMC proteins (known as Smc1–6) that form heterodimers in specific combinations: namely, Smc1-Smc3, Smc2-Smc4 and Smc5-Smc6. In vivo, SMC proteins associate with additional non-SMC subunits to form functional complexes. In eukaryotes, Smc1 and Smc3 form the core of the cohesin complex, Smc2 and Smc4 are components of the condensin complex, and Smc5 and Smc6, whose sequences are substantially divergent from those of Smc1–4 (Cobbe and Heck 2004), form a third complex known as the Smc5-Smc6 complex (Fig. 1).

Schematic representation of the three SMC complexes in budding yeast
Architecture of the Smc5-Smc6 complex
The Smc5 and Smc6 heterodimer is formed by interaction of the two proteins through their hinge domains. This interaction is essential for the function of the complex, since mutation of a conserved glycine residue in the Smc6 hinge causes temperature-sensitive growth in Schizosaccharomyces pombe (Sergeant et al. 2005). In the yeasts, at least six additional subunits are associated to Smc5 and Smc6; these are termed non-Smc elements (Nse1–6). In human cells, on the other hand, only four Nse proteins (hNse1–4) have been identified so far. Nse1 contains a RING finger domain that resembles those found in ubiquitin ligases (Fujioka et al. 2002). Although no enzymatic activity has been demonstrated for this subunit (Pebernard et al. 2008), mutation or deletion of the RING motif confers growth defects in Saccharomyces. cerevisiae (Santa Maria et al. 2007) and increased sensitivity to DNA damaging agents in S. pombe (Pebernard et al. 2008). In human cells, Nse1 is ubiquitinated in vivo (Taylor et al. 2008) and physically interacts with Nse3 (Sergeant et al. 2005; Palecek et al. 2006; Pebernard et al. 2006). Interestingly, Nse3 shows high homology to a class of proteins known as MAGE (melanoma antigen gene), which are overexpressed in certain types of cancers and whose function has been linked to cell cycle regulation, apoptosis and neuronal development (Barker and Salehi 2002). While only one MAGE gene exists in the genomes of plants, nematodes, insects and even non-mammalian vertebrates, mammalian cells carry 55 different MAGE genes. Human MAGEG1 was recently shown to be part of the Smc5-Smc6 complex (Taylor et al. 2008). Nse4 is a member of the kleisin protein family, presenting the characteristic winged helix motif (Palecek et al. 2006). Nse3 and Nse4 independently bind to the head domains of Smc5 and Smc6 (Palecek et al. 2006). A fourth subunit, Nse2, binds to Smc5-Smc6 independently of the Nse1-3-4 subunits, through interaction between its N-terminal half and the coiled-coil domain of Smc5 (Sergeant et al. 2005). Nse2, at least in human cells, is not necessary for the stability of the complex (Taylor et al. 2008). The C-terminal portion of Nse2 contains a conserved RING-like domain and shows SUMO-ligase activity both in vitro and in vivo (Andrews et al. 2005; Potts and Yu 2005; Zhao and Blobel 2005). A growing body of evidence suggests that sumoylation plays a key role in maintaining genomic stability (Geiss-Friedlander and Melchior 2007). The full list of targets of Nse2 ligase are still to be identified, but so far include the telomeric binding protein yKu70 and Smc5 in S. cerevisiae, Smc6 in S. pombe and Smc6, as well as other telomere binding proteins like TRAX, TRF1, TRF2, TIN2, and RAP1 in human cells (Andrews et al. 2005; Potts and Yu 2005; Zhao and Blobel 2005). While Nse2 is essential in all organisms analysed, mutation of the RING finger-like domain confers DNA damage sensitivity. Gel filtration experiments with complexes isolated from S. pombe or human cells have demonstrated that Nse1-4 co-fractionate and are mostly associated with the Smc5-Smc6 complex, while only residual levels of monomeric protein are observed. This suggests that Nse2 SUMO-ligase activity is executed within the Smc5-Smc6 complex context (Taylor et al. 2008). Nse5 and Nse6, in contrast to the rest of Nse subunits, show poor sequence homology between different species and have so far been identified only in yeast (Pebernard et al. 2006; Zhao and Blobel 2005).
Chromosomal localization
Microarray-analysed ChIP technology in budding yeast and chromatin fractionation in Xenopus laevis have been used to study the association of the Smc5-Smc6 complex with chromosomes. Like cohesin, the Smc5-Smc6 complex binds to distinct sites on chromosomes in a cell cycle-regulated manner (Lindroos et al. 2006). Loading of the complex occurs during S phase, in a process that is dependent on DNA replication (Lindroos et al. 2006). In Xenopus laevis, inhibition of replication elongation by aphidicolin or fork stalling by hydroxyurea (HU) strongly reduces the amount of protein associated with DNA (Tsuyama et al. 2006). In budding yeast, high binding of the complex is observed at centromeres, telomeres and the ribosomal gene cluster on chromosome XII (Lindroos et al. 2006; Torres-Rosell et al. 2005). In contrast to cohesin and condensin, the frequency of arm interaction increases with chromosomal length. Most of the Smc5-Smc6 peaks are detected in intergenic regions and co-localize with those of cohesin (Lengronne et al. 2004; Lindroos et al. 2006). Chromosome association of Smc5-Smc6 depends partially on the cohesin loading complex Scc2/4 (Lindroos et al. 2006). In sharp contrast, however, the strong Smc5-Smc6 binding in the region downstream the rDNA on chromosome XII is independent of Scc2/4 (Lindroos et al. 2006). Finally, the pattern of Smc5-Smc6 association is influenced by the presence of cohesin, since Smc6 binds to more arm sites and forms narrower binding peaks in scc1-73 mutants (Strom et al. 2007). After reaching its maximum levels in G2 phase, the Smc5-Smc6 complex dissociates from chromosomes during mitosis. In X. laevis, the removal of the complex is completed by the time condensin starts accumulating on chromosomes (Tsuyama et al. 2006). Similar observations have also been reported in human cells (Taylor et al. 2001). In S. cerevisiae, the kinetics of Smc5-Smc6 dissociation are unknown, but it is likely to occur only after metaphase, as cells arrested with the microtubule poison nocodazole show strong association of Smc5-Smc6 with chromosomes (Torres-Rosell et al. 2005; Lindroos et al. 2006).
The role of Smc5-Smc6 in DNA damage response
Our understanding of Smc5-Smc6 function is still in the early stages, and is mainly phenotypical. One of the processes in which the complex was first found to be involved is the DNA damage response. Two independent screens for genes required for DNA damage repair, carried out in the 1970s, characterized two subunits of the Smc5-Smc6 complex, Smc6 and Nse2, respectively named Rad18 and Mms21, based on the increased sensitivity manifested in mutants of these proteins to gamma radiation and the alkylating agent methyl methanesulfonate (MMS) (Nasim and Smith 1975; Prakash and Prakash 1977). Further studies have subsequently provided evidence to support the link between the complex and DNA damage: in S. pombe or S. cerevisiae, mutation of any of the subunits of the complex induces hypersensitivity to DNA damage agents ranging from double-strand break (DSB)-inducing ionizing radiation to the alkylating agent MMS and UV light. The sensitivity of smc5-smc6 mutants to DNA damage does not arise from an inability to activate the appropriate checkpoints: in budding and fission yeast, all smc5-smc6 mutants analysed to date show wild-type kinetics and intensity of the checkpoint response and arrest the cell cycle in a dose-dependent manner (Verkade et al. 1999; Andrews et al. 2005; Torres-Rosell et al. 2007a). smc5-smc6 mutants are defective in DNA repair and recovery after treatment with genotoxic agents, which results in cell death due to mitotic ‘cut’ cells in which the nucleus has been bisected by the septum (Verkade et al. 1999; Torres-Rosell et al. 2005). On the basis of this observation, it has been suggested that smc5-smc6 mutants in S. pombe might be defective in the maintenance of the DNA damage checkpoint (Verkade et al. 1999). However, the imposition of an artificial G2 phase arrest, while able to rescue the DNA damage sensitivity of checkpoint-defective rad3-136 cells (Rad3 is the S. pombe homologue of Mec1/ATR checkpoint kinase), fails to decrease the percentage of catastrophic mitoses in smc5-smc6 mutants, (Verkade et al. 1999) thus suggesting that smc5-smc6 mutants are checkpoint-competent.
A number of epistasis studies in yeasts have indicated that Smc5-Smc6 function lies within the Rad51-dependent homologous recombination pathway. In fission yeast, smc5-smc6 mutants are less sensitive to ionizing radiation than rhp51Δ, while the double mutant smc5-smc6 rhp51Δ shows similar sensitivity to rhp51Δ (Lehmann et al. 1995). However, smc5-smc6 mutants are more sensitive than rhp51Δ to HU and UV, while the double mutant rescues the sensitivity of smc5-smc6 (Lehmann et al. 1995; Ampatzidou et al. 2006; Pebernard et al. 2006). In budding yeast, rad52Δ partially suppresses the temperature sensitivity of smc6-9 mutants (Torres-Rosell et al. 2005), demonstrating that recombination processes are toxic in the absence of Smc5-Smc6 function. Consistent with this, exposure of budding yeast mms21 and fission yeast smc6 mutants to MMS causes the accumulation of toxic recombination intermediates (Ampatzidou et al. 2006; Branzei et al. 2006). Synthetic lethality has been observed when smc5-smc6 mutants are combined with mutations in the Sgs1 helicase and the endonuclease Mus81 complex (Torres-Rosell et al. 2005; Cost and Cozzarelli 2006). Although the involvement of Smc5-Smc6 in homologous recombination has long been predicted, various studies have failed to detect any defect in the execution of Rhp51 (Rad51) or Rad52 mediated processes. For instance, budding yeast smc5-smc6 mutants show wild-type efficiency in gene conversion at the mating type locus (De Piccoli et al. 2006). In both fission and budding yeast, recombination between heteroalleles on a chromosome arm as well as ectopic recombination between chromosome sequences and a plasmid is not significantly increased in smc5-smc6 mutants (Morikawa et al. 2004; Onoda et al. 2004; Pebernard et al. 2004). However, increased recombination between heteroalleles in diploid cells has been reported for smc5 mutants (Cost and Cozzarelli 2006). In S. cerevisiae, the smc5-smc6 mutant shows a reduction in heteroallelic recombination between homologous chromosomes when grown in the presence of MMS (Onoda et al. 2004). This result is difficult to interpret because of the concomitant drop in the viability of the mutant in the presence of MMS compared with wild-type cells.
Smc5-Smc6 at DNA double-strand breaks (DSBs)
DNA double-strand breaks (DSB) are one of the most threatening alterations of a cell’s genetic material. Left unrepaired, DSBs can cause cell death (Bennett et al. 1993) and, if misrepaired, they can lead to genomic instability and the development of cancer in multicellular organisms. Eukaryotic cells have evolved two main mechanisms for the repair of DSBs: non-homologous end-joining (NHEJ) (Daley et al. 2005) and homologous recombination (HR) (Pâques and Haber 1999; Prado et al. 2003). NHEJ entails the direct rejoining of the broken ends of DNA, whereas HR involves a genomic search for similar sequences to be used as a template for repair. During the HR reaction the location of the template in relation to the site of damage is flexible. Thus, HR can take place between sister chromatids (Gonzalez-Barrera et al. 2003), homologous chromosomes, or related DNA sequences in the genome regardless of their chromosomal location (Pâques and Haber 1999; Aylon and Kupiec 2004). In yeast and mammalian cells, the cell cycle stage also determines the repair pathway used; HR is favoured in late S and G2 phases and inhibited in G1, because it requires cyclin B-dependent kinase (Cdk) activity (Ira et al. 2004; Esashi et al. 2005). While NHEJ is favoured in G1 phase (when the sister chromatid is absent), recombination is favoured during the periods of the cell cycle when sister chromatids are present (Caspari et al. 2002).
Following induction of DNA DSBs, cells undergo a coordinated process to ensure repair of the break, which involves the function of checkpoint, repair, chromatin, and structural proteins. A general overview of this response is summarized in Fig. 2. Upon DSB induction, the ends of the broken chromosome remain associated (Melo et al. 2001; Lisby et al. 2003a; Kaye et al. 2004; Lobachev et al. 2004), and cohesin is eventually recruited to regions around the break to hold sister chromatids together (Strom et al. 2004; Ünal et al. 2004). Smc5-Smc6 is also recruited to DSBs, binding to regions that span at least 25 kb on each side (De Piccoli et al. 2006; Lindroos et al. 2006; Potts et al. 2006). The function of Smc5-Smc6 in the repair of DNA breaks, like that of cohesin (Cortes-Ledesma and Aguilera 2006), is to promote sister chromatid recombination (De Piccoli et al. 2006; Potts et al. 2006). A plasmid-based recombination assay that physically detects sister chromatid exchanges (Gonzalez-Barrera et al. 2003) showed a 4-fold reduction in the repair of DSBs by equal and unequal sister chromatid exchange in smc5-smc6 mutants (De Piccoli et al. 2006). Similar defects in sister chromatid recombination have also been observed in human cells depleted by RNA interference for the hSmc5 and hNse2/Mms21 subunits (Potts et al. 2006), suggesting that the function of the complex during DSB repair is conserved through evolution.

Schematic representation of the cellular checkpoint response to induced DNA double-stranded breaks. Following DNA double-strand break (DSB) formation, the Mre11–Rad50–Xrs2 (MRX) complex and Sae2 are recruited to the DNA ends. The MRX complex initially processes the DSB ends and then dissociates when resection takes place. Single-stranded DNA generated by resection is bound by RP-A, which recruits the Ddc2/Mec1, Rad24/Rfc2-5 (RFC-like) and Ddc1/Rad17/Mec3 complexes. Tel1 and Mec1 checkpoint kinases phosphorylate H2A Ser 129 over a 50 kb region. Various chromatin remodelling complexes like the INO80 complex are then recruited by phosphorylated H2A. Subsequent downstream kinases such Rad53, Rad9, and Chk1 are then loaded. Their downstream functions mediate checkpoint event such as cell cycle arrest and DNA repair. Cohesin and Smc5-Smc6 complexes are also recruited de novo to DSBs and are important for downstream repair by sister chromatid recombination
The relationship between cohesin and Smc5-Smc6 at DNA DSBs has also been explored. Defects in DSB repair observed in cells depleted of hNse2 and the cohesin subunit hScc1 are epistatic, suggesting that the two complexes are required for the same pathway of recombination repair (Potts et al. 2006). Interestingly, the depletion of hSmc5 or hNse2 abolishes the recruitment of cohesin to DNA breaks (Potts et al. 2006). These observations support a model in which Smc5-Smc6 promotes sister chromatid recombination by recruiting cohesin to break sites. The molecular mechanism might involve Nse2-dependent sumoylation, since hNse2 stimulates the sumoylation of the two cohesin subunits hScc1 and hSA2 (Potts et al. 2006), and hNse2 depletion prevents the recruitment of cohesin to DSBs (Potts et al. 2006). A model involving Smc5-Smc6-dependent cohesin recruitment to DSBs is particularly attractive and would provide an elegant explanation for the striking similarity of the DSB repair defects in mutants of both complexes. Nevertheless, to date it is not clear whether the absence of cohesin from the break in Smc5-Smc6 depleted cells is a direct or indirect effect. Interestingly, depletion of hNse2 also abolishes the recruitment of hSmc5 to DSBs. Since the Smc5-Smc6 complex is stable in the absence of hNse2 (Taylor et al. 2008), it is possible that sumoylation of a third factor or other hNse2-mediated activities might be required for the recruitment of both cohesin and Smc5-Smc6 independently. It would be very informative to test whether a version of Scc1 that cannot be sumoylated failed to be loaded to DSBs and whether a catalytic-dead Nse2 would prevent the recruitment of Smc5-Smc6 to the break.
Observations in fission yeast indicate that smc6-74 rad21-45 (scc1) double mutants are more sensitive to gamma radiation than either single mutant, suggesting that these complexes do not function in totally overlapping DNA repair pathways (Verkade et al. 1999). Analysis of the factors required for Smc5-Smc6 loading to DSBs in nocodazole-arrested cells suggests that Smc5-Smc6 and cohesin are loaded via different mechanisms. Cohesin loading requires the helicase-endonuclease Mre11, a subunit of the MRX (MRN in higher eukaryotes) complex, the checkpoint kinases Mec1 and Tel1, their activity on H2AX, and the effector kinase Rad53. Smc5-Smc6 loading depends on Mre11, but not Mec1 and Rad53 (Lindroos et al. 2006). However, recent reports in budding yeast support the view that Smc5-Smc6 is required for cohesion establishment at DSBs; the smc6-56 mutant fails to promote loading of cohesion-proficient cohesin on undamaged chromosomes in response to DNA damage checkpoint activation (Strom et al. 2007). In contrast to what is reported in human cells, the yeast smc6-56 mutant affects the activation of cohesin rather than its loading to break sites (Strom et al. 2007). This result is puzzling because other factors, like the loader Scc2/4 complex (Strom et al. 2007) or the acetyltransferase Eco1 (Ünal et al. 2007), produce cohesion defects (Ciosk et al. 2000; Lengronne et al. 2006).
An interesting question is the exact role of Smc5-Smc6 during sister chromatid recombination. In addition to cohesion establishment, Smc5-Smc6 could restrain the freedom of movement of the DSB ends and recruit them to specialized regions of the nucleus. This would shelter the break from regions of homology present elsewhere in the genome, thereby favouring repair by sister chromatid recombination. In the absence of such a re-localization, allelic or ectopic sequences could compete with the sister chromatid sequence, promoting alternative donor sequence choice. Although these hypotheses are very speculative, recent studies support the possibility that Smc5-Smc6 plays a role in nuclear organization during DSB repair. In telomerase-negative human cell lines, the Nse2-dependent sumoylation of multiple telomere binding proteins (TRF1, TRF2, RAP1, and TIN2) is required to promote the homologous recombination-mediated alternative lengthening of telomeres (ALT) pathway. This form of telomere maintenance regulates telomere length in the absence of telomerase, thus inhibiting senescence (Potts and Yu 2007). Interestingly, in cells depleted of subunits of the Smc5-Smc6 complex, telomeres fail to be recruited to ALT-associated PML bodies (APB) and telomere shortening is observed. Furthermore, lack of telomere localization to the APB is observed in cells carrying an unsumoylable version of TRF1 and TRF2 (Potts and Yu 2007). The role of Nse2 in telomere localization seems to be conserved, since yeast Nse2 sumo ligase mutants fail to localize telomeres to the nuclear envelope (Zhao and Blobel 2005).
Smc5-Smc6 also plays a role during DSBs repair in the rDNA region by controlling the localization of the break (Torres-Rosell et al. 2007b). In budding yeast, DSBs occurring inside the rDNA are re-localized outside the nucleolus, as observed by the extremely low levels of Rad52 foci formed inside the nucleolus following DSB production by endonucleases or gamma radiation (Torres-Rosell et al. 2007b). The re-localization of the break is thought to promote sister chromatid recombination over other mechanisms by moving the broken ends away from other rDNA copies of the array (thus preventing non-sister recombination events). Remarkably, smc6-9 mutants are defective in the re-localization of DSBs induced inside the rDNA to positions outside the nucleolus (Fig. 3), demonstrating that the Smc5-Smc6 complex carries out a nuclear organization/compartmentalization function during DSB repair (Torres-Rosell et al. 2007b). As a consequence, hypomorphic alleles of Smc5-Smc6 show decreased levels of equal sister chromatid recombination compared with unequal and/or intrachromatid recombination in the rDNA. While these defects could be seen as a failure to promote active movement of the break outside the nucleolus, it could also be an indirect consequence of a general loss of organization of the nucleolus region in the absence of Smc5-Smc6 (Zhao and Blobel 2005), a general slowdown of the repair process (Murray and Carr 2008), or a direct failure to prevent the binding of Rad52 to breaks still located inside the nucleolus.

Smc5-Smc6 is involved in the re-localization of DNA DSBs occurring inside the rDNA to regions outside the nucleolus. In S. cerevisiae, the nucleolus (yellow) forms a crescent-shaped structure at the nuclear periphery that contains the rDNA genes located in the middle of chromosome XII (black; top left diagram). DSBs generated in the rDNA array are relocated to the nucleus in order to be repaired by Rad52-mediated recombination. In the absence of Smc5-Smc6 function, the DSB is not relocated to the nuclear compartment and undergoes Rad52-mediated repair in the nucleolus. This can result in abnormal repair, due to the repetitive nature of the locus, and to rDNA instability
Presently, it is not known whether DSB targeting to specific nuclear compartments occurs in response to DSBs in regions other than telomeres and the rDNA. The observation that multiple breaks localize to a single repair centre, seen as a large focus of recombination proteins in the cell, suggests that DSB ends can move to different positions within the nucleus in order to be repaired (Lisby et al. 2003b). Interestingly, recent data indicate that DSB repair at subtelomeric regions is affected by deletion of the components of the nuclear pore complex, raising the possibility that the location of DSBs towards the periphery of the nucleus might be important for its repair (Loeillet et al. 2005; Therizols et al. 2006).
Smc5-Smc6 at recombination sites and replication forks
Exposure of S. pombe cells to high concentrations of MMS, HU, or UV, or the deletion of the recombination protein Rph51 increases the sensitivity of smc5-smc6 mutants. At low concentrations of the genotoxic agents, deletion of Rph51 partially suppresses the DNA damage sensitivity of these mutants, suggesting that recombination intermediates become toxic in the absence of a fully functional Smc5-Smc6 complex (Ampatzidou et al. 2006; Pebernard et al. 2006). In budding yeast, the temperature sensitivity of smc5-smc6 mutants is partially suppressed by RAD52 deletion (Torres-Rosell et al. 2005; Cost and Cozzarelli 2006). These results demonstrate that in smc5-smc6 mutants, recombination produces toxic intermediates even in the absence of damaging agents. Moreover, exposure of the nse1-101 mutant to UV damage generates postreplication repair defects that are epistatic with Rad52 (Santa Maria et al. 2007).
All smc5-smc6 mutants tested are very sensitive to the DNA damage agent MMS, which produces methylated bases that need to be repaired during the replication process. One possibility is that Smc5-Smc6 is required to prevent or resolve (in a process that depends on recombination) specific structures that occur in the presence of stalled forks after MMS treatment. Sister chromatid junctions (SCJs), which are believed to be topological links between sister chromatids formed at replication forks (Benard et al. 2001; Segurado et al. 2002; Lopes et al. 2003), accumulate in certain mutants, including sgs1 and the nse2-SUMO ligase mutant, in the presence of MMS (Liberi et al. 2005; Branzei et al. 2006). The accumulation of SCJs generates DNA intermediates, called hemicatenanes, that appear as X-shaped molecules by two-dimensional gel electrophoresis (Liberi et al. 2005). Importantly, these intermediates show different physical properties from Holliday junctions, such as resistance to HJ endonucleases and the presence of stretches of ssDNA. The formation of hemicatenanes is thought to be promoted through conversion of SCJs by template switching of the two newly synthesized strands. While establishment of SCJs is Rad52-independent (Lopes et al. 2003), the template switching process and the formation of hemicatenanes require recombination (Liberi et al. 2005). The observation that mutants of Nse2 accumulate hemicatenanes (Branzei et al. 2006) raises the possibility that Smc5-Smc6 is required for the resolution of these structures. Interestingly, Sgs1 is sumoylated in vivo in response to MMS, but its sumoylation is not dependent on Nse2. This suggests that Smc5-Smc6 and Sgs1 promote resolution and/or prevent the formation of X-shaped molecules through independent mechanisms (Branzei et al. 2006).
An alternative role for Smc5-Smc6 during replication of damaged templates would be in the processing of collapsed replication forks following dissociation of the replicative polymerase. Fork stalling is a transient event that does not cause replisome dissociation; in fact, it is believed that maintaining the integrity of the replisome-fork complex at stalled forks is a prerequisite for preventing abnormal replication at nascent chains, thus avoiding recombinogenic structures. Occasionally, the replisome might dissociate, thus causing fork collapse. Little is known about this process in eukaryotic cells. Fork collapsing has been observed only in checkpoint mutants treated with genotoxic agents. Electron microscopy experiments showed that replication fork collapse observed in rad53Δ cells arrested in HU leads to an accumulation of regressed forks, single-stranded gaps, and hemi-replicated intermediates (Sogo et al. 2002). This situation, however, might be far from physiological: fork collapse could occur even in RAD53 + cells but with less dramatic consequences. E. coli provides an important model for understanding what could happen to collapsed eukaryotic forks (Heller and Marians 2006).
In bacteria, collapsed forks are processed into regressed forks, through the pairing of the newly synthesized strands that are subsequently processed by (1) resolvase RuvABC cutting of the Holliday junction-like intermediate and recombination-dependent replication restart, (2) branch migration and replication restart, or (3) nascent leading and lagging strand degradation by an exonuclease and direct replication restart (for review see Michel et al. 2007). Smc5-Smc6 could thus be involved in some of these processes, promoting the processing of regressed forks or the resolution of DNA intermediates occurring after fork collapse. In support of this possible role, a robust accumulation of Smc5-Smc6 at forks in cells deleted of RAD53 and arrested in HU has been detected (Lindroos et al. 2006). Since arrested forks are not stabilized in the absence of Rad53 and thus undergo collapse, the accumulation of Smc5-Smc6 at these sites indicates that the complex is recruited to collapsed forks (Lindroos et al. 2006). Remarkably, observations in S. pombe indicate that smc6-X cds1 (rad53) and smc6-74 cds1Δ cells arrested in HU accumulate replication fork structures and X-shaped molecules (Ampatzidou et al. 2006).
These results are reminiscent of the behaviour observed in the exonuclease EXO1 deleted strains of budding yeast. Exo1 is a DNA nuclease recruited to collapsed forks, where it promotes the degradation of regressed forks. Indeed, the Y molecule arc is maintained and X-shaped molecules accumulate in exo1Δ cells after fork collapse (Cotta-Ramusino et al. 2005). Therefore, the Smc5-Smc6 complex might function at collapsed forks by promoting their processing either directly or indirectly. The complex could regulate different factors required for the remodelling of collapsed forks and/or recombinational repair/restart, or it could provide structural organization. Accordingly, smc5-smc6 mutations are lethal in S. pombe when combined with deletions of genes encoding other DNA exonuclease, like RAD2, SWI1, and APN2. This suggests that in smc5-smc6 mutants DNA accumulates intermediate structures that critically depend on exonuclease activity for their resolution (Lee et al. 2007). In addition, smc5-smc6 mutants also show synthetic growth defects when combined with deletion of genes encoding DNA endonucleases required for the processing of abnormal fork structures, such as MUS81, MMS4 (EME1 in S. pombe), SLX4-SLX1, and SGS1 (Morikawa et al. 2004; Pebernard et al. 2004; Torres-Rosell et al. 2005).
Smc5-Smc6 and the ribosomal DNA array
Several studies have focused on the consequences of Smc5-Smc6 defects for ribosomal DNA (rDNA) replication and stability. Smc5-Smc6 is enriched on the ribosomal gene array in budding and fission yeasts (Torres-Rosell et al. 2005; Ampatzidou et al. 2006). In S. cerevisiae, rDNA segregation is defective when Smc5-Smc6 function is compromised (Torres-Rosell et al. 2005). Surprisingly, the abolition of recombination does not suppress the segregation defects of smc5-smc6 mutants (Torres-Rosell et al. 2007a), suggesting that non-disjunction of the rDNA is independent of faulty recombination. Analysis of the rDNA in smc5-smc6 mutants by two-dimensional gel electrophoresis and chromosome re-entry by PFGE of metaphase-arrested cells demonstrated that these mutants are unable to finish replication of the rDNA before metaphase execution (Torres-Rosell et al. 2007a). Therefore, the observed non-disjunction phenotype is caused by incomplete replication of the rDNA locus (Torres-Rosell et al. 2007a). The non-disjunction phenotype can be suppressed using conditions that facilitate replication through the locus; e.g. inactivating the polar barrier protein Fob1, which prevents the arrest of leftward moving forks, and inactivating Pol-I transcription, which reduces the amount of protein complexes bound to the rDNA template (Torres-Rosell et al. 2007a). Since no collapsed forks are detected in the rDNA of smc5-smc6 cells (Torres-Rosell et al. 2007a), it is likely that the defect of these mutants is caused by the inability of forks to progress through obstacles such as tightly bound protein-DNA complexes, or a specific defect in the resolution/termination of converging and RFB-arrested forks. Surprisingly, DNA replication intermediates within the rDNA locus are not detected by the DNA damage or replication checkpoints in smc5-smc6 cells, despite the fact that these checkpoints are fully competent in the mutants (Torres-Rosell et al. 2007a).
So why do smc5-smc6 cells fail to terminate replication at the rDNA, yet no checkpoint is activated? It is generally agreed that checkpoint sensors require the presence of a DSB and/or primed ssDNA for the activation of the downstream kinases that enforce the cell cycle arrest and assist the repair response (Zou 2007). Since smc5-smc6 mutants show wild-type efficiency in checkpoint activation (Torres-Rosell et al. 2007a), it is reasonable to speculate that none of the elements recognized by the sensors are present in smc5-smc6 cells at the time of mitosis. Therefore, the forks present at the rDNA in these mutants are likely to be arrested with little ssDNA exposed, at least insufficient for the binding of multiple RPA proteins required for triggering the checkpoint. Mapping of the leftward-moving forks arrested at the RFB indicates that hardly any ssDNA is exposed (Gruber et al. 2000). The progression of the rightward moving fork could be affected by the topological stress associated with rRNA transcription; in this case, the unwinding of the DNA by the replicative helicase would be impaired and no long stretches of DNA would be exposed. As a consequence of defects in fork progression and/or replication termination, forks in smc5-smc6 cells might lack checkpoint triggering features, thus allowing these cells to enter mitosis before they complete replication of the rDNA locus. This will consequently impede chromosome segregation. The implications of these findings are that eukaryotic cells cannot block mitosis when only a few ongoing (without damage signals) forks are present (Torres-Rosell et al. 2007a).
Another possible explanation for the mitotic entry before replication completion observed in smc5-smc6 mutants is that this complex might be both required for resolution of replication intermediates and the activation of an uncharacterized checkpoint that responds to their presence. This is an interesting possibility that should be explored in the future.
One complex, too many functions?
From the literature on Smc5-Smc6, the complex appears to function in the context of DNA replication and recombination. It acts as a troubleshooter by assisting recombination at DSBs and regulating the stability of the rDNA, in both recombination-independent and -dependent manners. It also seems to be important to promote replication termination and/or process the resolution of DNA intermediates genome-wide in the presence of damaged templates. Moreover, Smc5-Smc6 might be involved in cohesin loading, chromosome condensation, telomere maintenance, and much more. Are these not too many functions for a single protein complex?
It is conceptually difficult to imagine how one complex could promote different activities according to the context. Since, thus far, the characterization of the Smc5-Smc6 complex has been mainly phenotypical, it is not possible to exclude that some of the defects detected in smc5-smc6 mutants might be indirect. Complexity could arise from properties that emerge from the action of a simple activity in different chromosomal contexts, more than a complex activity per se. Assuming that Smc5-Smc6, like the other SMC complexes, plays a structural role in chromosome maintenance, some of the defects observed might also stem from the disruption of local or global chromosome structure.
Nevertheless, it might be difficult to understand Smc5-Smc6 function if the complex regulates more than one pathway. Some clues indicate that the complex might indeed play a multidimensional role in chromosome metabolism by being involved in different pathways. Smc5-Smc6 exhibits a variety of enzymatic activities, including sumoylation and possibly ubiquitination, with a growing body of evidence indicating that these modifications play key roles in the regulation of chromosome stability (Zhao 2007). In this sense, Smc5-Smc6 might work as a polyfunctional association platform able to recruit different effector proteins, thus promoting alternative processing of recombination and replication intermediates. In addition, modification of target proteins not directly associated with the complex could promote and regulate other functions, explaining some of the seemingly unrelated phenotypes observed. It will be of great importance to dissect the different pathways affected in smc5-smc6 mutants, and careful biochemical and genetic analysis will be required to uncover the various functions of the Smc5-Smc6 complex. Moreover, future work will need to focus on the mechanisms controlling Smc5-Smc6 activity in different contexts. How are sumoylation/ubiquitination activated after DNA damage? How is Smc5-Smc6 loaded and removed from chromatin? What are the biochemical activities of the complex on chromatin? What are the targets for its enzymatic activities? Analysis of the Smc5-Smc6 complex is still at its early stages, but it looks like an exciting ride is waiting ahead.
Abbreviations
- ALT:
-
alternative lengthening of telomeres
- APB:
-
ALT-associated PML bodies
- ChIP:
-
chromatin immunoprecipitation
- DSB:
-
double-strand break
- HR:
-
homologous recombination
- HU:
-
hydroxyurea
- MAGE:
-
melanoma antigen gene
- MMS:
-
methyl methanesulfonate
- NHEJ:
-
non-homologous end joining
- NSE:
-
non-SMC elements
- PFGE:
-
pulsed-field gel electrophoresis
- rDNA:
-
ribosomal DNA
- RFB:
-
replication fork barrier
- RING:
-
Zn finger domain
- SCJ:
-
sister chromatid junctions
- SMC:
-
structural maintenance of chromosome
- ssDNA:
-
single-stranded DNA
- SUMO:
-
small ubiquitin-like modifier
- UV:
-
ultraviolet
References
Ampatzidou E, Irmisch A, O’Connell MJ, Murray JM (2006) Smc5/6 is required for repair at collapsed replication forks. Mol Cell Biol 26:9387–9401
Andrews EA, Palecek J, Sergeant J, Taylor E, Lehmann AR, Watts FZ (2005) Nse2:a component of the Smc5–6 complex, is a SUMO ligase required for the response to DNA damage. Mol Cell Biol 25:185–196
Aylon Y, Kupiec M (2004) DSB repair: the yeast paradigm. DNA Repair (Amst) 3:797–815
Barker PA, Salehi A (2002) The MAGE proteins: emerging roles in cell cycle progression, apoptosis, and neurogenetic disease. J Neurosci Res 67:705–712
Benard M, Maric C, Pierron G (2001) DNA replication-dependent formation of joint DNA molecules in Physarum polycephalum. Mol Cell 7:971–980
Bennett CB, Lewis AL, Baldwin KK, Resnick MA (1993) Lethality induced by a single site-specific double-strand break in a dispensable yeast plasmid. Proc Natl Acad Sci U S A 90:5613–5617
Branzei D, Sollier J, Liberi G et al (2006) Ubc9- and mms21-mediated sumoylation counteracts recombinogenic events at damaged replication forks. Cell 127:509–522
Caspari T, Murray JM, Carr AM (2002) Cdc2-cyclin B kinase activity links Crb2 and Rqh1-topoisomerase III. Genes Dev 16:1195–1208
Ciosk R, Shirayama M, Shevchenko A, Tanaka T, Toth A, Nasmyth K (2000) Cohesin’s binding to chromosomes depends on a separate complex consisting of Scc2 and Scc4 proteins. Mol Cell 5:243–254
Cobbe N, Heck MM (2004) The evolution of SMC proteins: phylogenetic analysis and structural implications. Mol Biol Evol 21:332–347
Cortes-Ledesma F, Aguilera A (2006) Double-strand breaks arising by replication through a nick are repaired by cohesin-dependent sister-chromatid exchange. EMBO Rep 7:919–926
Cost GJ, Cozzarelli NR (2006) Smc5p promotes faithful chromosome transmission and DNA repair in Saccharomyces cerevisiae. Genetics 172:2185–2200
Cotta-Ramusino C, Fachinetti D, Lucca C et al (2005) Exo1 processes stalled replication forks and counteracts fork reversal in checkpoint-defective cells. Mol Cell 17:153–159
Daley JM, Palmbos PL, Wu D, Wilson TE (2005) Nonhomologous end joining in yeast. Annu Rev Genet 39:431–451
De Piccoli G, Cortes-Ledesma F, Ira G et al (2006) Smc5-Smc6 mediate DNA double-strand-break repair by promoting sister-chromatid recombination. Nat Cell Biol 8:1032–1034
Esashi F, Christ N, Gannon J et al (2005) CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature 434:598–604
Fujioka Y, Kimata Y, Nomaguchi K, Watanabe K, Kohno K (2002) Identification of a novel non-structural maintenance of chromosomes (SMC) component of the SMC5-SMC6 complex involved in DNA repair. J Biol Chem 277:21585–21591
Geiss-Friedlander R, Melchior F (2007) Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol 8:947–956
Gonzalez-Barrera S, Cortes-Ledesma F, Wellinger RE, Aguilera A (2003) Equal sister chromatid exchange is a major mechanism of double-strand break repair in yeast. Mol Cell 11:1661–1671
Gruber M, Wellinger RE, Sogo JM (2000) Architecture of the replication fork stalled at the 3′ end of yeast ribosomal genes. Mol Cell Biol 20:5777–5787
Heller RC, Marians KJ (2006) Replisome assembly and the direct restart of stalled replication forks. Nat Rev Mol Cell Biol 7:932–943
Ira G, Pellicioli A, Balijja A et al (2004) DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431:1011–1017
Kaye JA, Melo JA, Cheung SK, Vaze MB, Haber JE, Toczyski DP (2004) DNA breaks promote genomic instability by impeding proper chromosome segregation. Curr Biol 14:2096–2106
Lee KM, Nizza S, Hayes T et al (2007) Brc1-mediated rescue of Smc5/6 deficiency: requirement for multiple nucleases and a novel Rad18 function. Genetics 175:1585–1595
Lehmann AR, Walicka M, Griffiths DJ et al (1995) The rad18 gene of Schizosaccharomyces pombe defines a new subgroup of the SMC superfamily involved in DNA repair. Mol Cell Biol 15:7067–7080
Lengronne A, Katou Y, Mori S et al (2004) Cohesin relocation from sites of chromosomal loading to places of convergent transcription. Nature 430:573–578
Lengronne A, Mc Intyre J, Katou Y et al (2006) Establishment of sister chromatid cohesion at the S. cerevisiae replication fork. Mol Cell 23:787–799
Liberi G, Maffioletti G, Lucca C et al (2005) Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev 19:339–350
Lindroos HB, Strom L, Itoh T, Katou Y, Shirahige K, Sjogren C (2006) Chromosomal association of the Smc5/6 complex reveals that it functions in differently regulated pathways. Mol Cell 22:755–767
Lisby M, Antunez de Mayolo A, Mortensen UH, Rothstein R (2003a) Cell cycle-regulated centers of DNA double-strand break repair. Cell Cycle 2:479–483
Lisby M, Mortensen UH, Rothstein R (2003b) Colocalization of multiple DNA double-strand breaks at a single Rad52 repair centre. Nat Cell Biol 5:572–577
Lobachev K, Vitriol E, Stemple J, Resnick MA, Bloom K (2004) Chromosome fragmentation after induction of a double-strand break is an active process prevented by the RMX repair complex. Curr Biol 14:2107–2112
Loeillet S, Palancade B, Cartron M et al (2005) Genetic network interactions among replication, repair and nuclear pore deficiencies in yeast. DNA Repair (Amst) 4:459–468
Lopes M, Cotta-Ramusino C, Liberi G, Foiani M (2003) Branch migrating sister chromatid junctions form at replication origins through Rad51/Rad52-independent mechanisms. Mol Cell 12:1499–1510
Melo JA, Cohen J, Toczyski DP (2001) Two checkpoint complexes are independently recruited to sites of DNA damage in vivo. Genes Dev 15:2809–2821
Michel B, Boubakri H, Baharoglu Z, Le Masson M, Lestini R (2007) Recombination proteins and rescue of arrested replication forks. DNA Repair (Amst) 6:967–980
Morikawa H, Morishita T, Kawane S, Iwasaki H, Carr AM, Shinagawa H (2004) Rad62 protein functionally and physically associates with the smc5/smc6 protein complex and is required for chromosome integrity and recombination repair in fission yeast. Mol Cell Biol 24:9401–9413
Murray JM, Carr AM (2008) Smc5/6: a link between DNA repair and unidirectional replication? Nat Rev Mol Cell Biol 9:177–182
Nasim A, Smith BP (1975) Genetic control of radiation sensitivity in Schizosaccharomyces pombe. Genetics 79:573–582
Nasmyth K, Haering CH (2005) The structure and function of SMC and kleisin complexes. Annu Rev Biochem 74:595–648
Onoda F, Takeda M, Seki M et al (2004) SMC6 is required for MMS-induced interchromosomal and sister chromatid recombinations in Saccharomyces cerevisiae. DNA Repair (Amst) 3:429–439
Palecek J, Vidot S, Feng M, Doherty AJ, Lehmann AR (2006) The Smc5-Smc6 DNA repair complex. bridging of the Smc5-Smc6 heads by the KLEISIN, Nse4:and non-Kleisin subunits. J Biol Chem 281:36952–36959
Pâques F, Haber JE (1999) Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 63:349–404
Pebernard S, Mc Donald WH, Pavlova Y, Yates JR 3rd, Boddy MN (2004) Nse1:Nse2:and a novel subunit of the Smc5-Smc6 complex, Nse3:play a crucial role in meiosis. Mol Biol Cell 15:4866–4876
Pebernard S, Wohlschlegel J, Mc Donald WH, Yates JR 3rd, Boddy MN (2006) The Nse5-Nse6 dimer mediates DNA repair roles of the Smc5-Smc6 complex. Mol Cell Biol 26:1617–1630
Pebernard S, Perry JJ, Tainer JA, Boddy MN (2008) Nse1 RING-like domain supports functions of the Smc5-Smc6 holocomplex in genome stability. Mol Cell Biol 19:4099–4109
Potts PR, Yu H (2005) Human MMS21/NSE2 is a SUMO ligase required for DNA repair. Mol Cell Biol 25:7021–7032
Potts PR, Yu H (2007) The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat Struct Mol Biol 14:581–590
Potts PR, Porteus MH, Yu H (2006) Human SMC5/6 complex promotes sister chromatid homologous recombination by recruiting the SMC1/3 cohesin complex to double-strand breaks. EMBO J 25:3377–3388
Prado F, Cortes-Ledesma F, Huertas P, Aguilera A (2003) Mitotic recombination in Saccharomyces cerevisiae. Curr Genet 42:185–198
Prakash S, Prakash L (1977) Increased spontaneous mitotic segregation in MMS-sensitive mutants of Saccharomyces cerevisiae. Genetics 87:229–236
Santa Maria SR, Gangavarapu V, Johnson RE, Prakash L, Prakash S (2007) Requirement of Nse1:a subunit of the Smc5-Smc6 complex, for Rad52-dependent postreplication repair of UV-damaged DNA in Saccharomyces cerevisiae. Mol Cell Biol 27:8409–8418
Segurado M, Gomez M, Antequera F (2002) Increased recombination intermediates and homologous integration hot spots at DNA replication origins. Mol Cell 10:907–916
Sergeant J, Taylor E, Palecek J et al (2005) Composition and architecture of the Schizosaccharomyces pombe Rad18 (Smc5–6) complex. Mol Cell Biol 25:172–184
Sogo JM, Lopes M, Foiani M (2002) Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297:599–602
Strom L, Lindroos HB, Shirahige K, Sjogren C (2004) Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol Cell 16:1003–1015
Strom L, Karlsson C, Lindroos HB et al (2007) Postreplicative formation of cohesion is required for repair and induced by a single DNA break. Science 317:242–245
Taylor EM, Moghraby JS, Lees JH, Smit B, Moens PB, Lehmann AR (2001) Characterization of a novel human SMC heterodimer homologous to the Schizosaccharomyces pombe Rad18/Spr18 complex. Mol Biol Cell 12:1583–1594
Taylor EM, Copsey AC, Hudson JJ, Vidot S, Lehmann AR (2008) Identification of the proteins, including MAGEG1:that make up the human SMC5–6 protein complex. Mol Cell Biol 28:1197–1206
Therizols P, Fairhead C, Cabal GG et al (2006) Telomere tethering at the nuclear periphery is essential for efficient DNA double strand break repair in subtelomeric region. J Cell Biol 172:189–199
Torres-Rosell J, Machin F, Farmer S et al (2005) SMC5 and SMC6 genes are required for the segregation of repetitive chromosome regions. Nat Cell Biol 7:412–419
Torres-Rosell J, De Piccoli G, Cordon-Preciado V et al (2007a) Anaphase onset before complete DNA replication with intact checkpoint responses. Science 315:1411–1415
Torres-Rosell J, Sunjevaric I, De Piccoli G et al (2007b) The Smc5-Smc6 complex, SUMO modification of Rad52 regulates recombinational repair at the ribosomal gene locus. Nat Cell Biol 9:923–931
Tsuyama T, Inou K, Seki M et al (2006) Chromatin loading of Smc5/6 is induced by DNA replication but not by DNA double-strand breaks. Biochem Biophys Res Commun 351:935–939
Ünal E, Arbel-Eden A, Sattler U et al (2004) DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol Cell 16:991–1002
Ünal E, Heidinger-Pauli JM, Koshland D (2007) DNA double-strand breaks trigger genome-wide sister-chromatid cohesion through Eco1 (Ctf7). Science 317:245–248
Verkade HM, Bugg SJ, Lindsay HD, Carr AM, O’Connell MJ (1999) Rad18 is required for DNA repair and checkpoint responses in fission yeast. Mol Biol Cell 10:2905–2918
Zhao J (2007) Sumoylation regulates diverse biological processes. Cell Mol Life Sci 64:3017–3033
Zhao X, Blobel G (2005) A SUMO ligase is part of a nuclear multiprotein complex that affects DNA repair and chromosomal organization. Proc Natl Acad Sci U S A 102:4777–4782
Zou L (2007) Single- and double-stranded DNA: building a trigger of ATR-mediated DNA damage response. Genes Dev 21:879–885
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Christian Haering
Rights and permissions
About this article
Cite this article
Piccoli, G.D., Torres-Rosell, J. & Aragón, L. The unnamed complex: what do we know about Smc5-Smc6?. Chromosome Res 17, 251–263 (2009). https://doi.org/10.1007/s10577-008-9016-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10577-008-9016-8