
Abstract
L-type Ca2+ channels (LTCCs) are key elements in electromechanical coupling in striated muscles and formation of neuromuscular junctions (NMJs). However, the significance of LTCCs in regulation of neurotransmitter release is still far from understanding. Here, we found that LTCCs can increase evoked neurotransmitter release (especially asynchronous component) and spontaneous exocytosis in two functionally different compartment of the frog NMJ, namely distal and proximal parts. The effects of LTCC blockage on evoked and spontaneous release as well as timing of exocytotic events were prevented by inhibition of either protein kinase C (PKC) or P2Y receptors (P2Y-Rs). Hence, endogenous signaling via P2Y-R/PKC axis can sustain LTCC activity. Application of ATP, a co-neurotransmitter able to activate P2Y-Rs, suppressed both evoked and spontaneous exocytosis in distal and proximal parts. Surprisingly, inhibition of LTCCs (but not PKC) decreased the negative action of exogenous ATP on evoked (only in distal part) and spontaneous exocytosis. Lipid raft disruption suppressed (1) action of LTCC antagonist on neurotransmitter release selectively in distal region and (2) contribution of LTCCs in depressant effect of ATP on evoked and spontaneous release. Thus, LTCCs can enhance and desynchronize neurotransmitter release at basal conditions (without ATP addition), but contribute to ATP-mediated decrease in the exocytosis. The former action of LTCCs relies on P2Y-R/PKC axis, whereas the latter is triggered by exogenous ATP and PKC-independent. Furthermore, relevance of lipid rafts for LTCC function as well as LTCCs for ATP effects is different in distal and proximal part of the NMJ.
Graphic Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neurotransmission relies on exocytotic neurotransmitter release due to fusion of synaptic vesicles (SVs) with presynaptic membrane. Neurotransmitter release is triggered by Ca2+ influx through voltage-gated Ca2+ channels (VGCCs) anchored in sites of SV exocytosis, active zones. Some VGCCs are located relatively far from active zones and Ca2+ influx from these channels can modulate neurotransmitter release via action on resting Ca2+ levels as well as Ca2+-dependent enzymes and channels.
L-type Ca2+ channels (LTCCs), also known as Cav1, are expressed in skeletal muscle, heart, retina, and brain. LTCCs (Cav1.3 and Cav1.4), characterized by slow inactivation, operate in triggering SV exocytosis in ribbon synapses in retina and inner ear. In contrast to conventional phasic synapses, these sensory synapses are designed for analogue neurotransmission when neurotransmitter release is proportional to the changes in membrane depolarization (Dolphin and Lee 2020). Additionally, LTCCs participate in catecholamine release from chromaffin cells of adrenal glands in response to splanchnic nerve stimulation or nicotinic acetylcholine (ACh) receptor activation (Marcantoni et al. 2007). Evidently, that in most synapses, LTCCs have a modulatory role in control of neurotransmitter release. Particularly, activity of LTCCs can contribute to enhancement of SV exocytosis after learning and induction of long-term potentiation in cortical neurons (Subramanian et al. 2013). In the mammalian neuromuscular junctions (NMJs), LTCCs can participate in maintenance of exocytosis at high-frequency activity via promoting formation of new SVs by endocytosis (Perissinotti et al. 2008). Furthermore, there are silent LTCCs whose participation in ACh release in response to intense motor nerve terminal depolarization is unmasked under the conditions of KCa channel inhibition (Flink and Atchison 2003). In addition, 4-beta-phorbol ester, an activator of protein kinase C (PKC), can increase quantal ACh release due to opening of quiescent LTCCs at resting potential without apparent depolarization in motor nerve terminals (Arenson and Evans 2001). Some important presynaptic receptors can affect neurotransmitter release partially via modulation of LTCC activity. LTCCs are involved in inhibitory effect of adenosine metabolite inosine on spontaneous and evoked ACh release (Cinalli et al. 2013) as well as in excitatory effects of adenosine A2A receptor activation and muscarinic M1 receptor agonist on neurotransmitter release in NMJs (Palma et al. 2011; Oliveira and Correia-de-Sa 2005). Importantly, Ca2+ influx through LTCCs is critical for clustering of ACh receptors in the center of muscle fiber at the onset of NMJ development (Kaplan et al. 2018) as well as formation cluster of SVs and prevention of neurotransmission in immature and regenerating presynaptic nerve terminals (Chipman et al. 2014). Loss of amyloid precursor protein, important for structure and function of developing NMJs, can facilitate aberrant activity of LTCCs contributing to changes in neurotransmitter release during repetitive stimulation in mouse neuromuscular junctions (Yang et al. 2007).
The complex character of LTCC involvement in neurotransmission can be related with location of the channels in presynaptic, postsynaptic, and glial cell membranes as well as changes in the channel functioning during ontogenesis (Robitaille et al. 1996; Dolphin and Lee 2020; Kaplan et al. 2018). Additionally, distribution of LTCCs in membrane microdomains (lipid rafts) (Park and Kim 2009) as well as inclusion of the channels into different complexes within one cell (Calin-Jageman and Lee 2008) can create functional diversity of LTCCs. Important unresolved aspects related to LTCCs are about existence of the channel functional heterogeneity along the nerve terminals and possible engagement of LTCCs in control of timing of the neurotransmitter release. In addition, the implication of lipid rafts in LTCC function as well as LTCC contribution to ATP-dependent regulation of spontaneous and evoked neurotransmitter release in different regions of NMJs are open questions. To address these issues, in the present study, the changes in quantum content, asynchrony of evoked ACh release as well as alteration in spontaneous release were detected in proximal and distal parts of the frog NMJs. These parts of the motor nerve terminal are characterized by higher and low neurotransmitter release probabilities, respectively, as well as have specific features in electrogenesis, SV exo-endocytosis and regulation of neurotransmission (Tsentsevitsky et al. 2020a, 2011; Ginebaugh et al. 2020; Zefirov et al. 2003). These properties make the long frog motor nerve terminals are suitable for investigation of LTCC compartmentalization-related processes in synapses.
Methods
Animals, Preparations, and Chemicals
Frogs (Rana ridibunda) were obtained from lakes and then placed into a pool with dechlorinated flowing water (4 °C). The pool was located in the dark room under temperature- and humidity-stable conditions. All experiments were performed within autumn–winter period. After euthanasia, the cutaneous pectoris muscles were excised.
Isolated cutaneous pectoris muscle with nerve stub was pinned into a Sylgard-coated thermostatically controlled chamber (total volume-5 ml), which was continuously perfused (5 ml*min−1) with the Ringer’s solution: 113.0 mM NaCl, 2.5 mM KCl, 4.0 mM MgCl2, 0.3 mM CaCl2, 1.5 mM NaHCO3, and 5 mM HEPES; pH was adjusted to 7.3. The bathing temperature was kept at 20 ± 0.3 °C. The following drugs were used: 5 µM nitrendipine (LTCC blocker) (Palma et al. 2011; Katz et al. 1996), 1 µM (−)-Bay K 8644 (agonist of LTCCs) (Flink and Atchison 2003; Gaydukov et al. 2009), 10 nM omega-conotoxin GVIA/CgTx (selective antagonist of N-type Ca2+ channel/NTCC) (Tsentsevitsky et al. 2020a), 100 µM ATP (De Lorenzo et al. 2006; Giniatullin et al. 2015, 2005; Sokolova et al. 2003; Tsentsevitsky et al. 2011), 100 µM suramin (P2Y receptor antagonist) (De Lorenzo et al. 2006; Giniatullin et al. 2005; Sokolova et al. 2003), 2 µM chelerythrine (PKC inhibitor) (Sokolova et al. 2003; Petrov et al. 2015; Zakyrjanova et al. 2021), 100 nM iberiotoxin (selective antagonist of Ca2+-dependent K+ channels/BK channels) (Gaydukov et al. 2009), 1 mM N-acetyl-l-cysteine/NAC (antioxidant) (Giniatullin et al. 2005; Tsentsevitsky et al. 2011, 2020a), 0.5 µM CdCl2 (Tsentsevitsky et al. 2020a). The concentrations of used chemicals were selected based on previous data (including our) obtained from studies on the NMJs. All reagents were purchased from Sigma. Application of the reagents lasted for 20–40 min. In the case of evaluation of effect of one “X” drug on a background of other “Y” reagent application, the “Y” reagent was added to the bathing saline 20–40 min prior to exposure to “X” drug and remained in the working solution throughout the experiment. In some experiments, muscles were pretreated with 0.1 mM methyl-β-cyclodextrin (MβCD, lipid raft disrupting agent) for 15 min (Zakyrjanova et al. 2021), and then MβCD was removed out of the bathing solution and the muscle were rinsed for 10 min before drug application.
Electrophysiological Assays
Recording of postsynaptic electrical events, namely end-plate potentials (EPPs) and miniature EPPs (MEPPs), was carried out extracellularly as described in (Tsentsevitsky et al. 2011). Nerve stimulation at 0.5 Hz (via a suction electrode) was used to evoke EPPs. Two Ringer’s saline-filled glass microelectrodes (with a resistance of 2–3 MΩ) were placed in proximal (~ 3 to 5 µm from the end of the last myelinated segment; 1st electrode) and distal region (~ 90 to 120 µm; 2nd electrode) of the nerve terminal. In the proximal regions, an action potential (AP) had a characteristic shape (Mallart 1984), which is distinguishable from that of AP in the distal region (Fig. 1). This was another hallmark of the correct microelectrode position. The consistency of microelectrode position was monitored throughout the experiment [see in Tsentsevitsky et al. (2011)].

Effect of LTCC modulators on evoked neurotransmitter release. Change in quantum content, m (a) and P90 parameter, an indicator of synchrony of neurotransmitter release (b) after treatment with LTCC inhibitor (nitrendipine; Nitr) or activator (Bay K). Shown the changes in distal and proximal parts of the NMJs. Data are represented as circles—values in individual animals, box range—SEM, whiskers—SD. Horizontal dash line is value (100%) before inhibitor/activator application. N = 12–16 for each group. ***P ˂ 0.001, **P ˂ 0.01, *P < 0.05—compared to value prior to LTCC modulator addition. c Superimposed uni-quantal EPPs recorded extracellularly (in response to 70 stimuli) in distal and proximal region of the same nerve terminal. Stimulus artifact, presynaptic AP (spike), and synaptic delay are denoted (for Control/”Cntr” traces). In distal and proximal parts, there is different shape of the spike. Quantal content was calculated from logarithmic ratio between the number of total stimuli and number of failures (the method of “failures”). Decrease in quantal content manifests as a decrease in number of uni-quantal EPPs in response to the same number of impulses (compare the number of EPP traces in Cntr, Nitr, and Bay K). EPPs appear with different synaptic delays after spikes. Synchronous exocytotic events are characterized by a close coupling to spike (short delay), whereas asynchronous release occurs with longer and variable delay. Bottom, shown calculation of P90 (for the individual experiments) from cumulative histogram of synaptic delays of the EPPs; vertical arrowed lines denote the 90% percentile (in ms). The decrease in P90 due to LTCC inhibition indicates on synchronization of exocytotic events with action potential, i.e., suppression of asynchronous component of the release. Agonist of LTCCs has opposite effect (desynchronization of the release/increase in asynchronous exocytosis)
The timing of the single exocytotic event (degree of synchronicity with presynaptic AP) can be precisely assessed by analysis of real synaptic delays of the uni-quantal EPPs (Katz and Miledi 1965). Therefore, experiments were carried out in the presence of 0.3 mM Ca2+ and 4.0 mM Mg2+; and quantal content (m) was determined using the equation (“method of failures”): m = lnN/N0, where N is the total number of stimuli and N0 is the number of stimuli unaccompanied by EPP (synaptic failures) (Tsentsevitsky et al. 2011, 2020b; Del Castillo and Katz 1954; Katz and Miledi 1965). To receive reliable measurements, in individual experiment, 1500–2500 stimuli were applied to motor nerve prior to and after drug application. The real synaptic delays were analyzed as previously described in detail (Katz and Miledi 1965; Tsentsevitsky et al. 2011). Initially, the synaptic delay, as time from the peak of the presynaptic sodium current spike to the point corresponding to 20% rise phase of the uni-quantal EPP, was estimated (Katz and Miledi 1965; Tsentsevitsky et al. 2011). Then, indicative value (90% percentile, in ms) for dispersion of the real synaptic delay (P90) was estimated from cumulative distribution of synaptic delays [described in detail (Tsentsevitsky et al. 2011; Bukcharaeva et al. 1999)]. P90 is the interval between the minimal synaptic delay and the point at which 90% of all uni-quantal EPPs had occurred (Fig. 1). Decrease and increase in value of P90 corresponds to a synchronization and desynchronization of the exocytotic event, respectively (Bukcharaeva et al. 1999).
Lipid Raft Labeling
Cholera toxin B subunit (CTxB) conjugated to fluorescent dye AlexaFluor 488 (ThermoFisher) was used to lipid raft visualization. Fluorescent-labeled CTxB specifically interacts with GM1 ganglioside clusters, predominantly resided into the lipid rafts (Margheri et al. 2014). The muscles were exposed to CTxB (1 μg/ml) for 15 min and then rinsed out for 30 min, and after that the fluorescence was detected (Kasimov et al. 2016). Additionally, tetramethylrhodamine-labeled αBtx (a marker for NMJ) was added simultaneously with CTxB to the bathing saline. Fluorescence of CTxB/αBTX was excited at a 488/555 nm and the emission was recorded with bandpass filter of a 505–545/610–650 nm. Fluorescent images were captured from 15 to 20 different NMJs in individual muscle. The values from the NMJs were pooled to calculate mean value in each muscle. BX51WI microscope (Olympus) equipped with spinning disk confocal unit (Olympus), DIC-optics, and LumPlanPF 100xw objective were used. Images were captured by DP71 camera (Olympus) under control of cellSens software (Olympus).
Statistics
Statistical analysis was carried out using OriginPro software. Data represent the mean ± SD, where sample size (n) is the number of experiments on individual animals. The sample size is indicated in figure legends. Statistical significance was assessed by Kolmogorov–Smirnov test (P90) or paired (for values before and after drug application) two-tail t-test and Mann–Whitney U test (for comparison of two unpaired groups); in case of multiple comparisons, one-way ANOVA was used. Values of P ≤ 0.05, P ≤ 0 0.01, P ≤ 0,001 were considered as significant.
Results
Effects of Modulation of L-Type Ca2+ Channel Activity on Neurotransmitter Release
We recorded EPPs in two functionally specific compartments of the same nerve terminal, in the distal and proximal parts (Fig. 1). Inhibitor of LTCCs (nitrendipine, 5 µM) decreased quantum content (m) of EPPs in both parts to the similar degree (Fig. 1a). Surprisingly, agonist of LTCCs (Bay K, 1 µM) had the same action (Fig. 1a). In contrast to the same direction of action of antagonist and agonist on the quantum content, nitrendipine decreased P90 parameter (an indicator for dispersion of synaptic delays and asynchrony of neurotransmitter release) in distal and proximal parts, but Bay K increased P90 in the distal part of the nerve terminal (Fig. 1b). In the proximal part, there were no significant changes in P90 in response to Bay K administration.
Accordingly, activation or inhibition of LTCCs could reduce quantum content. This suggest that “optimal” LTCC activity is required to set up the level of neurotransmitter release at the endogenous content of ATP/ADP (denoted in the text as basal conditions). At the same time, asynchrony of the neurotransmitter release (in the distal part) is increased or decreased due to activation or inhibition of LTCCs, respectively. Since, LTCC is a positive regulator of asynchronous exocytosis.
The suppression of asynchronous neurotransmitter release is a typical response to inhibition of VGCCs that control neurotransmitter release. Likely under these conditions, only “most active” SVs docked closely to the VGCC cluster preferably undergo exocytosis upon arriving AP (Tsentsevitsky et al. 2020a). Indeed, inhibition of main N-type of VGCCs (with 10 nM CgTx) in active zone decreased P90 parameter as well as quantum content of EPPs in both distal and proximal parts of the nerve terminal (S. Fig. 1A). Theoretically, the changes in both quantal content and timing of the neurotransmitter release in response to modulation of LTCCs can be directly Ca2+-dependent or rely on stimulation of Ca2+-dependent enzymes or channels. For example, BK channels can be activated by Ca2+ influx via VGCCs (including L-type) and regulate bidirectionally neurotransmitter release in synapses, including NMJs (Wang et al. 2020; Xu and Slaughter 2005; Flink and Atchison 2003; Liu et al. 2007). To test a possible link of LTCCs and BK channels, the latter were blocked with iberiotoxin (100 nM). Preliminary inhibition of BK channels with iberiotoxin suppressed the effects of nitrendipine on quantum content and P90 in both distal and proximal part of NMJs. At the same time, iberiotoxin itself had no significant influence on quantum content and neurotransmitter release timing (S. Fig. 1B, C). Hence, LTCC-dependent BK channel activation can contribute to facilitation of neurotransmitter release and its desynchronization.
These results focus on dual role of LTCCs in regulation of neurotransmitter release in the NMJs. Probably, activity of LTCCs might increase or decrease evoked quantum secretion in context-dependent manner and there are different population of LTCCs in NMJs.
Contribution of LTCCs to Depressant Effect of Exogenous ATP on Neurotransmitter Release
ATP is a main co-neurotransmitter in vertebrate NMJs. Application of ATP (100 µM) decreased quantum content of EPP in distal (less) and proximal (more profound) regions of the NMJ (Fig. 2a). To test a dependence of this effect on LTCCs, ATP was added into the bathing solution after preliminary inhibition or activation of LTCCs with nitrendipine or Bay K, respectively. Inhibition of LTCCs attenuated the depressant effect of ATP, whereas agonist of LTCCs potentiated the ATP-induced suppression of neurotransmitter release in the distal part (Fig. 2a). At the same time, modulation of LTCCs had no marked influence on ATP-mediated decrease in quantum content in the proximal part. ATP itself and under the conditions of preliminary LTCC activity modulation did not affect the timing of neurotransmitter release (Fig. 2b). Hence, LTCCs can markedly augment ATP-mediated depression of neurotransmitter release specifically in distal part. Probably, ATP-driven LTCC activation can suppress quantum release, without affecting timing of the release.

Crosstalk between LTCCs and ATP-sensitive P2Y-Rs in control of evoked neurotransmitter release in distal and proximal parts of NMJ. a, b The effects of exogenous ATP on quantum content (a) and timing of neurotransmitter release, P90 (b) in control (Cntr) and after pre-treatment with LTCC antagonist (Nitr) or agonist (Bay K). 100% is value before ATP application (dash line). Data pointing to inhibitory effect of LTCC activity on evoked secretion are highlighted with gray arrows (a). c, d Changes in quantum content (c) and P90 (d) due to P2Y-R inhibition with suramine (Sur) and in response to ATP or nitrendipine application on background of preliminary P2Y-R inhibition (ATP + Sur or Nitr + Sur). Data are expressed as circles—values in individual animals, box range—SEM, whiskers—SD. N = 5–12 for each group. ***P ˂ 0.001, **P ˂ 0.01, *P < 0.05—compared to value prior to drug application or between two groups
Notably, that selective blockage of N-type of VGCCs (a main type accountable for triggering evoked exocytosis) as well as pretreatment with Cd2+ at very low concentration (0.5 µM), in which it seems to affect the main VGCC subtype, did not modulate the depressant action of ATP in proximal part of NMJ and facilitated ATP effect in the distal region (S. Fig. 2a). This points to different regulation of neurotransmitter release in the distal and proximal regions as well as opposite roles of NTCCs and LTCCs in ATP action in the distal region of nerve terminal.
In the NMJs, endogenous and exogenous ATP act mainly via P2Y metabotropic receptors, particularly P2Y12/13 (Giniatullin et al. 2015; Guarracino et al. 2016). Indeed, an inhibitor of P2Y-R, suramin (100 µM), increased quantum content of EPPs and blocked the effect of ATP administration in both distal and proximal parts (Fig. 2c). This suggests occurrence of depressant action of endogenous ATP on evoked neurotransmitter release. Importantly, when P2Y-Rs were blocked with suramin, inhibitor of LTCCs lost ability to affect the quantum content of EPP in the distal and proximal parts (Fig. 2c). At the same time, when purine receptors were preliminarily stimulated by exogeneous ATP, nitrendipine still suppressed quantum release in both proximal and distal part of NMJs (S. Fig. 2b). These data indicate that stimulation of P2Y-Rs is required for maintenance of LTCC activity, and on background of P2Y-R inactivation, LTCCs do not contribute to neurotransmitter release significantly. Consistent with this possibility is that P2Y-R inhibition prevented the effect of LTCC antagonist on timing of neurotransmitter release (Fig. 2d). It should be noted that like ATP, suramin alone or in combination with ATP had no effect on timing of the neurotransmitter release (Fig. 2d).
The complexity of LTCC function manifests as opposite influence of these channel modulation on neurotransmitter release in different contexts. Obtained data suggest that in the basal conditions, stimulation of P2Y-Rs with endogenous ligands supports the overall activity of LTCCs, and the latter can maintain the certain level of neurotransmitter release along the entire nerve terminal as well as increase asynchronous release. At the same time, suppression of the neurotransmitter release by exogenous ATP partially relies on LTCC activation in the distal part of NMJ. In the latter case, LTCCs are negative regulator of neurosecretion, but they cannot modulate a timing of the neurotransmitter release under these conditions. Probably that different functional populations of LTCCs as well as receptors for ATP exist in the distal part of NMJs.
Protein Kinase C (PKC) in Effects of LTCC Blocker
Presynaptic effects of ATP and adenosine as well as ACh in NMJs can be partially related with PKC (Galkin et al. 2001; Santafe et al. 2006; Branisteanu et al. 1989). Moreover, this protein kinase can positively modulate LTCCs in NMJs (Fu and Huang 1994; Urbano et al. 2001). Hence, we compared the effects of PKC and LTCC inhibition. Antagonist of PKC (chelerythrine, 2 µM) had the same effect on quantum content of EPPs as LTCC blocker (Fig. 3a). Furthermore, preliminary blockage of PKC completely prevented effect of nitrendipine in both proximal and distal parts of NMJs (Fig. 3a). Importantly, chelerythrine synchronized neurotransmitter release (but only in the distal part of the NMJs) and abolished effects of nitrendipine on timing of the neurotransmitter release (Fig. 3b). Thus, PKC activity can be required for maintaining LTCC activity, which is important for regulation of the quantum content and timing of the neurotransmitter release.

PKC in maintenance of LTCC activity in distal and proximal parts of nerve terminal. a, b Changes in quantal content, m (a) and synchronicity of the release, P90 (b) due to PKC inhibition with chelerythrine (Chel) or in response to LTCC blockage after preliminary inhibition of PKC (Nitr + Chel). The effect of LTCC antagonist itself (Nitr; from Fig. 1) is shown on a and b. Data are expressed as circles—values in individual animals, box range—SEM, whiskers—SD. N = 5–14 for each group. ***P ˂ 0.001, **P ˂ 0.01, *P < 0.05—compared to value (100%) prior to drug application
Surprisingly, that in contrast to LTCC blockage, inhibition of PKC with chelerythrine did not significantly affect the reduction of neurotransmitter release due to exogenous ATP application in both the distal and proximal parts of NMJ (S. Fig. 3). At the same time, blockage of LTCCs on background of PKC inhibition attenuated the depressant effect of exogenous ATP. This suggests that exogenous ATP can decrease quantum content via PKC-independent and LTCC-dependent pathway (S. Fig. 3).
LTCCs in Spontaneous Neurotransmitter Release
Spontaneous and evoked modes of neurotransmitter release can be regulated by different mechanisms in NMJs (Petrov et al. 2014; Liu et al. 2019; Rodrigues et al. 2013). Particularly, spontaneous release can be Ca2+-independent or rely on bulk intracellular Ca2+ levels (Kaeser and Regehr 2014). Antagonist of LTCCs nitrendipine decreased frequency of MEPPs (indicator of spontaneous neurotransmitter release) in both proximal and distant part of the NMJs (Fig. 4a). At the same time, agonist of LTCCs had no influence on MEPP frequency (due to high variability in data). Notably, inhibitor of PKC decreased MEPP frequency (in distal part) and completely prevented additional action of the LTCC blocker on this parameter in both distal and proximal parts (S. Fig. 4A). Thus, like in case with evoked exocytosis, a tonic activity of PKC–LTCC axis may be required for maintenance of spontaneous release in the NMJs.

LTCC-dependent regulation of spontaneous release in distal and proximal part. a Effect of LTCC antagonist (Nitr) and agonist (Bay K) on frequency of MEPPs (in %). Right, typical MEPPs recorded extracellularly. b LTCC contribution in ATP and P2Y-R-driven control of spontaneous release. Shown changes in MEPP frequency in response to ATP administration in control (Cntr) and after preliminary inhibition (+ Nitr) or activation (+ Bay) of LTCCs. Also, influence of P2Y-R inhibition with suramine (Sur) and LTCC inhibition on background of pre-treatment with P2Y-R antagonist (Nitr + Sur) are displayed. Data pointing to inhibitory effect of LTCC activity on spontaneous secretion are highlighted with gray arrows (b). Data are expressed as circles—values in individual animals, box range—SEM, whiskers—SD. N = 4–15 for each group. **P ˂ 0.01, *P < 0.05—compared to value before drug application
Exogenous ATP suppressed spontaneous release in both parts of NMJs (Fig. 4b). If LTCCs were inhibited, ATP lost ability to suppress spontaneous neurotransmitter release, whereas the effect of ATP was completely preserved in conditions of LTCC agonist action (Fig. 4b). Furthermore, ATP increased spontaneous release in the distal part if LTCCs were blocked (Fig. 4b). This suggests that activity of LTCCs is engaged in ATP-driven suppression of spontaneous neurotransmitter release. It should be noted that inhibition of NTCCs or Cd2+ at low concentration decreased frequency of MEPPs (S. Fig. 4B), but did not prevent the depressant action of ATP on spontaneous release (S. Fig. 4C). The similar situation was observed on background of PKC inhibition (S. Fig. 4C). These results suggest that NTCCs as well as PKC can enhance spontaneous release, but, in contrast to LTCCs, they were not involved in the depressant action of ATP. This points to PKC-independent action of ATP on spontaneous exocytosis in the NMJ.
In contrast to ATP, inhibition of P2Y-Rs with suramin increased spontaneous neurotransmitter release in proximal part of the NMJs (Fig. 4b). Also, on background of P2Y-R inhibition, nitrendipine did not affect frequency of MEPPs in both distal and proximal parts (Fig. 4b). This also points to dependence of LTCC inhibitor effect on P2Y-R activation and the requirement of endogenous ATP/ADP-driven (basal) P2Y-R activity for maintenance of LTCC function.
Hence, as in case with evoked quantum secretion, LTCCs can have opposite influence on spontaneous release at basal conditions and in response to exogenous ATP. At endogenous levels of ATP, LTCCs could positively regulate spontaneous release, but ATP-driven activation of LTCCs can be engaged in attenuation of spontaneous exocytosis. Indeed, if spontaneous release was preliminarily suppressed with ATP, subsequent application of nitrendipine was able to further decrease the frequency of MEPPs, indicating that separate set of LTCCs can support spontaneous release under these conditions (S. Fig. 4C).
Previously, we have found that increase in reactive oxygen species production can partially mediate depressant effect of ATP on evoked release in the frog NMJs (Giniatullin et al. 2005). However, an antioxidant NAC (1 mM) was not able to attenuate effect of ATP on the spontaneous release in both proximal and distal parts of the NMJs (S. Fig. 4C). This makes LTCCs as a potential key element in ATP-mediated suppression of spontaneous release at the NMJs.
Role of Lipid Rafts in LTCC Function
A number of ion channels and receptors, including P2Y12, can reside in lipid rafts in synapses, including NMJs (Krivoi and Petrov 2019; Giniatullin et al. 2015; Petrov et al. 2011a). Relocation of channels or receptors from raft to non-raft membranes can markedly affect their functions. Different role of LTCCs (under basal condition vs exogenous ATP administration) in neurotransmitter release can be linked with distribution of the channel as well as P2Y-Rs into lipid rafts. To test lipid raft-dependence of the effects of LTCC inhibition by its own and in combination with ATP, pre-treatment with methyl-β-cyclodextrin (MβCD) at low concentration (0.1 mM) was used to lipid raft disruption. This approach is effective in vertebrate NMJs (Giniatullin et al. 2015; Kasimov et al. 2015; Mukhutdinova et al. 2018; Petrov et al. 2011b). Indeed, staining with a lipid raft marker CTxB was markedly decreased by 15-min exposure to 0.1 mM MβCD (S. Fig. 5A).
After MβCD-pretreatment, inhibition of LTCCs lost the ability to suppress quantum content selectively in the distal part of NMJs, whereas effect of nitrendipine was preserved in the proximal region (Fig. 5a). Surprisingly, synchronization of neurotransmitter release due to LTCC blockage was unaffected by the lipid raft disruption (Fig. 5a). Also, nitrendipine still suppressed spontaneous release in proximal part in MβCD-preexposed muscles (Fig. 5a). There was a trend to decrease in the depressant action of LTCC blocker on spontaneous release in distal part after lipid raft disruption. Accordingly, raft disruption can attenuate activity of LTCC specifically in distal part of nerve terminals. Along the same lines, pretreatment with raft disrupting agent prevented effect of LTCC blocker on quantum content on background of preliminary ATP administration, which normally might facilitate LTCC activity (S. Fig. 5B).

Role of lipid rafts in LTCC-dependent regulation of neurotransmitter release. a Effect of LTCC antagonist (Nitr) on parameters of evoked (m and P90) and spontaneous (MEPP frequency) release in control (Cntr; from Figs. 1 and 4a) and MβCD-pretreated muscles. b, c Influence of lipid raft disruption with MβCD on the effects of exogenous ATP in native sample (b) and muscle pretreated with LTCC antagonist (c). The effects of ATP under control conditions and on background of LTCC inhibition (from Figs. 2a, b, 4b) are denoted on b and c, respectively. Shown changes in quantal content (m), timing of the release (P90), and frequency of spontaneous exocytotic events. Data are expressed as circles—values in individual animals, box range—SEM, whiskers—SD. N = 6–14 for each group. ***P ˂ 0.001, **P ˂ 0.01, *P < 0.05—compared to value prior to Nitr (a) or ATP (b, c) application or between two groups
Lipid raft disruption did not markedly modify the effects of ATP on evoked and spontaneous release (Fig. 5b). Only tendency to enhancement of ATP-mediated depression of both evoked and spontaneous release was observed in distal part of MβCD-treated NMJs. As a result, the differences in expression of ATP effect on evoked release in distal vs proximal part disappeared in MβCD-pretreated muscles. Indeed, in control, ATP depressed quantum content more significantly in proximal vs distal part, whereas after lipid raft disruption, ATP reduced it to similar degree in both parts of the NMJ (Fig. 5b).
After lipid raft disruption, the LTCC blockage lost the ability to decrease the depressant effect of ATP on evoked release in distal part of the NMJs (Fig. 5c). Lipid raft disruption and LTCC inhibition, both individually and in combination, did not change the effect of ATP on quantum content in proximal part of the NMJs (Fig. 5c). So, raft integrity in distal part of the nerve terminal can be required for participation of LTCCs in the effect of purinoreceptor activation with ATP. Probably, if lipid rafts are disorganized, ATP can suppress neurotransmitter release via LTCC-independent (alternative) pathway.
In addition, when lipid raft integrity was perturbed, LTCC antagonist lost the ability to prevent the depressant action of exogenous ATP on spontaneous release in both distal and proximal parts. Indeed, in MβCD + nitrendipine-treated NMJs, ATP decreased MEPP frequency to the similar levels as in control samples, whereas ATP did not change and even increased MEPP frequency in proximal and distal parts, respectively, of nitrendipine-treated NMJs (Fig. 5c). These data indicate that lipid rafts are required for realization of ATP effect on spontaneous release via LTCC-dependent pathway.
Taken together, these results suggest that lipid rafts are important for LTCC-dependent regulation of evoked release under both basal conditions and ATP administration in distal (but not proximal) part of the NMJs. In addition, lipid rafts seem to participate in exogenous ATP/LTCC-mediated control of spontaneous release in both distal and proximal parts of the NMJs.
Discussion
The main findings of the present study (Fig. 6) are as follows: (1) expression of stimulatory effect of LTCCs on spontaneous and evoked (especially, asynchronous) release is dependent on basal (endogenous) activity of P2Y-Rs and PKC; (2) however, LTCC (in PKC-independent manner) can contribute to suppression of neurotransmitter release under condition of exogenous ATP administration; (3) engagement of LTCCs in neurotransmitter release as well as its timing have specific features in distal and proximal compartment of the nerve terminal. Specifically, (3a) depressant effect of exogenous ATP on evoked exocytosis partially relies on LTCC activity in distal part, whereas negative action of ATP on spontaneous release is linked with LTCCs both in distal and proximal parts; (3b) lipid rafts are required for involvement of LTCCs in basal and ATP-driven control of evoked neurotransmitter release and synchrony of exocytotic events specifically in the distal region, and at the same time, ATP-induced suppression of spontaneous release via LTCCs is lipid raft-dependent process in both distal and proximal compartments.

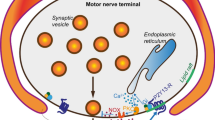
Putative role of LTCCs in regulation of neurotransmitter release in distal and proximal compartments of NMJs. Left side of cartoon: endogenous ATP released at basal conditions as co-neurotransmitter or from glial cell (not shown) stimulates P2Y-Rs which can maintain activity of LTCCs (in orange) via PKC-dependent signaling. In turn, LTCCs increase spontaneous exocytosis and evoked release (especially, asynchronous component). One possibility is that Ca2+ influx through LTCCs can simulate Ca2+-dependent enzymes and channels (e.g., Ca2+-activated K+ channels, BK), thereby affecting the neurotransmitter release in both distal and proximal parts. This LTCC-driven regulation of neurotransmitter release is dependent on lipid raft integrity specifically in distal (but not proximal) compartment. Right side of cartoon: LTCC (in blue) can negatively regulate neurotransmitter release under certain conditions. Indeed, stimulation of P2Y-Rs with exogenous ATP at high concentration may suppress neurotransmitter release partially via activation of LTCCs. Under this context, LTCCs attenuate both evoked exocytosis (in distal region) and spontaneous release (in distal and proximal compartments). ATP-triggered depressant action of LTCCs is dependent on lipid rafts. Note that if: (1) LTCCs are blocked, ATP can even increase in spontaneous release in distal compartment; (2) lipid raft are disrupted, then ATP may reduce evoked release in LTCC-independent manner in distal region. In proximal region, ATP attenuates evoked exocytosis in LTCCs and lipid rafts independent way (3). Also, exogenous ATP-mediated activation of LTCCs had no effect on timing of neurotransmitter release. Since, different functional population of LTCCs can operate in basal conditions (left side) and in response to exogenous ATP (right side). Distal compartment is characterized by more complex mechanisms which are engaged LTCCs, P2Y-Rs, and lipid rafts for bidirectional control of neurotransmitter release. AP, action potential; NTCC, N-type Ca2+ channel
SV exocytosis occurring in active zone is triggered by Ca2+ influx through NTCCs in frog NMJs (Dittrich et al. 2018). LTCCs can play a modulatory role in neurotransmission, including in NMJs (Dolphin and Lee 2020). Indeed, pharmacological activation of LTCCs with BAY K 8644 increased quantal content of EPPs at low-frequency stimulation in the rat NMJs (Atchison and O'Leary 1987). Generally, in mammalian NMJs, LTCCs contribute to neurotransmission under special conditions, including high-frequency stimulation (Perissinotti et al. 2008), intense nerve terminal depolarization (Flink and Atchison 2003), action of 4-beta-Phorbol 12-myristate 13-acetate, an activator of PKC and exocytotic protein Munc-13 (Arenson and Evans 2001), genetic deficit of amyloid precursor protein (Yang et al. 2007). Under all these conditions, activation of LTCC can increase neurotransmitter release. Here, we found that under lower level of [Ca2+]out, either inhibitor or activator of LTCCs can decrease evoked exocytosis, but spontaneous release is sensitive only to LTCC blockage. The appearance of marked effects of LTCC inhibition in low [Ca2+]out solution indicates a possible disinhibition of LTCCs by the decrease in external Ca2+. In addition, paradoxical depressant effect of pharmacological LTCC stimulation can be related with decreased extracellular Ca2+ levels and, as a consequence, a lesser influx of Ca2+, which can activate another signaling enzyme(s) able to depress ACh release. For example, Ca2+-activated phosphatase PP2B can depress quantal neurotransmitter release in mice NMJs (Gaydukov et al. 2013). In addition, increase in [Ca2+]in in terminal Schwann cells, which also express LTCCs (Robitaille et al. 1996), can negatively regulate neurotransmitter release in NMJs (Ko and Robitaille 2015). Importantly, inhibition of LTCCs decreased asynchronous release, whereas activation had reverse (in distal part) or no effect (in proximal part) on timing of the exocytotic events. Accordingly, optimal LTCC activity can maintain evoked neurotransmitter release at certain level and positively regulate asynchronous component of AP-induced exocytosis as well as spontaneous release.
LTCC can affect neurotransmitter release through multiple Ca2+-dependent mechanisms. One possibility is that LTCC-dependent Ca2+ influx could activate BK channels (Sun et al. 2003). The latter might bidirectionally control neurotransmitters release (Wang et al. 2020; Xu and Slaughter 2005; Flink and Atchison 2003; Liu et al. 2007; Gaydukov et al. 2009). Inhibition of BK channels precluded the effects of LTCC blockage on evoked neurotransmitter release (inducing asynchronous component) in both distal and proximal parts. This suggest that BK channels can be downstream component in LTCC-dependent regulation of the neurotransmitter release in the frog NMJs. According to this scenario, BK channel can be positive regulator of neurotransmitter release. Local increase in [K]out due to activity of BK channels in active zone can increase Ca2+ currents and neurotransmitter release (Xu and Slaughter 2005). Similarly, knockout of BK channels suppressed neurotransmitter release likely via interfering with Ca2+-induced release in mice NMJs (Wang et al. 2020). It should be noted that inhibition of BK channels itself had no effect on evoked neurotransmitter release in our experiments. This can reflect a silence of BK channels when extracellular Ca2+ level is low. In contrast, at 2.0 mM [Ca2+]out in bathing solution, BK channel antagonists increased quantum content of EPPs, which was dependent on subsequent LTCC activation in NMJs of mice diaphragm (Gaydukov et al. 2009; Flink and Atchison 2003). Although, under normal [Ca2+]out conditions, three BK channel blockers had no effect on neurotransmitter release in tibialis anterior muscle of adult mice (Wang et al. 2020).
Importantly, the effects of LTCC inhibition were completely prevented by preliminary inhibition of either P2Y-Rs or PKC. Taking into account that activation of P2Y-Rs by ATP and ADP can stimulate PKC in NMJs (Sokolova et al. 2003; De Lorenzo et al. 2006), and PKC can open quiescent LTCCs in motor nerve terminal (Arenson and Evans 2001), endogenous ATP/ADP can support activation of LTCCs via stimulation of P2Y-R/PKC signaling. Note that the ability of LTCCs to enhance asynchronous release suggests that these channels can be an important source of Ca2+ for triggering this type of exocytotic event, which play role in synaptic plasticity and coincidence detection (Kaeser and Regehr 2014). Furthermore, shift from synchronous to asynchronous exocytosis can occur under the pathological conditions, including models of amyotrophic lateral sclerosis and spinal muscular atrophy (Ruiz et al. 2010; Pagani et al. 2006).
Ca2+ influx via LTCCs is mainly considered as positive modulator of neurotransmitter release (Dolphin and Lee 2020; Perissinotti et al. 2008; Flink and Atchison 2003; Arenson and Evans 2001; Yang et al. 2007). Although, Ca2+-dependent enzymes and channels (e.g., protease calpain, Ca2+/calmodulin-dependent protein kinase II and BK channels) can also suppress neurotransmitter release (Ando et al. 2005; Liu et al. 2007). We found that in distal part of motor nerve terminal, depressant effect of exogenous ATP on evoked neurotransmitter release can be enhanced by LTCC agonist and suppressed by LTCC antagonist. This points to the ability of ATP to attenuate evoked neurotransmitter release via activation of some population of LTCCs specifically in distal part. In this scenario, LTCCs can serve as negative regulator of AP-induced exocytosis. Although agonist of LTCCs did not significantly modulate ATP-mediated decrease in spontaneous release, inhibitor of LTCC prevent and even invert this effect of ATP in proximal and distal parts, respectively. Hence, LTCC activation can suppress both spontaneous and evoked neurotransmitter release in the context of purino-receptor stimulation with exogenous ATP. Moreover, the contribution of LTCCs in the depressant action of ATP seems more in distal compartment of the nerve terminal. This compartment is characterized by specific mechanism of regulation as well as lower probability of neurotransmitter release (Tsentsevitsky et al. 2011, 2020a; Robitaille and Tremblay 1991; Tsentsevitsky and Petrov 2021). Furthermore, this part of nerve terminals is a key site for remodeling and growth. Accordingly, specific function of LTCCs in the distal part can be linked with an importance of LTCCs in early NMJ formation and axon branching (Kaplan et al. 2018) as well as suppression of neurotransmission in newly sprouted NMJs (Chipman et al. 2014). Indeed, LTCCs can suppress neurotransmitter release in newly formed frog and rodent NMJs (Balezina et al. 2007; Sugiura and Ko 1997). Interestingly, BK channel inhibition did not prevent LTCC blocker-mediated increase in quantal release in regenerating frog NMJs (Sugiura and Ko 1997).
Some portion of P2Y-Rs for ATP as well as LTCCs can reside in lipid rafts in synapses (Giniatullin et al. 2015; Mercer et al. 2011). In additional, lipid rafts and membrane cholesterol are essential for axonal growth and regeneration; these processes are dependent on signaling in axonal distal region (Rosello-Busquets et al. 2019; Petrov and Zefirov 2013). Accordingly, specific functioning of LTCCs in the distal vs proximal compartment as well as different role of LTCCs under basal conditions and in response to exogenous ATP can be partially linked with cholesterol-rich membrane microdomains, lipid rafts. Indeed, in distal compartment, lipid raft disruption precluded participation of LTCCs in regulation evoked release (including its timing) either under basal condition or ATP application. At the same time, there was no effect of lipid raft disruption on LTCC-related control of evoked release in proximal part of the same nerve terminal. This suggests that lipid rafts of the distal parts are required for LTCC functioning as well as their coupling to ATP receptors. Probably some specific lipid components, cytoskeletal, or scaffold proteins can be responsible to more close interactions between LTCCs, lipid rafts, and sites of evoked exocytosis in distal part of the motor nerve terminal. For example, scaffold-like protein MARCKS is essential for axonal development and enriched in tip of axonal branches (Sosa et al. 2016). In chromaffin cells, MARCKCs along with LTCC and PKCε can form a lipid raft-associated complex which selectively regulates exocytosis of large dense-core vesicles (Park and Kim 2009). Interestingly, participation of LTCCs in effect of exogenous ATP on spontaneous exocytosis was sensitive to lipid raft disruption in both distal and proximal parts of the nerve terminal. This indicates that there are different mechanisms of LTCC-dependent regulation of spontaneous and evoked release.
Conclusion
In the present study, using low Ca2+ external solution allowed to reveal that LTCCs can bidirectionally control the neurotransmitter release and this LTCC-mediated regulation occurs in compartment, lipid raft, and purinoreceptor-dependent manner in the NMJs. Endogenous activity of P2Y-R-PKC axis can sustain LTCC activity, which is required for maintenance of evoked release and enhancement of asynchronous and spontaneous release. At the same time, overstimulation of the purinoreceptors with exogenous ATP can suppress evoked and spontaneous release partially via activation of LTCCs. This role of LTCCs in depressant effects of ATP is more pronounced in distal part of the nerve terminal. Also, in the distal compartment, LTCC-dependent regulation of evoked release as well as engagement of LTCCs in the action of exogenous ATP on evoked exocytosis specifically depend on lipid raft integrity. Hence, distal region of the axon represents a unique presynaptic compartment, where activity of LTCCs can be regulated by specific mechanism. Physiological significance of such complex LTCC-mediated regulation of presynaptic function as well as whether this regulation operates in normocalcium external solution are topics for further studies. The main limitations of the current work are use of pharmacological approaches to interfere with the channel function, so genetic and molecular studies are needed to understand the precise role of LTCCs in control of presynaptic processes in NMJs.
Data Availability
All mentioned data are represented in the main manuscript figures and supplementary figures. Other additional data will be made available on reasonable request.
Abbreviations
- ACh:
-
Acetylcholine
- AP:
-
Action potential
- CgTx:
-
Omega-conotoxin GVIA
- CTxB:
-
Cholera toxin B subunit
- EPP:
-
End-plate potential
- MEPP:
-
Miniature EPP
- LTCC:
-
L-type Ca2+ channel
- NMJ:
-
Neuromuscular junction
- NTCC:
-
N-type Ca2+ channel
- PKC:
-
Protein kinase C
- P2Y-R:
-
P2Y-receptor
- SV:
-
Synaptic vesicle
- VGCC:
-
Voltage-gated Ca2+ channel
References
Ando K, Kudo Y, Takahashi M (2005) Negative regulation of neurotransmitter release by calpain: a possible involvement of specific SNAP-25 cleavage. J Neurochem 94(3):651–658. https://doi.org/10.1111/j.1471-4159.2005.03160.x
Arenson MS, Evans SC (2001) Activation of protein kinase C increases acetylcholine release from frog motor nerves by a direct action on L-type Ca(2+) channels and apparently not by depolarisation of the terminal. Neuroscience 104(4):1157–1164. https://doi.org/10.1016/s0306-4522(01)00114-2
Atchison WD, O’Leary SM (1987) BAY K 8644 increases release of acetylcholine at the murine neuromuscular junction. Brain Res 419(1–2):315–319. https://doi.org/10.1016/0006-8993(87)90599-3
Balezina OP, Bogacheva PO, Orlova TY (2007) Effect of L-type calcium channel blockers on activity of newly formed synapses in mice. Bull Exp Biol Med 143(2):171–174. https://doi.org/10.1007/s10517-007-0041-y
Branisteanu DD, Branisteanu DD, Covic A, Brailoiu E, Serban DN, Haulica ID (1989) Adenosine effects upon the spontaneous quantal transmitter release at the frog neuromuscular junction in the presence of protein kinase C-blocking and -activating agents. Neurosci Lett 98(1):96–100. https://doi.org/10.1016/0304-3940(89)90380-7
Bukcharaeva EA, Kim KC, Moravec J, Nikolsky EE, Vyskocil F (1999) Noradrenaline synchronizes evoked quantal release at frog neuromuscular junctions. J Physiol 517(Pt 3):879–888. https://doi.org/10.1111/j.1469-7793.1999.0879s.x
Calin-Jageman I, Lee A (2008) Ca(v)1 L-type Ca2+ channel signaling complexes in neurons. J Neurochem 105(3):573–583. https://doi.org/10.1111/j.1471-4159.2008.05286.x
Chipman PH, Schachner M, Rafuse VF (2014) Presynaptic NCAM is required for motor neurons to functionally expand their peripheral field of innervation in partially denervated muscles. J Neurosci 34(32):10497–10510. https://doi.org/10.1523/JNEUROSCI.0697-14.2014
Cinalli AR, Guarracino JF, Fernandez V, Roquel LI, Losavio AS (2013) Inosine induces presynaptic inhibition of acetylcholine release by activation of A3 adenosine receptors at the mouse neuromuscular junction. Br J Pharmacol 169(8):1810–1823. https://doi.org/10.1111/bph.12262
De Lorenzo S, Veggetti M, Muchnik S, Losavio A (2006) Presynaptic inhibition of spontaneous acetylcholine release mediated by P2Y receptors at the mouse neuromuscular junction. Neuroscience 142(1):71–85. https://doi.org/10.1016/j.neuroscience.2006.05.062
Del Castillo J, Katz B (1954) The effect of magnesium on the activity of motor nerve endings. J Physiol 124(3):553–559. https://doi.org/10.1113/jphysiol.1954.sp005128
Dittrich M, Homan AE, Meriney SD (2018) Presynaptic mechanisms controlling calcium-triggered transmitter release at the neuromuscular junction. Curr Opin Physiol 4:15–24. https://doi.org/10.1016/j.cophys.2018.03.004
Dolphin AC, Lee A (2020) Presynaptic calcium channels: specialized control of synaptic neurotransmitter release. Nat Rev Neurosci 21(4):213–229. https://doi.org/10.1038/s41583-020-0278-2
Flink MT, Atchison WD (2003) Iberiotoxin-induced block of Ca2+-activated K+ channels induces dihydropyridine sensitivity of ACh release from mammalian motor nerve terminals. J Pharmacol Exp Ther 305(2):646–652. https://doi.org/10.1124/jpet.102.046102
Fu WM, Huang FL (1994) L-type Ca2+ channel is involved in the regulation of spontaneous transmitter release at developing neuromuscular synapses. Neuroscience 58(1):131–140. https://doi.org/10.1016/0306-4522(94)90160-0
Galkin AV, Giniatullin RA, Mukhtarov MR, Svandova I, Grishin SN, Vyskocil F (2001) ATP but not adenosine inhibits nonquantal acetylcholine release at the mouse neuromuscular junction. Eur J Neurosci 13(11):2047–2053. https://doi.org/10.1046/j.0953-816x.2001.01582.x
Gaydukov AE, Melnikova SN, Balezina OP (2009) Facilitation of acetylcholine secretion in mouse motor synapses caused by calcium release from depots upon activation of L-type calcium channels. Bull Exp Biol Med 148(2):163–166. https://doi.org/10.1007/s10517-009-0678-9
Gaydukov AE, Tarasova EO, Balezina OP (2013) Calcium-dependent phosphatase calcineurin downregulates evoked neurotransmitter release in neuromuscular junctions of mice. Neurochem J 7(1):29–33. https://doi.org/10.1134/s1819712413010030
Ginebaugh SP, Cyphers ED, Lanka V, Ortiz G, Miller EW, Laghaei R, Meriney SD (2020) The frog motor nerve terminal has very brief action potentials and three electrical regions predicted to differentially control transmitter release. J Neurosci. https://doi.org/10.1523/JNEUROSCI.2415-19.2020
Giniatullin AR, Grishin SN, Sharifullina ER, Petrov AM, Zefirov AL, Giniatullin RA (2005) Reactive oxygen species contribute to the presynaptic action of extracellular ATP at the frog neuromuscular junction. J Physiol 565(Pt 1):229–242. https://doi.org/10.1113/jphysiol.2005.084186
Giniatullin A, Petrov A, Giniatullin R (2015) The involvement of P2Y12 receptors, NADPH oxidase, and lipid rafts in the action of extracellular ATP on synaptic transmission at the frog neuromuscular junction. Neuroscience 285:324–332. https://doi.org/10.1016/j.neuroscience.2014.11.039
Guarracino JF, Cinalli AR, Fernandez V, Roquel LI, Losavio AS (2016) P2Y13 receptors mediate presynaptic inhibition of acetylcholine release induced by adenine nucleotides at the mouse neuromuscular junction. Neuroscience 326:31–44. https://doi.org/10.1016/j.neuroscience.2016.03.066
Kaeser PS, Regehr WG (2014) Molecular mechanisms for synchronous, asynchronous, and spontaneous neurotransmitter release. Annu Rev Physiol 76:333–363. https://doi.org/10.1146/annurev-physiol-021113-170338
Kaplan MM, Sultana N, Benedetti A, Obermair GJ, Linde NF, Papadopoulos S, Dayal A, Grabner M, Flucher BE (2018) Calcium influx and release cooperatively regulate AChR patterning and motor axon outgrowth during neuromuscular junction formation. Cell Rep 23(13):3891–3904. https://doi.org/10.1016/j.celrep.2018.05.085
Kasimov MR, Giniatullin AR, Zefirov AL, Petrov AM (2015) Effects of 5alpha-cholestan-3-one on the synaptic vesicle cycle at the mouse neuromuscular junction. Biochim Biophys Acta 1851(5):674–685. https://doi.org/10.1016/j.bbalip.2015.02.012
Kasimov MR, Zakyrjanova GF, Giniatullin AR, Zefirov AL, Petrov AM (2016) Similar oxysterols may lead to opposite effects on synaptic transmission: olesoxime versus 5alpha-cholestan-3-one at the frog neuromuscular junction. Biochim Biophys Acta 1861(7):606–616. https://doi.org/10.1016/j.bbalip.2016.04.010
Katz B, Miledi R (1965) The measurement of synaptic delay, and the time course of acetylcholine release at the neuromuscular junction. Proc R Soc Lond B Biol Sci 161:483–495. https://doi.org/10.1098/rspb.1965.0016
Katz E, Ferro PA, Weisz G, Uchitel OD (1996) Calcium channels involved in synaptic transmission at the mature and regenerating mouse neuromuscular junction. J Physiol 497(Pt 3):687–697. https://doi.org/10.1113/jphysiol.1996.sp021800
Ko CP, Robitaille R (2015) Perisynaptic schwann cells at the neuromuscular synapse: adaptable, multitasking glial cells. Cold Spring Harb Perspect Biol 7(10):a020503. https://doi.org/10.1101/cshperspect.a020503
Krivoi II, Petrov AM (2019) Cholesterol and the safety factor for neuromuscular transmission. Int J Mol Sci. https://doi.org/10.3390/ijms20051046
Liu Q, Chen B, Ge Q, Wang ZW (2007) Presynaptic Ca2+/calmodulin-dependent protein kinase II modulates neurotransmitter release by activating BK channels at Caenorhabditis elegans neuromuscular junction. J Neurosci 27(39):10404–10413. https://doi.org/10.1523/JNEUROSCI.5634-06.2007
Liu Y, Sugiura Y, Sudhof TC, Lin W (2019) Ablation of all synaptobrevin vSNAREs blocks evoked but not spontaneous neurotransmitter release at neuromuscular synapses. J Neurosci 39(31):6049–6066. https://doi.org/10.1523/JNEUROSCI.0403-19.2019
Mallart A (1984) Presynaptic currents in frog motor endings. Pflugers Arch 400(1):8–13. https://doi.org/10.1007/bf00670529
Marcantoni A, Baldelli P, Hernandez-Guijo JM, Comunanza V, Carabelli V, Carbone E (2007) L-type calcium channels in adrenal chromaffin cells: role in pace-making and secretion. Cell Calcium 42(4–5):397–408. https://doi.org/10.1016/j.ceca.2007.04.015
Margheri G, D’Agostino R, Trigari S, Sottini S, Del Rosso M (2014) The beta-subunit of cholera toxin has a high affinity for ganglioside GM1 embedded into solid supported lipid membranes with a lipid raft-like composition. Lipids 49(2):203–206. https://doi.org/10.1007/s11745-013-3845-8
Mercer AJ, Chen M, Thoreson WB (2011) Lateral mobility of presynaptic L-type calcium channels at photoreceptor ribbon synapses. J Neurosci 31(12):4397–4406. https://doi.org/10.1523/JNEUROSCI.5921-10.2011
Mukhutdinova KA, Kasimov MR, Giniatullin AR, Zakyrjanova GF, Petrov AM (2018) 24S-hydroxycholesterol suppresses neuromuscular transmission in SOD1(G93A) mice: a possible role of NO and lipid rafts. Mol Cell Neurosci 88:308–318. https://doi.org/10.1016/j.mcn.2018.03.006
Oliveira L, Correia-de-Sa P (2005) Protein kinase A and Ca(v)1 (L-Type) channels are common targets to facilitatory adenosine A2A and muscarinic M1 receptors on rat motoneurons. Neurosignals 14(5):262–272. https://doi.org/10.1159/000088642
Pagani MR, Reisin RC, Uchitel OD (2006) Calcium signaling pathways mediating synaptic potentiation triggered by amyotrophic lateral sclerosis IgG in motor nerve terminals. J Neurosci 26(10):2661–2672. https://doi.org/10.1523/JNEUROSCI.4394-05.2006
Palma AG, Muchnik S, Losavio AS (2011) Excitatory effect of the A2A adenosine receptor agonist CGS-21680 on spontaneous and K+-evoked acetylcholine release at the mouse neuromuscular junction. Neuroscience 172:164–176. https://doi.org/10.1016/j.neuroscience.2010.10.015
Park Y, Kim KT (2009) Dominant role of lipid rafts L-type calcium channel in activity-dependent potentiation of large dense-core vesicle exocytosis. J Neurochem 110(2):520–529. https://doi.org/10.1111/j.1471-4159.2009.06148.x
Perissinotti PP, Giugovaz Tropper B, Uchitel OD (2008) L-type calcium channels are involved in fast endocytosis at the mouse neuromuscular junction. Eur J Neurosci 27(6):1333–1344. https://doi.org/10.1111/j.1460-9568.2008.06113.x
Petrov AM, Zefirov AL (2013) Cholesterol and lipid rafts in the biological membranes. Role in the release, reception and ion channel functions. Usp Fiziol Nauk 44(1):17–38
Petrov AM, Kudryashova KE, Odnoshivkina YG, Zefirov AL (2011a) Cholesterol and lipid rafts in the plasma membrane of nerve terminal and membrane of synaptic vesicles. Neurochem J 5(1):13–19. https://doi.org/10.1134/s1819712411010089
Petrov AM, Naumenko NV, Uzinskaya KV, Giniatullin AR, Urazaev AK, Zefirov AL (2011b) Increased non-quantal release of acetylcholine after inhibition of endocytosis by methyl-beta-cyclodextrin: the role of vesicular acetylcholine transporter. Neuroscience 186:1–12. https://doi.org/10.1016/j.neuroscience.2011.04.051
Petrov AM, Yakovleva AA, Zefirov AL (2014) Role of membrane cholesterol in spontaneous exocytosis at frog neuromuscular synapses: reactive oxygen species-calcium interplay. J Physiol 592(22):4995–5009. https://doi.org/10.1113/jphysiol.2014.279695
Petrov AM, Zakyrjanova GF, Yakovleva AA, Zefirov AL (2015) Inhibition of protein kinase C affects on mode of synaptic vesicle exocytosis due to cholesterol depletion. Biochem Biophys Res Commun 456(1):145–150. https://doi.org/10.1016/j.bbrc.2014.11.049
Robitaille R, Tremblay JP (1991) Non-uniform responses to Ca2+ along the frog neuromuscular junction: effects on the probability of spontaneous and evoked transmitter release. Neuroscience 40(2):571–585. https://doi.org/10.1016/0306-4522(91)90142-b
Robitaille R, Bourque MJ, Vandaele S (1996) Localization of L-type Ca2+ channels at perisynaptic glial cells of the frog neuromuscular junction. J Neurosci 16(1):148–158
Rodrigues HA, Lima RF, Fonseca Mde C, Amaral EA, Martinelli PM, Naves LA, Gomez MV, Kushmerick C, Prado MA, Guatimosim C (2013) Membrane cholesterol regulates different modes of synaptic vesicle release and retrieval at the frog neuromuscular junction. Eur J Neurosci 38(7):2978–2987. https://doi.org/10.1111/ejn.12300
Rosello-Busquets C, de la Oliva N, Martinez-Marmol R, Hernaiz-Llorens M, Pascual M, Muhaisen A, Navarro X, Del Valle J, Soriano E (2019) Cholesterol depletion regulates axonal growth and enhances central and peripheral nerve regeneration. Front Cell Neurosci 13:40. https://doi.org/10.3389/fncel.2019.00040
Ruiz R, Casanas JJ, Torres-Benito L, Cano R, Tabares L (2010) Altered intracellular Ca2+ homeostasis in nerve terminals of severe spinal muscular atrophy mice. J Neurosci 30(3):849–857. https://doi.org/10.1523/JNEUROSCI.4496-09.2010
Santafe MM, Lanuza MA, Garcia N, Tomas J (2006) Muscarinic autoreceptors modulate transmitter release through protein kinase C and protein kinase A in the rat motor nerve terminal. Eur J Neurosci 23(8):2048–2056. https://doi.org/10.1111/j.1460-9568.2006.04753.x
Sokolova E, Grishin S, Shakirzyanova A, Talantova M, Giniatullin R (2003) Distinct receptors and different transduction mechanisms for ATP and adenosine at the frog motor nerve endings. Eur J Neurosci 18(5):1254–1264. https://doi.org/10.1046/j.1460-9568.2003.02835.x
Sosa LJ, Malter JS, Hu J, Bustos Plonka F, Oksdath M, Nieto Guil AF, Quiroga S, Pfenninger KH (2016) Protein interacting with NIMA (never in mitosis A)-1 regulates axonal growth cone adhesion and spreading through myristoylated alanine-rich C kinase substrate isomerization. J Neurochem 137(5):744–755. https://doi.org/10.1111/jnc.13612
Subramanian J, Dye L, Morozov A (2013) Rap1 signaling prevents L-type calcium channel-dependent neurotransmitter release. J Neurosci 33(17):7245–7252. https://doi.org/10.1523/JNEUROSCI.5963-11.2013
Sugiura Y, Ko CP (1997) Novel modulatory effect of L-type calcium channels at newly formed neuromuscular junctions. J Neurosci 17(3):1101–1111
Sun X, Gu XQ, Haddad GG (2003) Calcium influx via L- and N-type calcium channels activates a transient large-conductance Ca2+-activated K+ current in mouse neocortical pyramidal neurons. J Neurosci 23(9):3639–3648
Tsentsevitsky AN, Petrov AM (2021) Synaptic mechanisms of cadmium neurotoxicity. Neural Regen Res 16(9):1762–1763. https://doi.org/10.4103/1673-5374.306067
Tsentsevitsky A, Nikolsky E, Giniatullin R, Bukharaeva E (2011) Opposite modulation of time course of quantal release in two parts of the same synapse by reactive oxygen species. Neuroscience 189:93–99. https://doi.org/10.1016/j.neuroscience.2011.05.033
Tsentsevitsky AN, Zakyrjanova GF, Petrov AM (2020a) Cadmium desynchronizes neurotransmitter release in the neuromuscular junction: key role of ROS. Free Radic Biol Med 155:19–28. https://doi.org/10.1016/j.freeradbiomed.2020.05.017
Tsentsevitsky AN, Zakyrjanova GF, Petrov AM, Kovyazina IV (2020b) Breakdown of phospholipids and the elevated nitric oxide are involved in M3 muscarinic regulation of acetylcholine secretion in the frog motor synapse. Biochem Biophys Res Commun 524(3):589–594. https://doi.org/10.1016/j.bbrc.2020.01.112
Urbano FJ, Depetris RS, Uchitel OD (2001) Coupling of L-type calcium channels to neurotransmitter release at mouse motor nerve terminals. Pflugers Arch 441(6):824–831. https://doi.org/10.1007/s004240000489
Wang X, Burke SRA, Talmadge RJ, Voss AA, Rich MM (2020) Depressed neuromuscular transmission causes weakness in mice lacking BK potassium channels. J Gen Physiol. https://doi.org/10.1085/jgp.201912526
Xu JW, Slaughter MM (2005) Large-conductance calcium-activated potassium channels facilitate transmitter release in salamander rod synapse. J Neurosci 25(33):7660–7668. https://doi.org/10.1523/JNEUROSCI.1572-05.2005
Yang L, Wang B, Long C, Wu G, Zheng H (2007) Increased asynchronous release and aberrant calcium channel activation in amyloid precursor protein deficient neuromuscular synapses. Neuroscience 149(4):768–778. https://doi.org/10.1016/j.neuroscience.2007.08.025
Zakyrjanova GF, Tsentsevitsky AN, Kuznetsova EA, Petrov AM (2021) Immune-related oxysterol modulates neuromuscular transmission via non-genomic liver X receptor-dependent mechanism. Free Radic Biol Med 174:121–134. https://doi.org/10.1016/j.freeradbiomed.2021.08.013
Zefirov AL, Grigor’ev PN, Petrov AM, Minlebaev MG, Sitdikova GF (2003) Analysis of living motor nerve ending of a frog by endocytotic fluorescent marker FM 1–43. Tsitologiia 45(12):1163–1171
Funding
This study was supported in part by the Russian Science Foundation Grant #21-14-00044, https://rscf.ru/project/21-14-00044/ (3.2–3.5), and partially the government assignment for FRC Kazan Scientific Center of RAS (3.1).
Author information
Authors and Affiliations
Contributions
ANT produced and analyzed data. A.N.T. and A.M.P. interpreted results of experiments, designed, and supervised the research. A.M.P. wrote the manuscript. All the authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. All authors consent to publish the article.
Ethical Approval
The present study was conducted in compliance with the NIH Guide for the Care and Use of Laboratory Animals and the requirements of the EU Directive 2010/63/EU. The protocol of experiments was approved by the Ethical Committee of Kazan Medical University.
Informed Consent
This is not applicable. This manuscript does not contain any studies with human participants.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tsentsevitsky, A.N., Petrov, A.M. L-type Ca2+ Channels at Low External Calcium Differentially Regulate Neurotransmitter Release in Proximal–Distal Compartments of the Frog Neuromuscular Junction. Cell Mol Neurobiol 42, 2833–2847 (2022). https://doi.org/10.1007/s10571-021-01152-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-021-01152-w