
Abstract
In Huntington’s disease (HD) the imperfect expanded CAG repeat in the first exon of the HTT gene leads to the generation of a polyglutamine (polyQ) protein, which has some neuronal toxicity, potentially mollified by formation of aggregates. Accumulated research, reviewed here, implicates both the polyQ protein and the expanded repeat RNA in causing toxicity leading to neurodegeneration in HD. Different theories have emerged as to how the neurodegeneration spreads throughout the brain, with one possibility being the transport of toxic protein and RNA in extracellular vesicles (EVs). Most cell types in the brain release EVs and these have been shown to contain neurodegenerative proteins in the case of prion protein and amyloid-beta peptide. In this study, we used a model culture system with an overexpression of HTT-exon 1 polyQ-GFP constructs in human 293T cells and found that the EVs did incorporate both the polyQ-GFP protein and expanded repeat RNA. Striatal mouse neural cells were able to take up these EVs with a consequent increase in the green fluorescent protein (GFP) and polyQ-GFP RNAs, but with no evidence of uptake of polyQ-GFP protein or any apparent toxicity, at least over a relatively short period of exposure. A differentiated striatal cell line expressing endogenous levels of Hdh mRNA containing the expanded repeat incorporated more of this mRNA into EVs as compared to similar cells expressing this mRNA with a normal repeat length. These findings support the potential of EVs to deliver toxic expanded trinucleotide repeat RNAs from one cell to another, but further work will be needed to evaluate potential EV and cell-type specificity of transfer and effects of long-term exposure. It seems likely that expanded HD-associated repeat RNA may appear in biofluids and may have use as biomarkers of disease state and response to therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Overview
This article includes both a Background Review section describing current understanding of the role of the expanded trinucleotide repeat RNA and polyglutamine (polyQ) huntingtin (HTT) protein in the pathology of Huntington’s disease (HD), and a Research Evaluation section describing novel studies in a cell culture model of incorporation and intercellular transfer of these two potentially toxic molecules via extracellular vesicles (EVs).
Background Review
Huntington’s disease is a dominantly inherited neurodegenerative disease caused by a trinucleotide (imperfect CAG repeat) in the first coding exon of the HTT gene (The Huntington Disease Collaborative Research Group 1993). The mutant HTT protein includes an expanded polyglutamine (polyQ) repeat and forms aggregates in both the nucleus (Kegel et al. 2002) and cytoplasm (Davies et al. 1997; DiFiglia et al. 1997). Neuronal degeneration begins in regions of the brain where expression of the protein is relatively high and spreads from there to adjacent regions, progressing in a spatially circumscribed area along the gradients (Vonsattel and DiFiglia 1998; Bayram-Weston et al. 2012). Several recent articles suggest that progression of neuropathology may result from transfer of the toxic mutant HTT protein from cell-to-cell within the brain (Costanzo and Zurzolo 2013; Bellingham et al. 2012; Aguzzi and Rajendran 2009). This toxicity could be mediated/influenced by release and uptake of mutant protein fibrils or aggregates (Ren et al. 2009), direct cell-to-cell transfer of these toxic proteins via nanotubes (Costanzo et al. 2013), or horizontal transfer via extracellular vesicles (EVs; Schneider and Simons 2013).
Potential Toxicity of Protein and RNA in HD
Neurons expressing high levels of HTT appear especially vulnerable to cell death when harboring an allele with an expanded CAG repeat (Vonsattel and DiFiglia 1998). The cause of cell death may be related to the expanded polyQ protein/peptides and/or the CAG-repeat RNA encoding it. The mutant HTT protein is known to form inclusion bodies, including amyloid-like fibrils (Hoffner and Djian 2014) and protein aggregates together with ubiquitin and proteases (Finkbeiner and Mitra 2008). Interestingly, the aggregated forms of mutant mouse HTT appear to form as a protective coping response, with the diffuse forms apparently being responsible for toxicity (Arrasate et al. 2004). How mutant HTT proteins and/or RNA cause toxicity is still unknown. By virtue of multiple hydrophobic alpha helices, HTT protein appears to regulate many protein–protein interactions. One mechanism may involve its association with proteins critical to ciliogenesis, which, in turn, is important in neuronal development and cerebral spinal fluid (CSF) flow (Keryer et al. 2011). A widely held view is that the propensity of mutant HTT to accumulate, misfold, and aggregate indicates that the capacity of neurons to refold or clear mutant HTT is exceeded in HD. From this perspective, abnormal deposition of aggregated protein is an indicator of a fundamental stress on neuronal protein homeostasis that could cause neurodegeneration through several mechanisms. Misfolded forms of mutant HTT may present non-native surfaces that will associate with other mutant HTT molecules or cellular targets if levels of chaperones are insufficient to buffer them. The excess load of malfolded proteins may deplete critical capacity of the cellular chaperone and clearance systems, leading to the misfolding of other, possibly unrelated metastable proteins and unmitigated proteotoxicity through both gain- and loss-of-function mechanisms (Finkbeiner 2012). In addition, one of HTT’s wild-type functions may be to regulate autophagy, such that the polyQ expansion leads to dysregulation of autophagy-dependent protein and organelle homeostasis and loss of normal autophagy function (Ochaba et al. 2014).
The expanded CAG-repeat RNA may also form hairpin loops with protein binding properties that could disrupt dynamic protein complexes (Fiszer and Krzyzosiak 2013). Several other disruptive CAG-repeat RNA mechanisms have also been implicated, including binding to proteins critical to splicing of precursor mRNAs for different genes within or exiting from the nucleus (for reviews see Fiszer and Krzyzosiak 2013; Nalavade et al. 2013). Fragments of CAG repeats, which exist at higher concentrations in cells with expanded repeats, may also act as siRNAs to decrease the translation of mRNA with complementary targets with a minimum of 7 CTG repeat sequence in their 3′UTRs (Bañez-Coronel et al. 2012). Accumulation of these repeat RNAs in the nucleolus can interfere with production of ribosomal RNA (Tsoi and Chan 2014). Another intriguing RNA mechanism is “Repeat-associated non-ATG translation” (RAN), which can initiate novel transcripts within the long hairpin structures formed by CAG repeats leading to the production of polyQ, polyS, or polyA proteins, with potentially disruptive functions (Zu et al. 2011). Toxic polyA, polyS, poly L, and polyC proteins have been found to accumulate in HD brains (Bañez-Coronel et al. 2015).
Extracellular Vesicles as Potential Transport Vehicles in HD
EVs carry select cell-specific cargos, including lipids, proteins, DNA, and RNA packaged by the “donor” cells within a bilipid membrane similar in orientation to the plasma membrane (Colombo et al. 2014; Kalra et al. 2012; Balaj et al. 2011). These vesicles can be released by multivesicular bodies derived from late endosomes, termed exosomes, or by budding from the plasma membrane, termed microvesicles or ectosomes (Cocucci and Meldolesi 2015). Uptake of all these different types of vesicles into recipient cells occurs typically through endocytosis. EVs, a generic term which includes all types of shedded vesicles, have been implicated both in communication between cells and in the elimination of unwanted products in the nervous system and other tissues (Schneider and Simons 2013; Zappulli et al. 2015). In other neurodegenerative diseases, including prion disease and Alzheimer’s disease, there is strong evidence that the scrapie form of prion and the Aβ peptide, respectively, can be transferred via EVs, potentially contributing to the spread of toxicity within the brain (Alais et al. 2008; Joshi et al. 2015; Saman et al. 2014). However, other studies suggest that neuronal EVs may actually degrade Aβ peptides (Rajendran et al. 2006; Aguzzi and Rajendran 2009). The potential spread of alpha-synuclein through EVs in Parkinson’s disease is still under investigation (Lööv et al. 2015—this issue).
The potential for transmission of the mutant HTT protein between cells is supported by findings of aggregates of mutant protein within grafts of normal fetal tissue in the brains of HD patients (Cicchetti et al. 2014). However, since these aggregates were present in the extracellular space, it is not clear whether they were taken up by normal cells that then went onto die releasing the aggregates, or were produced by endogenous HD cells and brought into the region via the vasculature. Further, aggregates of expanded polyQ HTT peptides were found to be transferred from cell-to-cell in culture and to recruit endogenous normal HTT peptides into the aggregates (Ren et al. 2009). The potential for incorporation of mutant HTT into EVs is consistent with its association with membranes and vesicles in fibroblasts from HD patients (Velier et al. 1998). Additional studies support the intercellular transfer of mutant HTT aggregates via tunneling nanotubes (Costanzo et al. 2013; Herrera et al. 2011), phagocytic glia (Pearce et al. 2015), and synaptic connections between neurons involving endocytotic uptake (Pecho-Vrieseling et al. 2014; Babcock and Ganetzky 2015). The potential for generation of EVs from the tips of nanotubes is suggested by images of cells in culture in which membranes are labeled with fluorescent proteins (Lai et al. 2015) and by release from membrane filopodia-like protrusions (Rilla et al. 2014). Both nanotubes and EVs are emerging as mechanisms whereby cells can exchange information through proteins/nucleic acids which cannot be secreted per se, but are only released in association with vesicles surrounded by cell membranes (Agnati and Fuxe 2014).
Research Evaluation: HTT Protein and CAG-Repeat RNA EV Loading and Transfer in Cell Culture
Summary
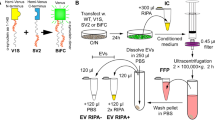
We evaluated incorporation of normal or expanded repeat CAG RNA and normal or polyQ-GFP HTT-exon 1 (Httex1) protein into EVs and their transfer to striatal neural cells in culture. Human 293T cells were either transfected with an expression cassette or infected with a lentiviral vector encoding exon 1 fragments of the human HTT gene containing either a normal CAG-repeat length or an expanded CAG repeat fused to GFP, with the expanded repeat protein-forming aggregates (Arrasate et al. 2004). EVs released by these cells were found to contain both CAG-repeat RNA and normal and mutant Httex1-GFP peptides. Mouse striatal cell lines derived from knock-in mice bearing either a homozygous normal or CAG-expanded repeat of the human Hdh exon 1 were found to take up labeled EV from these 293T cells. The uptake of the EV resulted in some transfer of unique GFP and polyQ-GFP RNA sequences, but no evidence was found for transfer of GFP+ proteins or of toxicity over the relatively short (72 and 96 h) EV exposure period. A differentiated striatal cell line with the expanded repeat in exon 1 of the mouse Hdh gene incorporated more of this mRNA into EVs as compared to a similar line with normal repeat length. These studies demonstrate that EVs from cells expressing mutant HTT contain CAG-repeat RNA and can potentially transfer this RNA to neural cells under sustained conditions of EV exposure.
Methods
Constructs and Vectors
DNA expression constructs corresponding to exon 1 of the human HTT gene with either 25 (normal) or 97 (mutant) copies of the imperfect CAG-repeat encoding polyQ fused in-frame with GFP (pGW1-Httex1-(Q25/Q97)-GFP) (Arrasate et al. 2004) were cloned into a lentivirus plasmid FUGW (Lois et al. 2002) using restriction sites BamH1 and Age1 and used for transfection of DNA or transduction after packaging into lentiviral vectors.
Cell Culture and Infection/Transfection
Human embryonic kidney 293T cells obtained from Dr. D. Baltimore (Rockefeller University, New York) were used as the donor cells for the EVs in experiments as they are efficiently transfected. 293T cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10 % fetal bovine serum (FBS) and penicillin/streptomycin (Corning, NY USA). Cells were transfected with normal and expanded CAG plasmid constructs using polyethylenimine (PEI) solution (Polyscience, Warrington, PA USA) at a final concentration of 5 μg/ml with 15 µg of DNA per 100 cm plate overnight.
293T cells were also stably transduced with a lentiviral vector encoding palmitoylated (palm) tdTomato (Lai et al. 2015). Lentiviral infections were also performed to generate 293T cells expressing either GFP, Httex1-25Q-GFP, or Httex1-97Q GFP. Cells were plated at a density of 6 × 105 cells per well in a 6-well plate 1 day before infection and lentiviral vectors: FUGW-GFP, FUGW- Httex1-25Q-GFP, and FUGW- Httex1-97Q-GFP were used at multiplicity of infection (M.O.I.) of 20 with 5 μg/ml polybrene. GFP signal was detected 72 h after infection.
Immortalized mouse striatal cell lines, STHdhQ111/Q111 and STHdhQ7/Q7 (Trettel et al. 2000) were obtained from the Coriell Biorepository (Camden, NJ USA). These cells were generated from striatal neurons of knock-in transgenic mouse embryos (E14) containing homozygous Hdh loci with exon 1 containing either 7 (normal) or 111 (expanded) imperfect CAG repeats derived from human sequence. These cells were cultured in high glucose DMEM (Corning) plus 10 % FBS and 40 mg/ml of G418. Cells were grown at 33 °C and neuronal differentiation was induced by incubation with a cocktail of α-fibroblast growth factor (FGF; 10 ng/ml), 3-isobutyl-1-methylxanthine (IBMX; 240 μM), forskolin (48.6 μM), and dopamine (5 μM) (Sigma, St. Louis, MO USA) in DMEM/F12 for 12 h.
Isolation of EV Pellet
HEK293T cells were transfected or infected with lentiviral constructs or vectors, respectively, expressing different lengths of CAG repeat in HTT-exon 1 (above) overnight to maximize levels of expression and then fresh medium with 5 % vesicle-depleted FBS (centrifuged at 100,000g for 18 h in a 70Ti Rotor overnight at 4 °C to pellet EVs) and 1 % penicillin/streptomycin were added onto cells. Conditioned media were collected 48 h later and centrifuged at 300g for 10 min and 2000g for 15 min followed by 0.8 μm filtration (Millipore, Billerica, MA USA) to remove cells and large debris. The supernatant was then ultra-centrifuged at 100,000g for 2 h at 4 °C in a 70Ti rotor. The pelleted EVs were resuspended in PBS, which had been double filtered through 0.22 μm filter (Millipore).
Monitoring of EV Number and Uptake by Mouse Striatal Cell Lines
The concentration of purified EVs were measured as particles using an LM10 nanoparticle analyzer (NanoSight, Duxbury, MA USA) with NTA software version 2.2. An aliquot of palm-td-Tomato EVs (Lai et al. 2015) generated by 293T cells were incubated with striatal cell lines for 24 h and EV uptake was visualized by Nikon Eclipse TE2000-U fluorescent microscopy.
Cell Viability Assay
STHdhQ111 and STHdhQ7 cells were plated at density of 10,000 cells/well in 48-well plates. Six hours after plating, neuronal differentiation was induced over 12 h. Isolated EVs from 293T cells transfected with DNA or transduced 5 days earlier with different vectors were added at a concentration of 2 x 1010 EVs, unless otherwise noted, to each well every 24 h over a 72-h period. Then cell viability was assessed using the CellTiter-Glo assay (Promega, Madison, WI USA), as per manufacturer’s instructions.
Western Blot Analysis and Sucrose Density Gradient of EVs
Both cells and EVs were lysed in ice-cold RIPA buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1 % NP-40, 1.5 % Na-deoxycholate, 1 % SDS) supplemented with EDTA-protease inhibitor (Roche Applied Science, Indianapolis, IN USA) and protein concentration was measured using a Bio-Rad assay (Bio-Rad, Hercules, CA USA). Twenty μg protein (unless otherwise specified) was loaded on a 4–12 % gradient gel (Invitrogen, Grand Island, NY USA) for each condition and the gel was transferred using Xcell Surelock blot module (Invitrogen) on a nitrocellulose membrane (Bio-rad), the membrane was then blocked with 5 % non-fat milk (Lab Scientific, Highlands, NJ USA) followed by the primary antibody incubation. Monoclonal antibodies 1C2 antibody (Millipore) and 3B5H10 (Sigma) to the polyQ repeat (1:5000) (Millipore) and anti-GFP (Invitrogen) were used to detect normal and mutant HTT–GFP fusion proteins; Alix (Santa Cruz, Dallas, TX USA) was used as an EV marker, and GAPDH was used to normalize protein loading (Millipore). The proteins on blots were detected using enhanced chemiluminescence (ECL, Thermal Fisher Scientific, Lafayette, CO USA) and exposed to HyBlot CL Autoradiography film (Denville Scientific Inc., Metuchen, NJ USA) after incubation with horseradish peroxide (HRP)-conjugated secondary antibody (Molecular Probes, Eugene, OR USA).
Isolated EVs were layered on a sucrose gradient (60, 45, 30, 8 % from bottom to top) and ultra-centrifuged at 5000 rpm for 38 min at 4 °C in a tabletop ultracentrifuge (Beckman, Pasadena, CA USA). The top 500 µl fraction was labeled as the first fraction, then every 350 µl of a total of 10 fractions were collected and each fraction was spun down again at 100,000g for 1 h to collect EVs and then analyzed by Western blot analysis.
Fluorescence Microscopy
293T cells, which had been transfected with DNA constructs or transduced with lentiviral vectors for FUGW-GFP, FUGW- Httex1-25Q-GFP, or FUGW- Httex1-97Q-GFP, were plated on coverslips at 80 % confluency and cells were fixed using 4 % paraformaldehyde for 10 min, 24 h after seeding. The nuclei were counterstained with TO-PRO-3 (Invitrogen), and slides were coverslipped using Dako fluorescent mounting solution (Dako, Carpinteria, CA USA). Cells were then imaged with Zeiss confocal microscopy.
PCR for Poly CAG and Target mRNAs
RNA and DNA were isolated from lentivirus-transduced cells and EVs collected from them. RT-PCR to monitor expression of the HTT-exon 1 cassettes was carried out using the following set of HTT primers: HTT Forward/HTT Reverse (5′-CTGCAGGTCGACTCTAGAGGAT-3′/5′-AAGTCGATGCCCTTCAGCTC-3′), which cover exon sequence at the 5′ end and GFP sequence at the 3′ end on either side of the repeat elements, and GAPDH Forward/GAPDH Reverse (5′-GAAGGTGAAGGTCGGAGTC-′3/5′-GAAGATGGTGATGGGATTTC-′3), using both transfected and stable 293T cells and EVs collected from them. The PCR product along with Quick-Load 1 Kb DNA ladder (NEB, Ipswich, MA USA) were then run on a 2 % agarose gel (Apex Bioresearch Product, Whitmore Lake, MI USA) to check if they were the right size.
Expression of Hdh mRNA in wild-type and mutant mouse striatal cells (7/7 and 111/111) and EVs was carried with qRT-PCR out using mHdh Forward/mHdh reverse (5′-CAGATGTCAGAATGGTGGCT-3′/5′-GCCTTGGAAGATTAGAATCCA-3′). hprt and beta-actin mRNAs were used to normalize the expression (mHPRT Forward/mHPRT Reverse (5′-GGTTAAGCAGTACAGCCCCA-3′/5′-AGAGGTCCTTTTCACCAGCA-3′); mActB Forward/mActB Reverse (5′-GCTTCTTTGCAGCTCCTTCGT/5′-CCAGCGCAGCGATATCG-3′). Reverse transcription was done using Sensiscript (Qiagen, Valencia, CA USA) and qPCR was carried out with Power SYBR green (Thermal Fisher Scientific) according to manufacture protocol.
Statistical Analysis
The unpaired 2-sample t test was used to compare groups. One-way ANOVA, followed by Bonferroni’s test, was conducted to test for significance among multiple groups, comparing all pairs of columns.
Results
Expression of GFP and GFP-repeat constructs in 293T cells
In initial experiments we transduced 293T cells with lentiviral vectors encoding versions of exon 1 of the human HTT gene fused in-frame with GFP with a homomeric polyQ stretch of 25 (control) or 97 (disease-related) glutamines. Five days after infection we evaluated the expression of GFP in the cells (Fig. 1). Cells transduced with the GFP control or Httex1-25Q-GFP vectors showed green fluorescence throughout the cytoplasm, while those transfected with Httex197Q-GFP showed GFP+ aggregates within the cytoplasm. Western blot analysis revealed the presence of GFP and polyQ immunoreactive proteins of the appropriate sizes in cells transduced with the Httex1-25Q-GFP (approximately 48 kD, open arrow head) or Httex1-97Q-GFP (approximately 63 kD, closed arrowhead) vectors (Fig. 2a). Interestingly, the amount of GFP+/polyQ+ protein appeared to be greater in Httex1-97Q-GFP transduced cells than in Httex1-25Q-GFP transduced cells, using either the GFP and polyQ antibodies normalized to levels of GAPDH, possibly due to the aggregation of the Httex1-97Q-GFP protein in cells. We also observed higher amounts of the Httex1-97Q-GFP protein, as compared to Httex1-25Q-GFP protein in EVs derived from these transduced cells, when normalized to the EV marker, Alix (Fig. 2b), supporting the incorporation of this expanded polyQ protein into the EV pellet. The apparently higher levels of Httex1-97Q-GFP compared to Httex1-25Q-GFP in EVs could reflect, to some extent, the higher affinity of this antibody (1C2) for long polyQ repeat regions (Trottier et al. 1995), and was seen to a lesser extent with GFP antibodies.

Confocal images of GFP+ aggregates in 293T cells transduced Httex1-25Q & 97Q-GFP lentiviral vectors. 293T cells were transduced with lentiviral vectors expressing FUGW-GFP (a), FUGW- Httex1-25Q-GFP (b), or FUGW- Httex1-97Q-GFP (c). Cells were imaged 96 h after infection following fixation in 4 % PFA with TO-PRO nuclear staining. These are representative images with aggregates indicated by arrows. Magnification bar 50 μm

Western blot of protein content of 293T cells non-transduced or transduced with FUGW- Httex1-25Q-GFP or FUGW- Httex1-97Q-GFP lentiviral vectors. Cells were replated 72 h after infection with lentiviral vectors, and then both cells (a) and EVs (b) were harvested 48 h after replating. Proteins were resolved by SDS-PAGE and immunoblotted for GFP and polyQ (1C2), using GAPDH to normalize for protein in cells and Alix for EV protein. Solid arrowheads indicate predicted size of 97Q-GFP protein and open arrowheads indicates position of Httex1-25Q-GFP protein. EVs were not collected from control cells as there was little-to-no GFP protein expressed
RT-PCR and gel electrophoretic analysis of Httex1-25Q-GFP and Httex1-97Q-GFP mRNA in lentiviral vector-transduced cells showed the latter transcript to be absent, with expression possibly lost due to toxicity (Supplementary Fig. S1). For this reason, we changed our protocol to carry out transient DNA transfections of 293T cells using Httex1-25Q-GFP or Httex1-97Q-GFP plasmids. This resulted in clear expression of all the correct size transcripts (GFP = 435 bp; Httex1-25Q-GFP = 711 bp and Httex1-97Q-GFP = 927 bp) in cells and in the EV pellets derived from them 64 h after transfection (Fig. 3). These data support the incorporation of these unique transcripts into the EVs. Western blot analysis of GFP and polyQ immunoreactive proteins in transfected cell confirmed the presence of appropriate MW GFP +/polyQ + proteins in transfected cells and EVs derived from them (Fig. 4).

RT-PCR of HTT-GFP RNA with no or different repeat lengths from transfected 293T cells and EVs. Cells and EVs were harvested 64 h after transfection with FUGW-GFP, FUGW- Httex1-25Q-GFP, or FUGW- Httex1-97Q-GFP plasmid vectors and RNA was extracted using miRNeasy minikit. RT-PCR products of HTT-GFP RNA with no or different repeat lengths from transfected 293T cells and EVs were analyzed on 1 % agarose gel using 1 kb Quick-Load ladder and 100 bp DNA ladder. [Open arrowhead 25Q-GFP, solid arrowhead 97Q-GFP, gray arrowhead GFP

Western blot of protein content of 293T cells non-transfected or transfected with FUGW-GFP, FUGW- Httex1-25Q-GFP, or FUGW- Httex1-97Q-GFP plasmid vectors. Cells (a) and EVs (b) were harvested 48 h after transfection and proteins resolved by SDS-PAGE and immunoblotted for GFP and polyQ (1C2), using GAPDH to normalize for protein in cells and Alix for EV protein. Open arrowhead indicates predicted size of Httex1-25Q-GFP protein and solid arrowhead indicates position of Httex1-97Q-GFP protein, gray arrowhead indicates the approximate size of GFP protein
The EV pellets generated by differential centrifugation can contain both EVs and protein aggregates. In order to evaluate whether the GFP/polyQ proteins were in EVs, the pellet was resuspended and resolved by sucrose density gradient centrifugation, using Alix to mark the EV fractions (Fig. 5). Highest levels of Alix were found in fractions 3–5 in control cells (Fig. 5a) and in cells transfected with the GFP (B) or polyQ-GFP constructs (Fig. 5c, d). In Httex1-25Q-GFP transfected cells (Fig. 5c), GFP+/1C2+ bands of the appropriate size were highest in fractions 3–7, suggesting the distribution is primarily in EVs, but may also be in pelleted protein aggregates. In the Httex1-97Q-GFP transfected cells (Fig. 5d), a GFP+/1C2+ band of the appropriate size was highest in fractions 3–7, again indicating that a substantial portion was in EVs. Other minor GFP bands seen on these blots are considered to be breakdown products, SDS-insoluble complexes, or post-translationally modified proteins. Thus, for GFP, Httex1-25Q-GFP and Httex1-97Q-GFP a substantial portion of these proteins are incorporated into EVs, with the rest assumed to be in protein aggregates in the conditioned medium that pellet at 100,000 x g.

Western blot of sucrose density fractions of EVs from transfected 293T cells analyzed for polyQ, GFP, and Alix. Pellets of EVs derived from 293T cell which had not been transfected or transfected with FUGW-GFP, FUGW- Httex1-25Q-GFP, or FUGW- Httex1-97Q-GFP plasmid vectors were resuspended and layered onto a sucrose density gradient. Following centrifugation, 8 fractions of the gradient were collected from top to bottom. These fractions were pelleted by ultracentrifugation and pellets were lysed for SDS-PAGE and immunoblotting, using antibodies to GFP, polyQ (1C2), and Alix. Open arrowhead indicates predicted size of Httex1-25Q-GFP protein and solid arrowhead indicates position of Httex1-97Q-GFP protein
Exposure of Striatal Cells to EVs from polyQ-GFP Transfected 293T Cells
In order to model control and HD neurons, we used differentiated mouse striatal neural cells that expressed either normal 7/7Q or expanded 111/111Q repeats within the mouse Hdh gene (Trettel et al. 2000). These cells grew at similar rates, with the 111/111Q having longer processes than the 7/7Q cells both in the differentiated and undifferentiated state (Supplementary Fig. S2). First, we evaluated uptake of 293T EVs labeled with palm-td-Tomato (Lai et al. 2015) and saw extensive uptake into striatal cells with normal (Fig. 6a) and expanded repeats (Fig. 6b) at 24 h after EV exposure. Then these striatal cells were either not treated or exposed to the EVs pelleted from conditioned medium generated by 293T cells, which had been non-transfected or transfected with GFP, Httex1-25Q-GFP, and Httex1-97Q-GFP plasmid vectors. Exposure was every 24 h over a 72-h period using 1 × 1010 EVs per 5 × 104 cells. After this relatively brief EV exposure there was no sign of GFP+ aggregates in the striatal cells (data not shown). Analysis of GFP-specific sequences by RT-PCR in EV-exposed striatal cells revealed some transfer of the GFP and Httex1-25Q-GFP mRNAs to both 7/7Q and 111/111Q cells (Fig. 7a). Transfer of the Httex1-97Q-GFP mRNA was very faint in 7/7Q cells and not detectable in 111/111Q cells. Given that in general, smaller RNAs are more efficiently packaged in EVs (Crescitelli et al. 2013), this transfer efficiency may reflect in part the size differences in these transcripts: GFP = 435 bp; Httex1-25Q-GFP = 711 bp and Httex1-97Q-GFP = 927 bp, as well as the relatively short time of exposure of striatal cells to EVs. Western blot analysis of the striatal cells exposed to these EVs did not reveal any GFP+ staining and the IC2 staining of polyQ did not reveal the 25Q-GFP or 97Q-GFP proteins (data not shown). This apparent transfer of the CAG-repeat RNA and not the polyQ-GFP proteins may reflect the higher detection sensitivity of RT-PCR for RNA, compared to Western blots for proteins. Further, this exposure to EVs containing CAG-repeat RNA and polyQ-GFP proteins did not result in any apparent toxicity to the striatal cells over 72 and 96 h time period (Fig. 7b).

Uptake of 293T vesicles by striatal cell lines over 24 h. EVs were harvested from 293T cells stably expressing palm-td-Tomato and incubated with differentiated striatal cells expressing a normal polyQ—STHdhQ7/Q7, or b expanded repeat polyQ—STHdhQ111/Q111. Fluorescence microscopy was carried out 24 h after EV exposure. Magnification bar 100 μm

Transfer of polyQ-GFP protein and RNA via EVs from transfected 293T cells to striatal cells over 72 h. Striatal cells were allowed to attach for 6 h before being differentiated for 12 h. EVs were harvested from non-transfected 293T cells (293T) or 293T cells transfected with FUGW-GFP, FUGW- Httex1-25Q-GFP, or FUGW- Httex1-97Q-GFP plasmid vectors. Striatal cells were not treated with EVs—designated NT, or treated with EVs added at a density of 2 × 1010 EVs per well (10,000 cells per well) in a 48-well plate (unless otherwise noted) every 24 h for 72 h in total. a PCR of RNA isolated from non-treated and EV-treated striatal cells after a 72 h exposure using primers specific to GFP (gray arrowhead), Httex1-25Q-GFP (open arrowhead), and Httex1-97Q-GFP (solid arrowhead) transcripts. b Viability of striatal cells under these different conditions was determined using CellTiter-Glo assay after EV treatment for either 72 h or 96 h
PolyQ-Related Contents in EVs from Striatal Cell Lines
Given that overexpression of RNA/proteins in cells favors their incorporation into EVs (Squadrito et al., 2014), we also explored the presence of normal and polyQ HTT protein and mRNA in EVs from 7/7Q and 111/111Q striatal cells that express these proteins/mRNAs at endogenous levels under their own promoter. We were not able to detect HTT protein or fragments using a monoclonal antibody 3B5H10 (Supplementary Fig. S3), but found that the mRNA with an expanded CAG-repeat element was incorporated to a greater extent into EVs from 111/111Q cells as compared to those from 7/7Q cells (Fig. 8).

Incorporation of HTT RNA with extended CAG repeat into EVs. Loading of HTT RNA into EV was analyzed by qRT-PCR from differentiated mouse striatal cells expressing wild-type HTT (Q7/Q7) or mutant HTT (Q111/Q111) under the mouse endogenous Hdh promoter. Loading of RNA messages into EVs was calculated by comparing the levels of Q7/Q7 and Q111/Q111 in EVs normalized to cellular levels and plotted using the comparative \(2^{{ - \varDelta \varDelta C_{\text{t}} }}\) method. All Ct values were normalized to averages of hprt and beta-actin mRNA. Value of Q7/Q7 was set to 1. (C t values of samples are provided in Supplementary Table 1.)
Conclusions
Our research findings are consistent with fragments of the expanded CAG-repeat RNA and polyQ protein associated with HD being incorporated into EVs. This finding should be considered with the caveats that in this culture study, where 293T cells were transfected with DNA expression constructs, the CAG-repeat RNA and polyQ protein were overexpressed in shorter forms that would occur for the HTT mRNA or HTT protein, which favored their chance of being incorporated into vesicles. However, we also found that in a differentiated striatal cell line in which HTT was expressed under its own promoter with either a normal or expanded CAG repeat in exon 1, the expanded repeat mRNA was preferentially incorporated into EVs compared to the expression level in cells.
It has been established that neurons release EVs (Fauré et al. 2006) and that the neurons that initially degenerate in HD are those expressing relatively high level of HTT (Vonsattel and DiFiglia 1998). The ability of CAG-repeat RNA/polyQ-loaded EVs to deliver this cargo to neural cells was not conclusive in this study. Using a sensitive RT-PCR assay of RNA in EVs produced by transfected cells, there did appear to be transfer of the shorter GFP+ and CAG-repeat RNAs to striatal cells, but transfer of protein was not demonstrated, possibly due to decreased sensitivity of the protein detection method or the protein being degraded in the recipient cells. Further, no toxicity was found after exposure of striatal cells to CAG-repeat RNA/polyQ-loaded EVs for 72 and 96 h. This could reflect the fairly short experimental exposure period, as compared to the EV bombardment that would occur in neurons in an HD brain over years. Another possibility is that the cells overexpressing these GFP-polyQ proteins use EVs as a means of eliminating them from cells, and thereby reducing their toxicity, as is seen with aggregates of the polyQ proteins (Arrasate et al. 2004). These preliminary studies support further work both on the transfer of CAG-repeat RNA and polyQ proteins via EVs over longer time intervals and on in vivo models in the brain to assess the fate of transferred RNA/proteins and potential toxicity to recipient cells. In addition, these data evoke the potential to evaluate the levels of expanded repeat RNA and polyQ proteins in EVs in biofluids of HD patients as biomarkers of disease progression and response to therapy.
Abbreviations
- CSF:
-
Cerebral spinal fluid
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- EVs:
-
Extracellular vesicles
- FBS:
-
Fetal bovine serum
- FGF:
-
Fibroblast growth factor
- GFP:
-
Green fluorescent protein
- HRP:
-
Horseradish peroxide
- IBMX:
-
Isobutyl-1-methylxanthine
- M.O.I.:
-
Multiplicity of infection
- palm:
-
Palmitoylated
- PEI:
-
Polyethylenimine
- polyQ:
-
Polyglutamine
References
Agnati LF, Fuxe K (2014) Extracellular-vesicle type of volume transmission and tunnelling-nanotube type of wiring transmission add a new dimension to brain neuro-glial networks. Philos Trans R Soc Lond B Biol Sci 369(1652). doi:10.1098/rstb.2013.0505
Aguzzi A, Rajendran L (2009) The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 64(6):783–790. doi:10.1016/j.neuron.2009.12.016
Alais S, Simoes S, Baas D, Lehmann S, Raposo G, Darlix JL, Leblanc P (2008) Mouse neuroblastoma cells release prion infectivity associated with exosomal vesicles. Biol Cell 100(10):603–615. doi:10.1042/BC2008002
Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431(7010):805–810
Babcock DT, Ganetzky B (2015) Transcellular spreading of huntingtin aggregates in the Drosophila brain. Proc Natl Acad Sci USA 112(39):E5427–5433. doi:10.1073/pnas.1516217112
Balaj L, Lessard R, Dai L, Cho Y-J, Pomeroy SL, Breakefield XO, Skog J (2011) Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat Commun 2:180. doi:10.1038/ncomms1180
Bañez-Coronel M, Porta S, Kagerbauer B, Mateu-Huertas E, Pantano L, Ferrer I, Guzmán M, Estivill X, Martí E (2012) A pathogenic mechanism in Huntington’s disease involves small CAG-repeated RNAs with neurotoxic activity. PLoS Genet 8(2):e1002481. doi:10.1371/journal.pgen.1002481
Bañez-Coronel M, Ayhan F, Tarabochia AD, Zu T, Perez BA, Tusi SK, Pletnikova O, Borchelt DR, Ross CA, Margolis RL, Yachnis AT, Troncoso JC, Ranum LP (2015) RAN translation in Huntington disease. Neuron 88(4):667–677. doi:10.1016/j.neuron.2015.10.038
Bayram-Weston Z, Jones L, Dunnett SB, Brooks SP (2012) Light and electron microscopic characterization of the evolution of cellular pathology in HdhQ92 Huntington’s disease knock-in mice. Brain Res Bull 88(2–3):171–181. doi:10.1016/j.brainresbull.2011.03.013
Bellingham SA, Guo BB, Coleman BM, Hill AF (2012) Exosomes: vehicles for the transfer of toxic proteins associated with neurodegenerative diseases? Front Physiol 3:124. doi:10.3389/fphys.2012.00124
Cicchetti F, Lacroix S, Cisbani G, Vallières N, Saint-Pierre M, St-Amour I, Tolouei R, Skepper JN, Hauser RA, Mantovani D, Barker RA, Freeman TB (2014) Mutant huntingtin is present in neuronal grafts in Huntington disease patients. Ann Neurol 76(1):31–42. doi:10.1002/ana.24174
Cocucci E, Meldolesi J (2015) Ectosomes and exosomes: shedding the confusion between extracellular vesicles. Trends Cell Biol 25(6):364–372. doi:10.1016/j.tcb.2015.01.004
Colombo M, Raposo G, Théry C (2014) Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 30:255–289. doi:10.1146/annurev-cellbio-101512-122326
Costanzo M, Zurzolo C (2013) The cell biology of prion-like spread of protein aggregates: mechanisms and implication in neurodegeneration. Biochem J 452(1):1–17. doi:10.1042/BJ20121898
Costanzo M, Abounit S, Marzo L, Danckaert A, Chamoun Z, Roux P, Zurzolo C (2013) Transfer of polyglutamine aggregates in neuronal cells occurs in tunneling nanotubes. J Cell Sci 126(Pt 16):3678–3685. doi:10.1242/jcs.126086
Crescitelli R, Lässer C, Szabó TG, Kittel A, Eldh M, Dianzani I, Buzás EI, Lötvall J (2013) Distinct RNA profiles in subpopulations of extracellular vesicles: apoptotic bodies, microvesicles and exosomes. J Extracell Vesicles 2. doi:10.3402/jev.v2i0.20677
Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90(3):537–548
DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277(5334):1990–1993
Fauré J, Lachenal G, Court M, Hirrlinger J, Chatellard-Causse C, Blot B, Grange J, Schoehn G, Goldberg Y, Boyer V, Kirchhoff F, Raposo G, Garin J, Sadoul R (2006) Exosomes are released by cultured cortical neurones. Mol Cell Neurosci 31(4):642–648
Finkbeiner S (2012) Huntington’s disease. In: Morimoto RI, Kelly JW, Selkoe DJ (eds) Protein homeostasis: the chemistry and biology of diseases of protein conformation. The cold spring harbor perspectives in biology series. Cold Spring Harbor Press, New York, pp 211–234. doi:10.1101/cshperspect.a007476
Finkbeiner S, Mitra S (2008) The ubiquitin-proteasome pathway in Huntington’s disease. ScientificWorldJournal 8:421–433. doi:10.1100/tsw.2008.1160
Fiszer A, Krzyzosiak WJ (2013) RNA toxicity in polyglutamine disorders: concepts, models, and progress of research. J Mol Med (Berl) 91(6):683–691. doi:10.1007/s00109-013-1016-2
Herrera F, Tenreiro S, Miller-Fleming L, Outeiro TF (2011) Visualization of cell-to-cell transmission of mutant huntingtin oligomers. PLoS Curr 3:RRN1210. doi:10.1371/currents.RRN1210
Hoffner G, Djian P (2014) Polyglutamine aggregation in huntington disease: does structure determine toxicity? Mol Neurobiol 52(3):1297–1314
Joshi P, Benussi L, Furlan R, Ghidoni R, Verderio C (2015) Extracellular vesicles in Alzheimer’s disease: friends or foes? Focus on aβ-vesicle interaction. Int J Mol Sci 16(3):4800–4813. doi:10.3390/ijms16034800
Kalra H, Simpson RJ, Ji H, Aikawa E, Altevogt P, Askenase P, Bond VC, Borràs FE, Breakefield X, Budnik V, Buzas E, Camussi G, Clayton A, Cocucci E, Falcon-Perez JM, Gabrielsson S, Gho YS, Gupta D, Harsha HC, Hendrix A, Hill AF, Inal JM, Jenster G, Krämer-Albers EM, Lim SK, Llorente A, Lötvall J, Marcilla A, Mincheva-Nilsson L, Nazarenko I, Nieuwland R, Nolte-’t Hoen EN, Pandey A, Patel T, Piper MG, Pluchino S, Prasad TS, Rajendran L, Raposo G, Record M, Reid GE, Sánchez-Madrid F, Schiffelers RM, Siljander P, Stensballe A, Stoorvogel W, Taylor D, Thery C, Valadi H, van Balkom BW, Vázquez J, Vidal M, Wauben MH, Yáñez-Mó M, Zoeller M, Mathivanan S (2012) Vesiclepedia: a compendium for extracellular vesicles with continuous community annotation. PLoS Biol 10(12):e1001450. doi:10.1371/journal.pbio.1001450
Kegel KB, Meloni AR, Yi Y, Kim YJ, Doyle E, Cuiffo BG, Sapp E, Wang Y, Qin ZH, Chen JD, Nevins JR, Aronin N, DiFiglia M (2002) Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription. J Biol Chem 277(9):7466–7476
Keryer G, Pineda JR, Liot G, Kim J, Dietrich P, Benstaali C, Smith K, Cordelières FP, Spassky N, Ferrante RJ, Dragatsis I, Saudou F (2011) Ciliogenesis is regulated by a huntingtin-HAP1-PCM1 pathway and is altered in Huntington disease. J Clin Invest 121(11):4372–4382. doi:10.1172/JCI57552
Krauss S, Griesche N, Jastrzebska E, Chen C, Rutschow D, Achmüller C, Dorn S, Boesch SM, Lalowski M, Wanker E, Schneider R, Schweiger S (2013) Translation of HTT mRNA with expanded CAG repeats is regulated by the MID1-PP2A protein complex. Nat Commun 4:1511. doi:10.1038/ncomms2514
Lai CP, Kim EY, Badr CE, Weissleder R, Mempel TR, Tannous BA, Breakefield XO (2015) Visualization and tracking of tumour extracellular vesicle delivery and RNA translation using multiplexed reporters. Nat Commun 6:7029. doi:10.1038/ncomms8029
Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D (2002) Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295(5556):868–872
Lööv C, Hyman BT, Breakefield XO, Ingelsson M (2015) Alpha-synuclein in extracellular vesicles—functional implications and diagnostic opportunities. Cell Mol Neurobiol (in press)
Nalavade R, Griesche N, Ryan DP, Hildebrand S, Krauss S (2013) Mechanisms of RNA-induced toxicity in CAG repeat disorders. Cell Death Dis 4:e752. doi:10.1038/cddis.2013.276
Ochaba J, Lukacsovich T, Csikos G, Zheng S, Margulis J, Salazar L, Mao K, Lau AL, Yeung SY, Humbert S, Saudou F, Klionsky DJ, Finkbeiner S, Zeitlin SO, Marsh JL, Housman DE, Thompson LM, Steffan JS (2014) Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc Natl Acad Sci U S A 111:16889–16894. doi:10.11073/pnas.1420103111
Pearce MM, Spartz EJ, Hong W, Luo L, Kopito RR (2015) Prion-like transmission of neuronal huntingtin aggregates to phagocytic glia in the Drosophila brain. Nat Commun 6:6768. doi:10.1038/ncomms7768
Pecho-Vrieseling E, Rieker C, Fuchs S, Bleckmann D, Esposito MS, Botta P, Goldstein C, Bernhard M, Galimberti I, Müller M, Lüthi A, Arber S, Bouwmeester T, van der Putten H, Di Giorgio FP (2014) Transneuronal propagation of mutant huntingtin contributes to non-cell autonomous pathology in neurons. Nat Neurosci 17(8):1064–1072. doi:10.1038/nn.3761
Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, Simons K (2006) Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci U S A 103(30):11172–11177
Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR (2009) Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol 11(2):219–225. doi:10.1038/ncb1830
Rilla K, Siiskonen H, Tammi M, Tammi R (2014) Hyaluronan-coated extracellular vesicles–a novel link between hyaluronan and cancer. Adv Cancer Res 123:121–148. doi:10.1016/B1978-0-12-800092-2.00005-8
Saman S, Lee NC, Inoyo I, Jin J, Li Z, Doyle T, McKee AC, Hall GF (2014) Proteins recruited to exosomes by tau overexpression implicate novel cellular mechanisms linking tau secretion with Alzheimer’s disease. J Alzheimers Dis 40(S1):S47–70. doi:10.3233/JAD-132135
Schneider A, Simons M (2013) Exosomes: vesicular carriers for intercellular communication in neurodegenerative disorders. Cell Tissue Res 352(3):33–47. doi:10.1007/s00441-00012-01428-00442
Squadrito ML, Baer C, Burdet F, Maderna C, Gilfillan GD, Lyle R, Ibberson M, De Palma M (2014) Endogenous RNAs modulate microRNA sorting to exosomes and transfer to acceptor cells. Cell Rep 8(5):1432–1446. doi:10.1016/j.celrep.2014.07.035
The Huntington’s Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72(6):971–983
Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME (2000) Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet 9(19):2799–2809
Trottier Y, Lutz Y, Stevanin G, Imbert G, Devys D, Cancel G, Saudou F, Weber C, David G, Tora L (1995) Polyglutamine expansion as a pathological epitope in Huntington’s disease and four dominant cerebellar ataxias. Nature 378(6555):403–406
Tsoi H, Chan HY (2014) Roles of the nucleolus in the CAG RNA-mediated toxicity. Biochim Biophys Acta 1842(6):779–784. doi:10.1016/j.bbadis.2013.11.015
Velier J, Kim M, Schwarz C, Kim TW, Sapp E, Chase K, Aronin N, DiFiglia M (1998) Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways. Exp Neurol 152(1):34–40
Vonsattel JP, DiFiglia M (1998) Huntington disease. J Neuropathol Exp Neurol 57(5):369–384
Zappulli V, Friis KP, Fitzpatrick Z, Maguire CA, Breakefield XO (2015) Extracellular vesicles and intercellular communication within the nervous system. J Clin Invest (in press)
Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA, Nan Z, Forster C, Low WC, Schoser B, Somia NV, Clark HB, Schmechel S, Bitterman PB, Gourdon G, Swanson MS, Moseley M, Ranum LP (2011) Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A 108(1):260–265. doi:10.1073/pnas.1013343108
Acknowledgments
We thank Ms. Suzanne McDavitt for skilled editorial assistance. Drs. Marian DiFiglia, Ellen Sapp, and Neal Aronin for insightful comments on HD pathophysiology. This work was supported by the NIH Common Fund through the Office of Strategic Coordination/Office of the NIH Director, NCI U19 CA179563 (XOB), and NIH NRSA NIA postdoctoral training grant, 2T32AG000222 (JSR). Additional support was from NIH 3R01 NS039074 (SF). Lentiviral vectors were produced by the MGH Vector Core supported by NIN/NINDS P30 NS045775 (XOB and Dr. Bakhos Tannous).
Author information
Authors and Affiliations
Corresponding author
Additional information
Xuan Zhang, Erik R. Abels, and Jasmina S. Redzic have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10571_2016_350_MOESM1_ESM.tiff
RT-PCR of polyQ-GFP RNA with different repeat lengths from transduced 293T cells and EVs. 293T cells were transduced using lentiviral vectors encoding GFP (not shown), Httex1-25Q-GFP, and Httex1-97Q-GFP. Two weeks later (equivalent to about 6 passages) RNA from cells and EVs was extracted using the miRNeasy mini kit. RT-PCR products were analyzed on 1 % agarose gel using 1 kb Quick-Load ladder. [Open arrowhead—25Q-GFP; solid arrowhead—97Q-GFP; gray arrowhead—GFP RNAs.] (TIFF 11720 kb)
10571_2016_350_MOESM2_ESM.tiff
Bright field image showing striatal neuron-like STHdhQ7/Q7 and STHdhQ111/Q111 cells 12 h after differentiation. Immortalized mouse striatal cell lines, STHdhQ111/Q111 and STHdhQ7/Q7 were normally cultured in high glucose DMEM (Corning) plus 10 % FBS and 40 mg/ml of G418 at 33oC. After neuronal differentiation, which was induced by incubation with a dopamine cocktail of α-FGF (10 ng/ml), 3-IBMX (240 μM), forskolin (48.6 μM), and dopamine (5 μM) (Sigma) in DMEM/F12 for 12 h, cells develop long neuron-like processes with STHdhQ7/Q7 appearing to have larger soma than the STHdh Q111/Q111 cells. (TIFF 11720 kb)
10571_2016_350_MOESM3_ESM.tiff
HTT protein was detected in the STHdh111/111 cells but not in the EVs from these cells. Striatal cells were allowed to attach for 6 h before being differentiated. Cells and EVs in conditioned media were harvested from both striatal cell lines 48 h after differentiation. Western blots were performed for both striatal cells and EV lysates using monoclonal 3B5H10 antibody. The HTT protein (approx. 350 kDa) was found in STHdh111/111 cells, but was not detectable in STHdh7/7 cells, consistent with the low levels of HTT protein in the latter (Krauss et al. 2013). No HTT immunoreactive proteins were found in the EVs from these cell lines. Open circle = non-specific protein; solid circle = HTT protein. (TIFF 11720 kb)
Rights and permissions
About this article

Cite this article
Zhang, X., Abels, E.R., Redzic, J.S. et al. Potential Transfer of Polyglutamine and CAG-Repeat RNA in Extracellular Vesicles in Huntington’s Disease: Background and Evaluation in Cell Culture. Cell Mol Neurobiol 36, 459–470 (2016). https://doi.org/10.1007/s10571-016-0350-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-016-0350-7