
Abstract
Perampanel is a novel α-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor (AMPAR) antagonist, approved in over 35 countries as an adjunctive therapy for the treatment of seizures. Recently, it was found to exert protective effects against ischemic neuronal injury in vitro. In the present study, we investigated the potential protective effects of perampanel in a traumatic brain injury (TBI) model in rats. Oral administration with perampanel at a dose of 5 mg/kg exerted no major organ-related toxicities. We found that perampanel significantly attenuated TBI-induced brain edema, brain contusion volume, and gross motor dysfunction. The results of Morris water maze test demonstrated that perampanel treatment also improved cognitive function after TBI. These neuroprotective effects were accompanied by reduced neuronal apoptosis, as evidenced by decreased TUNEL-positive cells in brain sections. Moreover, perampanel markedly inhibited lipid peroxidation and obviously preserved the endogenous antioxidant system after TBI. In addition, enzyme-linked immunosorbent assay (ELISA) was performed at 4 and 24 h after TBI to evaluate the expression of inflammatory cytokines. The results showed that perampanel suppressed the expression of pro-inflammatory cytokines TNF-α and IL-1β, whereas increased the levels of anti-inflammatory cytokines IL-10 and TGF-β1. These data show that the orally active AMPAR antagonist perampanel affords protection against TBI-induced neuronal damage and neurological dysfunction through anti-oxidative and anti-inflammatory activity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Traumatic brain injury (TBI) is broadly defined as an alteration in brain function, or other evidence of brain pathology, caused by an external force (Chang et al. 2015). It remains a serious public health problem, and the World Health Organization (WHO) has placed TBI as a 21st century epidemic equivalent only to malaria and HIV/AIDS (Chua et al. 2007; Chen et al. 2011). Typically, TBI is divided into primary and secondary injury. The primary injury is followed by a variety of physiological, cellular, and molecular responses, which leads to sustained and progressive neuronal degeneration, resulting in permanent functional deficits (Davis 2000). In spite of dramatic improvements in the management of TBI, no multicenter phase 3 randomized controlled trial of a neuroprotective agent in patients with TBI has shown an improvement in a patient-centered outcome (Nichol et al. 2015).
Most excitatory transmission in the brain is mediated by the ionotropic glutamate receptors (iGluRs), such as N-methyl-d-aspartic receptor (NMDAR) and α-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor (AMPAR). Changed expression and function of these receptors are commonly implicated in pathological signaling occurring in several disease states, including traumatic and ischemic brain injury (Arundine and Tymianski 2004; Lea and Faden 2001). The AMPAR is composed of various combinations of four possible subunits, GluR1, GluR2, GluR3, and GluR4, and its Ca2+ conductance differs markedly according to whether the GluR2 subunit is present or not (Seeburg et al. 2001). Although alterations in intracellular calcium levels are a proximal event for AMPAR activation, several downstream signaling cascades, such as protein kinases, apoptotic factors, and inflammatory cytokines, have been found to be involved in AMPAR-mediated neuronal injury (Spaethling et al. 2012; Henley and Wilkinson 2013; Turetsky et al. 2005). Therefore, targeting to the AMPAR can be considered as a practical strategy for neuronal injury therapy. Schumann et al. found that the expression of GluR1 subunit of the AMPAR was increased in the hippocampus, but decreased in the cortex after TBI (Schumann et al. 2008). The selective competitive AMPAR antagonist NBQX was shown to exert robust neuroprotective effects in models of focal and global ischemia (Smith and Meldrum 1993; Sheardown et al. 1990). However, the effect of AMPAR blocking on neuronal injury and neurological function after TBI has not been determined. The aim of this study was to assess the potential neuroprotective activity and related mechanism of perampanel, an orally active, noncompetitive AMPAR antagonist in an in vivo TBI model.
Materials and Methods
Animals
Adult male Sprague–Dawley (SD) rats weighing 250–300 g were obtained from the Laboratory Animal Center of the Fourth Military Medical University. The animals had continuous access to food and water and were housed in cages in a room maintained at 20–22 °C with a 12 h light/12 h dark cycle. All experimental protocols and animal handling procedures were performed in accordance with the National Institutes of Health (NIH) guidelines for the use of experimental animals and approved by the Institutional Animal Care and Use Committee of the Fourth Military Medical University.
TBI Model
TBI was induced using a controlled cortical impact (CCI) model in accordance with previously detailed methods (Israelsson et al. 2008; Chen et al. 2011). Briefly, animals were anesthetized using 2 % isoflurane in oxygen and placed in the stereotaxic frame. Rectal temperature was monitored and maintained between 37.0 and 38.0 °C with a thermostatically controlled heating pad. A 6-mm-diameter craniotomy was performed using a portable drill over the right sensorimotor cortex, midway between lambda and bregma sutures. The dura mater was kept intact over the cortex. To induce injury, a pneumatic piston impactor device with a 5-mm diameter and rounded tip was used to impact the brain at a depth of 2 mm (velocity 5 m/s). Sham animals underwent an identical surgery with the exception of TBI.
Measurement of Brain Edema
Brain edema was determined with the wet-dry method 24 h after injury. Rats were killed by decapitation under deep anesthesia, and the brain was quickly removed. Tissue samples from injured hemispheres were dissected and weighed immediately to get wet weight. Dry weight was determined after heating the tissue for 48 h at 100 °C. Brain water content was then calculated using the following formula: \( \% \,{\text{H}}_{ 2} {\text{O}} = \left( {1 - {\text{dry weight/wet weight}}} \right) \times 100\,\% \)
Quantification of Contusion Volume
Contusion volume was measured 7 days after TBI by morphometric image analysis. Rats were anesthetized with 4 % isoflurane in oxygen and decapitated. The brains were removed and frozen in nitrogen vapor. A series of 40-mm-thick coronal brain sections were cut on a cryostat at 500-μm intervals. Brain sections were stained with hematoxylin and eosin for lesion volume calculation. The areas of the contusions were integrated, and the results were represented as a volume percentage of the contusion compared with the contralateral hemisphere to avoid the interference from brain edema in the ipsilateral hemisphere.
Behavioral Assessments
We used beam-balance and beam-walk tasks to assess gross and fine motor function (Jiang et al. 2013). The beam-balance task consists of placing the animal on an elevated (90-cm) narrow wooden beam (1.5 cm wide) and recording the duration. The animal remains on the beam for a maximum of 60 s. The beam-walk task allows for the assessment of refined locomotor activity. Briefly, the task consists of training or assessing animals using a negative-reinforcement paradigm to escape ambient light and high-decibel white noise by traversing an elevated (90-cm) narrow wooden beam (2.5 × 100 cm) and entering a darkened goal box at the opposite end. We assessed performance by the time to traverse the beam. Animals were pre-trained on both motor tasks 1 day before the operation and assessed on the day of operation to determine baseline performance. Postoperative testing occurred on days 1–5 and consisted of three trials (60 s allotted time) per day on each task. We used the average daily scores for each subject in statistical analysis.
Morris Water Maze Test
The Morris water maze (MWM) test was used to evaluate spatial learning and memory function as previously described with slight modifications (Bermpohl et al. 2007). A circular pool (150 cm diameter, 60 cm deep) was filled with opaque water to a depth of 45 cm and a platform 12 cm in diameter was placed 1 cm below the water’s surface. The rats were pre-trained for 4 days prior to TBI and then assessed 3 days after TBI. Each animal was given four trials per day, 120 s per trial. For each training trial, a rat was randomly placed in 1 of the 4 quadrants and allowed to swim freely for 120 s or until it found the platform. If the rat was unable to find the platform within 120 s, it was gently guided to the platform by the experimenter. The latency time to reach the hidden platform was recorded. 2 hours after the training trials, a probe trial was performed to test spatial memory function. The platform was removed and the rat was allowed to search freely for 120 s. The number of times each rat crossed the former platform location was measured to assess spatial memory.
TUNEL Staining
Neuronal apoptosis was measured by TUNEL staining, a method used to observe DNA strand breaks in nuclei. In brief, sections of 4 μm thick were cut and mounted on poly-l-lysine-coated slides, and treated with proteinase K solution (20 μg/ml) for 10 min at room temperature to permeabilize tissues. TUNEL staining was performed by labeling with fluorescein TUNEL reagent mixture for 60 min at 37 °C according to the manufacturer’s suggested protocol (Promega, Madison, WI, USA), and examined under a fluorescence microscopy. The number of TUNEL-positive cells in each section in 10 microscopic fields (at ×600 magnification) was counted by an investigator blinded to the grouping.
Evaluation of Lipid Peroxidation
After various treatments, the ipsilateral cortical tissues were dissected from the rats and homogenized in chilled PBS, and then centrifuged at 10,000g at 4 °C for 10 min. The supernatants were collected, and the protein content was determined using a BCA protein assay kit (Pierce, Rockford, IL, USA). The levels of MDA and 4-HNE were detected by commercial kits according to the manufacturer’s suggested protocol (Nanjing Jiancheng Bioengineering Institute, China).
Measurement of Antioxidant Enzymatic Activity
The brain tissue samples obtained above were also used for detecting the antioxidant enzymatic activities. The enzyme activities of catalase (CAT), superoxide dismutase (SOD), and glutathione transferase (GST) in tissue homogenates were measured according to the technical manual of the detection kits (Cayman Chemical, USA). The activities were expressed as the percentage of sham.
Estimation of Inflammatory Cytokines
To detect the expression of inflammation-related cytokines, rats were sacrificed at 4 and 24 h after TBI and the brain tissue homogenates were obtained from the injured cerebral hemisphere. The concentrations of TNF-α, IL-1β, IL-10, and TGF-β1 were measured using specific ELISA kits according to the manufacturers’ instructions (Boster Biological Technology, Wuhan, China).
Western Blot Analysis
The homogenates obtained above were also used for Western blot analysis. Forty μg of protein was resolved on 10 % SDS-PAGE gel and transferred onto polyvinylidene difluoride (PVDF) membranes. Membranes were blocked with 5 % nonfat milk and incubated with the Bcl-2 (1:800; Cell Signaling Technology, No. 2876), Bax (1:600; Cell Signaling Technology, No. 2772), Cleaved-caspase-3 (1:200; Cell Signaling Technology, No. 9661), or β-actin (1:800; Sigma, A5316) primary antibodies. Membranes were then washed and incubated for 1 h at room temperature with secondary antibodies. The Image J software was used to quantify the optical density of each band.
Statistical Analysis
Statistical analysis was performed using SPSS 16.0, a statistical software package. Statistical evaluation of the data was performed by one-way analysis of variance (ANOVA) followed by Bonferroni’s multiple comparisons or unpaired t test (two groups). A value of p < 0.05 was considered statistically significant.
Results
Perampanel Protects Against TBI-Induced Neuronal Damage
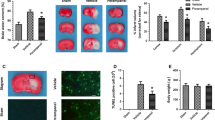
We first determined the cytotoxic effects of perampanel on normal tissues. H&E staining of the organs (heart, lung, liver, and kidney) collected from our experiments suggested no major organ-related toxicities (data not shown). Treatment with perampanel significantly decreased the brain edema after TBI, as evidenced by decreased brain water content (Fig. 1a). We also determined brain contusion volume at 7 days TBI, and the results showed that perampanel markedly attenuated the brain contusion volume after TBI (Fig. 1b).

Perampanel protects against TBI-induced neuronal damage. The rats were orally administered with saline water or perampanel (5 mg/kg) at 5 min after TBI, and brain water content at 24 h (a) and contusion volume at 7 days (b) were assayed, respectively. Values are expressed as mean ± SEM (n = 6/group). # p < 0.05 versus Sham. *p < 0.05 versus vehicle
Perampanel Preserves Motor Function After TBI
Next, we used the beam-balance task to evaluate gross motor function. The TBI-injured rats showed significantly worse performance on day 1 after TBI, and improved over time up to day 5 after injury (Fig. 2a). Treatment with perampanel significantly improved the beam-balance performance compared with vehicle group. In addition, beam-walk task was used to assess fine motor function. As shown in Fig. 2b, perampanel treatment obviously decreased the time to traverse the beam in TBI-injured animals. These results indicated that perampanel exert protective effects against TBI-induced motor dysfunction.

Perampanel preserves motor function after TBI. The rats were orally administered with saline water or perampanel (5 mg/kg) at 5 min after TBI. Beam-balance test (a time to maintain balance on an elevated narrow beam) and beam-walk test (b time to traverse an elevated narrow beam) were assayed up to 5 days after TBI. Values are expressed as mean ± SEM (n = 6/group)
Perampanel Improves Cognitive Function After TBI
We used water maze task to assess the effects of perampanel treatment on TBI-induced cognitive dysfunction, and during the trial, the path traveled by rats in each group was monitored by Ethovision software (Fig. 3a). The results showed that the average latency to the hidden platform in TBI-injured animals was significantly reduced by perampanel treatment (Fig. 3b). In the probe trial, the mean number of platform location crossing was significantly higher in the perampanel group compared with vehicle animals (Fig. 3c).

Perampanel improves cognitive function after TBI. The rats were orally administered with saline water or perampanel (5 mg/kg) at 5 min after TBI. Water maze task was performed to measure cognitive function, and computer tracking of the path traveled to reach the hidden platform was shown (a). Escape latency to the platform (b) and the number of platform location crossing (c) were recorded. Values are expressed as mean ± SEM (n = 6/group). # p < 0.05 versus Sham. *p < 0.05 versus Vehicle
Perampanel Attenuates TBI-Induced Neuronal Apoptosis
To investigate the effect of perampanel on neuronal apoptosis after TBI, TUNEL staining was performed on brain sections. As shown in Fig. 4a, there were few TUNEL-positive cells in the cortex of the sham group, and the number of apoptotic cells in the cortex surrounding the primary injury significantly increased at 24 h after TBI. Treatment with perampanel significantly attenuated the TBI-induced apoptosis in brain sections (Fig. 4b). Furthermore, we also detected the expression of apoptotic factors by Western blot analysis (Fig. 4c). As shown in Fig. 4d, perampanel treatment significantly increased Bcl-2 expression, but decreased the expression of Bax and cleaved-caspase-3.

Perampanel attenuates TBI-induced neuronal apoptosis. The rats were orally administered with saline water or perampanel (5 mg/kg) at 5 min after TBI. TUNEL staining was used to detect apoptotic cell death in brain sections (a), and the number of TUNEL-positive cells was calculated (b). The expression of apoptosis-related factors was detected by Western blot (c) and calculated (d). Values are expressed as mean ± SEM (n = 6/group). # p < 0.05 versus Sham. *p < 0.05 versus Vehicle
Perampanel Reduces TBI-Induced Oxidative Stress
To define whether perampanel treatment inhibits TBI-induced oxidative stress, lipid peroxidation was determined by measuring the expression of MDA and 4-HNE. As shown in Fig. 5a, b, TBI significantly evaluated the levels of MDA and 4-HNE in the ipsilateral hemisphere, which were both partially prevented by perampanel treatment. We also measured the enzymatic activities of SOD, CAT, and GST to test the effects of perampanel on the endogenous antioxidant system (Fig. 5b). The results showed that TB-induced decreases in these enzymes activities were significantly attenuated by perampanel.

Perampanel reduces TBI-induced oxidative stress. The rats were orally administered with saline water or perampanel (5 mg/kg) at 5 min after TBI. Lipid peroxidation was assayed by measuring the expression of MDA (a) and 4-HNE (b). The enzymatic activities of SOD, CAT, and GST were detected (c). Values are expressed as mean ± SEM (n = 6/group). # p < 0.05 versus Sham. *p < 0.05 versus Vehicle
Perampanel Inhibits TBI-Induced Inflammatory Responses
To determine whether perampanel-induced protection against TBI was mediated by anti-inflammatory activity, the expression of pro-inflammatory (TNF-α and IL-1β) and anti-inflammatory (IL-10 and TGF-β1) cytokines was measured by ELISA at 4 and 24 h after injury. The levels of TNF-α and IL-1β were significantly increased at 24 h, but not 4 h, after TBI. Treatment with perampanel attenuated the expression of TNF-α and IL-1β at 24 h, whereas there was no statistical difference in TNF-α and IL-1β levels at 4 h between each group (Fig. 6a, b). The concentrations of IL-10 and TGF-β1 at 24 h in vehicle group were lower than that in sham animals. As shown in Fig. 6c, d perampanel treatment evaluated the expression of IL-10 and TGF-β1 at both 4 and 24 h after TBI.

Perampanel inhibits TBI-induced inflammatory responses. The rats were orally administered with saline water or perampanel (5 mg/kg) at 5 min after TBI. The expression of TNF-α (a), IL-1β (b), IL-10 (c), and TGF-β1 (d) were detected at 4 h and 24 h after TBI, respectively. Values are expressed as mean ± SEM (n = 6/group). # p < 0.05 versus Sham. *p < 0.05 versus Vehicle
Discussion
TBI triggers acute neuronal damage and followed neurological deficits in patients. One of the important mechanisms of this process is the activation of excitotoxicity induced by excitatory amino acid. Several lines of evidence have linked excitotoxicity to the pathogenesis of both primary and secondary brain injury after TBI (Obrenovitch and Urenjak 1997; Hwabejire et al. 2013). Glutamate is the principal excitatory neurotransmitter in the CNS, and excessive release of glutamate can lead directly to excitotoxicity in neurons via its ionotropic receptors (Durand et al. 2008; Calabresi et al. 2003; Stone and Addae 2002). On activation by exogenous and endogenous ligands, glutamate receptors recruit their adapter proteins, activate downstream kinases, and thereby induce the expression of pro-apoptotic genes. Previous studies showed that AMPAR-mediated signaling was activated in ischemic brain, and AMPAR antagonists were shown to exert robust neuroprotective effects in models of focal and global ischemia (Smith and Meldrum 1993; Sheardown et al. 1990). However, whether blocking AMPAR has neuroprotective effects on TBI remains unknown. Thus, we evaluated TBI-induced oxidative stress, inflammatory responses and subsequent brain damage in rats treated with perampanel, an orally active, noncompetitive AMPAR antagonist.
In view of the heterogeneous nature of the clinical situation in TBI, numerous animal models of TBI have been developed (Xiong et al. 2013). Here, we used CCI model to mimic TBI in SD rats. This model uses a pneumatic impact device to drive a rigid impactor onto the exposed intact dura, and mimics cortical tissue loss, acute subdural hematoma, brain edema, and even coma (Dixon et al. 1991; Lighthall et al. 1990). The advantage of this injury model is that it can control mechanical factors, such as time, velocity, and depth of impact (Cernak 2005; Mao et al. 2006). Body temperature, heart rate, blood PH, and partial pressure of oxygen were monitored during surgery, and there was no significant difference in these parameters between the perampanel-treated and the -untreated animals (data not shown). Results from beam-balance, beam-walk, and water maze task showed that our injury model induced obvious motor and cognitive deficits in injured rats, which confirmed the success of the model of TBI.
Among the postsynaptic targets for neuroprotective drug research, the AMPAR has received particular attention for many years. However, many barriers have hindered the use of AMPAR antagonists as valuable drug candidates, such as poor blood–brain barrier (BBB) penetration, short half-lives and, importantly, side effects associated with pharmacological mechanisms (Weiser 2005; Walters et al. 2005; Grossman et al. 2009). To overcome these challenges, perampanel was discovered by high-throughout screening using the [3H]AMPA-binding assay and AMPA-induced cell death assays in primary cultured neurons (Hibi et al. 2012). It has been approved in over 35 countries as an adjunctive therapy for the treatment of seizures. Perampanel was shown to inhibit AMPAR-mediated responses during neurotransmission at an IC50 of 230 nM in vivo and 93 nM under in vitro conditions (Ceolin et al. 2012; Hanada et al. 2011). In addition, perampanel did not inhibit kainate- and NMDAR-mediated responses in neurotransmission or in NMDA-induced Ca2+ influx, making it more safer with low risk of the psychotomimetic side effects that are known to be elicited by inhibition of NMDAR (Olney et al. 1991). The results of our present study also demonstrated that perampanel was an orally active AMPAR antagonist with little organ-related toxicities. More importantly, we investigated the effect of AMPAR blocking on traumatic neuronal damage and extended the protective effects of perampanel into TBI.
Previous researches on mechanisms of TBI largely focus on the cascade of excitotoxicity, but increasing evidence supports the important role of inflammatory response in neuronal cell death and functional deficits (Correale and Villa 2004). Our data also showed that the levels of TNF-α and IL-1β were significantly increased, while the expressions of TNF-α and IL-1β were decreased after TBI. Inflammation-related cytokines are usually classified into pro- and anti-inflammatory mediators based on their ability to promote or suppress immune activation (Ziebell and Morganti-Kossmann 2010; Morganti-Kossmann et al. 2001). For example, increased expression of TNF-α and IL-1β have been detected in the CSF and brain parenchyma after brain injury in both humans and rodents (Winter et al. 2002; Woodroofe et al. 1991; Chao et al. 1995). In contrast, exogenous administration or gene transfer of IL-10 and TGF-β1 was shown to exert neuroprotection after brain injury (Ooboshi et al. 2005; Pang et al. 2001; Frenkel et al. 2003). In the present study, we found that perampanel treatment oppositely regulated the expression of pro-inflammatory and anti-inflammatory cytokines, which might contribute to its effect on TBI-induced lipid peroxidation and its anti-oxidative activity. Intriguingly, perampanel treatment was shown to increase the expression of IL-10 and TGF-β1 in both 4 and 24 h after injury TBI-injured animals. The AMPA and kainate positioning of subunits in their respective receptor complex is similar to the arrangement present in the NMDA receptor (Bolton and Paul 2006). Previous studies showed that noncompetitive NMDAR antagonists attenuate BCR and Toll-like receptor 4 (TLR4) B cell signaling and effector function and can foster IL-10 production (Simma et al. 2014). Furthermore, CNS-TGF-β1-deficient mice showed aberrant synaptic plasticity in the CA1 area of the hippocampus in a GluN2B-dependent manner and these mice were highly sensitive to excitotoxic injury (Koeglsperger et al. 2013). It is speculated that perampanel might promote activity of anti-inflammatory cytokines through indirect effects on NMDAR using similar arrangement of subunits. In addition, perampanel may also regulate neuronal inflammation after TBI through crosstalk between pro- and anti-inflammatory cytokines, which needs to be further determined.
A limitation of our study is that the tissues for assays on the concentrations of inflammatory cytokines and the enzymatic activities of anti-oxidative enzymes were harvested from entire cerebral hemisphere instead of specific brain tissues, such as cortex, striatum, and hippocampus. If the measurements were performed in the individual cerebral structures, more detailed information about the effect of perampanel on oxidative stress and inflammatory responses in specific cerebral areas would be provided.
In conclusion, our findings in a rat model of CCI-induced TBI support the potential use of the AMPAR antagonist perampanel as a therapeutic agent in the therapy of conditions is associated with TBI. Through its anti-oxidative and anti-inflammatory activities, this orally active, noncompetitive AMPAR antagonist might be widely used in medical applications as safe and effective neuroprotective drug.
References
Arundine M, Tymianski M (2004) Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol Life Sci 61(6):657–668. doi:10.1007/s00018-003-3319-x
Bermpohl D, You Z, Lo EH, Kim HH, Whalen MJ (2007) TNF alpha and Fas mediate tissue damage and functional outcome after traumatic brain injury in mice. J Cereb Blood Flow Metab 27(11):1806–1818. doi:10.1038/sj.jcbfm.9600487
Bolton C, Paul C (2006) Glutamate receptors in neuroinflammatory demyelinating disease. Mediat Inflamm 2:93684. doi:10.1155/MI/2006/93684
Calabresi P, Centonze D, Cupini LM, Costa C, Pisani F, Bernardi G (2003) Ionotropic glutamate receptors: still a target for neuroprotection in brain ischemia? Insights from in vitro studies. Neurobiol Dis 12(1):82–88
Ceolin L, Bortolotto ZA, Bannister N, Collingridge GL, Lodge D, Volianskis A (2012) A novel anti-epileptic agent, perampanel, selectively inhibits AMPA receptor-mediated synaptic transmission in the hippocampus. Neurochem Int 61(4):517–522. doi:10.1016/j.neuint.2012.02.035
Cernak I (2005) Animal models of head trauma. NeuroRx J Am Soc Exp NeuroTher 2(3):410–422. doi:10.1602/neurorx.2.3.410
Chang J, Phelan M, Cummings BJ (2015) A meta-analysis of efficacy in pre-clinical human stem cell therapies for traumatic brain injury. Exp Neurol 273:225–233. doi:10.1016/j.expneurol.2015.08.020
Chao CC, Hu S, Ehrlich L, Peterson PK (1995) Interleukin-1 and tumor necrosis factor-alpha synergistically mediate neurotoxicity: involvement of nitric oxide and of N-methyl-D-aspartate receptors. Brain Behav Immun 9(4):355–365. doi:10.1006/brbi.1995.1033
Chen T, Liu W, Chao X, Zhang L, Qu Y, Huo J, Fei Z (2011) Salvianolic acid B attenuates brain damage and inflammation after traumatic brain injury in mice. Brain Res Bull 84(2):163–168. doi:10.1016/j.brainresbull.2010.11.015
Chua KS, Ng YS, Yap SG, Bok CW (2007) A brief review of traumatic brain injury rehabilitation. Ann Acad Med, Singap 36(1):31–42
Correale J, Villa A (2004) The neuroprotective role of inflammation in nervous system injuries. J Neurol 251(11):1304–1316. doi:10.1007/s00415-004-0649-z
Davis AE (2000) Cognitive impairments following traumatic brain injury. Etiologies and interventions. Crit Care Nurs Clin North Am 12(4):447–456
Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL (1991) A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods 39(3):253–262
Durand D, Pampillo M, Caruso C, Lasaga M (2008) Role of metabotropic glutamate receptors in the control of neuroendocrine function. Neuropharmacology 55(4):577–583. doi:10.1016/j.neuropharm.2008.06.022
Frenkel D, Huang Z, Maron R, Koldzic DN, Hancock WW, Moskowitz MA, Weiner HL (2003) Nasal vaccination with myelin oligodendrocyte glycoprotein reduces stroke size by inducing IL-10-producing CD4+ T cells. J Immunol 171(12):6549–6555
Grossman SA, Ye X, Chamberlain M, Mikkelsen T, Batchelor T, Desideri S, Piantadosi S, Fisher J, Fine HA (2009) Talampanel with standard radiation and temozolomide in patients with newly diagnosed glioblastoma: a multicenter phase II trial. J Clin Oncol 27(25):4155–4161. doi:10.1200/JCO.2008.21.6895
Hanada T, Hashizume Y, Tokuhara N, Takenaka O, Kohmura N, Ogasawara A, Hatakeyama S, Ohgoh M, Ueno M, Nishizawa Y (2011) Perampanel: a novel, orally active, noncompetitive AMPA-receptor antagonist that reduces seizure activity in rodent models of epilepsy. Epilepsia 52(7):1331–1340. doi:10.1111/j.1528-1167.2011.03109.x
Henley JM, Wilkinson KA (2013) AMPA receptor trafficking and the mechanisms underlying synaptic plasticity and cognitive aging. Dialogues Clin Neurosci 15(1):11–27
Hibi S, Ueno K, Nagato S, Kawano K, Ito K, Norimine Y, Takenaka O, Hanada T, Yonaga M (2012) Discovery of 2-(2-oxo-1-phenyl-5-pyridin-2-yl-1,2-dihydropyridin-3-yl)benzonitrile (perampanel): a novel, noncompetitive alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropanoic acid (AMPA) receptor antagonist. J Med Chem 55(23):10584–10600. doi:10.1021/jm301268u
Hwabejire JO, Jin G, Imam AM, Duggan M, Sillesen M, Deperalta D, Jepsen CH, Lu J, Li Y, deMoya MA, Alam HB (2013) Pharmacologic modulation of cerebral metabolic derangement and excitotoxicity in a porcine model of traumatic brain injury and hemorrhagic shock. Surgery 154(2):234–243. doi:10.1016/j.surg.2013.04.008
Israelsson C, Bengtsson H, Kylberg A, Kullander K, Lewen A, Hillered L, Ebendal T (2008) Distinct cellular patterns of upregulated chemokine expression supporting a prominent inflammatory role in traumatic brain injury. J Neurotrauma 25(8):959–974. doi:10.1089/neu.2008.0562
Jiang X, Huang Y, Lin W, Gao D, Fei Z (2013) Protective effects of hydrogen sulfide in a rat model of traumatic brain injury via activation of mitochondrial adenosine triphosphate-sensitive potassium channels and reduction of oxidative stress. J Surg Res 184(2):e27–e35. doi:10.1016/j.jss.2013.03.067
Koeglsperger T, Li S, Brenneis C, Saulnier JL, Mayo L, Carrier Y, Selkoe DJ, Weiner HL (2013) Impaired glutamate recycling and GluN2B-mediated neuronal calcium overload in mice lacking TGF-beta1 in the CNS. Glia 61(6):985–1002. doi:10.1002/glia.22490
Lea PMt, Faden Al (2001) Traumatic brain injury: developmental differences in glutamate receptor response and the impact on treatment. Ment Retard Dev Disabil Res Rev 7(4):235–248. doi:10.1002/mrdd.1033
Lighthall JW, Goshgarian HG, Pinderski CR (1990) Characterization of axonal injury produced by controlled cortical impact. J Neurotrauma 7(2):65–76
Mao H, Zhang L, Yang KH, King AI (2006) Application of a finite element model of the brain to study traumatic brain injury mechanisms in the rat. Stapp Car Crash J 50:583–600
Morganti-Kossmann MC, Rancan M, Otto VI, Stahel PF, Kossmann T (2001) Role of cerebral inflammation after traumatic brain injury: a revisited concept. Shock 16(3):165–177
Nichol A, French C, Little L, Haddad S, Presneill J, Arabi Y, Bailey M, Cooper DJ, Duranteau J, Huet O, Mak A, McArthur C, Pettila V, Skrifvars M, Vallance S, Varma D, Wills J, Bellomo R (2015) Erythropoietin in traumatic brain injury (EPO-TBI): a double-blind randomised controlled trial. Lancet. doi:10.1016/S0140-6736(15)00386-4
Obrenovitch TP, Urenjak J (1997) Is high extracellular glutamate the key to excitotoxicity in traumatic brain injury? J Neurotrauma 14(10):677–698
Olney JW, Labruyere J, Wang G, Wozniak DF, Price MT, Sesma MA (1991) NMDA antagonist neurotoxicity: mechanism and prevention. Science 254(5037):1515–1518
Ooboshi H, Ibayashi S, Shichita T, Kumai Y, Takada J, Ago T, Arakawa S, Sugimori H, Kamouchi M, Kitazono T, Iida M (2005) Postischemic gene transfer of interleukin-10 protects against both focal and global brain ischemia. Circulation 111(7):913–919. doi:10.1161/01.CIR.0000155622.68580.DC
Pang L, Ye W, Che XM, Roessler BJ, Betz AL, Yang GY (2001) Reduction of inflammatory response in the mouse brain with adenoviral-mediated transforming growth factor-ss1 expression. Stroke 32(2):544–552
Schumann J, Alexandrovich GA, Biegon A, Yaka R (2008) Inhibition of NR2B phosphorylation restores alterations in NMDA receptor expression and improves functional recovery following traumatic brain injury in mice. J Neurotrauma 25(8):945–957. doi:10.1089/neu.2008.0521
Seeburg PH, Single F, Kuner T, Higuchi M, Sprengel R (2001) Genetic manipulation of key determinants of ion flow in glutamate receptor channels in the mouse. Brain Res 907(1–2):233–243
Sheardown MJ, Nielsen EO, Hansen AJ, Jacobsen P, Honore T (1990) 2,3-Dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline: a neuroprotectant for cerebral ischemia. Science 247(4942):571–574
Simma N, Bose T, Kahlfuss S, Mankiewicz J, Lowinus T, Luhder F, Schuler T, Schraven B, Heine M, Bommhardt U (2014) NMDA-receptor antagonists block B-cell function but foster IL-10 production in BCR/CD40-activated B cells. Cell Commun Signal 12:75. doi:10.1186/s12964-014-0075-5
Smith SE, Meldrum BS (1993) Cerebroprotective effect of a non-N-methyl-D-aspartate antagonist, NBQX, after focal ischaemia in the rat. Funct Neurol 8(1):43–48
Spaethling J, Le L, Meaney DF (2012) NMDA receptor mediated phosphorylation of GluR1 subunits contributes to the appearance of calcium-permeable AMPA receptors after mechanical stretch injury. Neurobiol Dis 46(3):646–654. doi:10.1016/j.nbd.2012.03.003
Stone TW, Addae JI (2002) The pharmacological manipulation of glutamate receptors and neuroprotection. Eur J Pharmacol 447(2–3):285–296
Turetsky D, Garringer E, Patneau DK (2005) Stargazin modulates native AMPA receptor functional properties by two distinct mechanisms. J Neurosci 25(32):7438–7448. doi:10.1523/JNEUROSCI.1108-05.2005
Walters MR, Kaste M, Lees KR, Diener HC, Hommel M, De Keyser J, Steiner H, Versavel M (2005) The AMPA antagonist ZK 200775 in patients with acute ischaemic stroke: a double-blind, multicentre, placebo-controlled safety and tolerability study. Cerebrovasc Dis 20(5):304–309. doi:10.1159/000087929
Weiser T (2005) AMPA receptor antagonists for the treatment of stroke. Curr Drug Targets CNS Neurol Disord 4(2):153–159
Winter CD, Iannotti F, Pringle AK, Trikkas C, Clough GF, Church MK (2002) A microdialysis method for the recovery of IL-1beta, IL-6 and nerve growth factor from human brain in vivo. J Neurosci Methods 119(1):45–50
Woodroofe MN, Sarna GS, Wadhwa M, Hayes GM, Loughlin AJ, Tinker A, Cuzner ML (1991) Detection of interleukin-1 and interleukin-6 in adult rat brain, following mechanical injury, by in vivo microdialysis: evidence of a role for microglia in cytokine production. J Neuroimmunol 33(3):227–236
Xiong Y, Mahmood A, Chopp M (2013) Animal models of traumatic brain injury. Nat Rev Neurosci 14(2):128–142. doi:10.1038/nrn3407
Ziebell JM, Morganti-Kossmann MC (2010) Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics 7(1):22–30. doi:10.1016/j.nurt.2009.10.016
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 81371447 and 81301037).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
There is no conflict of interest.
Additional information
Tao Chen, Shu-Hui Dai and Zhi-Quan Jiang have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Chen, T., Dai, SH., Jiang, ZQ. et al. The AMPAR Antagonist Perampanel Attenuates Traumatic Brain Injury Through Anti-Oxidative and Anti-Inflammatory Activity. Cell Mol Neurobiol 37, 43–52 (2017). https://doi.org/10.1007/s10571-016-0341-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-016-0341-8