
Abstract
In this research work, a series of thermoset polyurethane (tPU) nanocomposites and a thermoplastic PU (TPPU) were synthesized using semi-crystalline polyols. Poly(ε-caprolactone) (PCL), poly(tetramethylene glycol) (PTMG), and different architecture and compositions of PCL and PTMG were used as diols. PCL was used to synthesize a TPPU (PCL-TPPU) as the control specimen. Cellulose nanowhisker (CNW), with 1.0 wt% content, was used for synthesis of the tPU nanocomposites. The aim of this research was to investigate the impact of the thermosetting using CNWs and tuning crystallization of the polyols on the shape memory performance (SMP) of PUs. The analysis of the polyols’ crystallization and crystallization of the soft segments in the PU structures was studied to evaluate the impact of structural changes on the SMP of the PUs. The results of the dynamic mechanical thermal analysis showed that the tPUs have a large elastic modulus (E′) at temperatures around ~ 25–40 °C (~ 10 MPa) and very small tanδ height (below 0.15), which was attributed to their thermoset nature and presence of CNW with high E′ value (~ 110–220 GPa). The PCL-TPPU, as the control specimen, showed E′ in the range of 200–400 kPa. The PUs have also proved to be highly biocompatible.
Graphical abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Thermoplastic polyurethanes (TPPUs) and thermoset polyurethanes (tPUs) are a family of polymers that possess inherent thermal shape memory performance (SMP) (Jiu et al. 2016; Joo et al. 2018; Shirole et al. 2018). They can be thermally programmed through different heating and cooling cycles to undergo a reversible shape switching process between a permanent shape (L1) and a strain-induced temporary shape (L2). In a thermally programmable polymer there exists two main characteristic temperatures: switching temperature (Ts) and recovery temperature (Tr); and there exists two structural components: switching component (SC) and permanent component (PC) (Joo et al. 2018; Wang et al. 2018b; Kausar 2019). In a TPPU system, the hard domains play the role of the PC. In TPUs, the physical cross-link nature of the hard domains prohibits the slippage of the SC chains (Cao and Jana 2007; Wang et al. 2018a; Urbina et al. 2019). For TPPUs, the curing condition, including time and temperature, and the nature of hard segments would impact the ability of the hard segments to form segregated domains, which consequently impacts their SMP (Choi et al. 2012; Kausar 2019). However, for a tPU, the possibility of hard segment–soft segment mixing is eliminated and the final shape recovery improves to a large degree (Jiu et al. 2016). In a tPU structure, the soft domain plays the role of SC, and the cross-link points act as the PC (Jafari et al. 2020; Noormohammadi et al. 2021; Ranjbar et al. 2021). The SMP of PUs is defined by their ability to fix a temporary shape (shape fixity) and the ability to recover the original shape (shape recovery) (Jafari et al. 2020). It is known that elastic modulus plays a key role in tuning SMP. Thermosetting and the use of high modulus nanoparticles are two main procedures to improve elastic modulus (Ranjbar et al. 2021). In recent years, cellulose nanowhisker (CNW) has been the focus of numerous researches (Lu and Hsieh 2010) as the CNW is considered as a highly elastic nanorod particle, with an elastic modulus in the range of \(\approx\) 110–220 GPa. Upon ideal dispersion in polymer matrices, they can act as an efficient mediator to transfer the external applied energy in the direction of applied force and also increase the elastic modulus of the composites (Lahiji et al. 2010; Domingues et al. 2014; Tang et al. 2017).
CNWs are extracted from natural resources including cotton, wood, rice straw, and other cellulose-rich plants (George and Sabapathi 2015; Roman et al. 2009). Different extraction procedures were developed to isolate CNWs from their raw materials including acid-hydrolysis, oxidization method, ionic liquid and enzymatic hydrolysis (Trache et al. 2017; Vanderfleet and Cranston 2021). Each of these four main procedures have their own advantages and disadvantages. However, regarding the cost and time of the extraction process, acid-hydrolysis was considered as the main choice available for efficient extraction (Mariano et al. 2014). Cotton was also considered as the main renewable resource of CNWs (Xie et al. 2018; Du et al. 2019). The CNWs isolated from cotton by acid-hydrolysis had a crystallinity of 79–83% (SaifulAzry et al. 2021), an yield in the range of 57–69% with a diameter and length of 9–19 nm and 157–290 nm, respectively (Reid et al. 2017; Mu et al. 2019). CNWs have abundant -OH groups, which can be used for further chemical or physical modification (Roman et al. 2009; Tingaut et al. 2012). According to the literature, nanocomposites of thermoplastic polyurethanes (TPPUs) and CNWs have shown an improvement in mechanical properties including Young’s modulus (Rueda et al. 2013; Fortunati et al. 2017; Prataviera et al. 2018). Despite substantial research in this field, application of CNWs as an active agent in the field of thermoset polymers is limited (Pei et al. 2021). Their role as a cross-linking agent and their impact on the thermomechanical properties of the final thermoset polymer nanocomposites has not been fully investigated(Park et al. 2013).
In this work, different microstructures of polyols were prepared using PCL and poly(tetramethylene glycol) (PTMG). The polyols were composed of neat PCL and PTMG, with molecular weights of 2000 g/mol, their 50:50 w:w blends, the tri-block of PCL1000-PTMG2000-PCL1000 and the 100:50 w:w ratio of the blend and the block copolymer. Their crystallization behavior was studied. These specimens were further used to prepare thermoset PU/CNW nanocomposites. A thermoplastic PU of PCL (Mw: 2000 D) (PCL-TPPU) was synthesized as the control specimen to compare the different between the thermoplastic and thermoset PUs. The crystallization and dynamic behavior of the synthesized PUs were also studied. The mechanical properties, the shape memory performance of the PUs and the impact of the change in elastic modulus on shape fixity and recovery was thoroughly investigated. The cytocompatibility of the PUs was studied to evaluate their potential for biomedical applications.
Experimental section
Materials
Poly(ε-caprolactone) with molecular weight of 2000 D (PCL2000), Hexamethylene diisocyanate (HDI), Stannous Octoate (Sn(Oct)2), Dibutyl tindilaurate, and butane-diol (BDO) were purchased from Sigma and were used as received. Dimethylformamide (DMF, Sigma), Sulfuric acid (98%, sigma) were also used as received. Refined cotton (Iran, Golestan) was used as the source for extraction of cellulose nanocrystals. Poly(tetramethylene glycol)-diol (Mitsubishi chemical co.) with a molecular weight (Mw) of 2000 D (PTMG2000). DMEM and FBS (Gibco, UK), and penicillin/ streptomycin (Sigma). Culture plates and flasks (United Scientific F1004 Plastic Well Plate).
CNW extraction
The CNW extraction from purified cotton is fully described in our previous work (Noormohammadi et al. 2021). Here, we describe the procedure in brief. The sulfuric acid (98%) hydrolysis procedure was used for hydrolysis of cotton. 5 gr cotton and 100 mL acid solution in distilled water with a relative acid to water content of 50 mL: 50 mL was used for the hydrolysis procedure. The final suspension was centrifuged at 5000 rpm to extract the CNWs. Dialysis tube was used for increasing the pH of the CNWs to a range of 6.5–7.0. The surface-charge neutralized CNWs were further freeze-dried.
Block copolymer synthesis
The PCL1000-PTMG2000-PCL1000 block copolymer (Block 1:1) was synthesized using a pseudo-anionic bulk polymerization at 120 °C. Stannous Octoate (Sn(Oct)2) with a ratio of 0.2 mol% relative to PTMG2000 was used as the catalyst. In the first step of the polymerization, 10 gr of PTMG with a molecular weight of 2000 D was added to a 50 ml round bottom 3-neck flask and heated to 120 °C. Then, vacuum was applied to the melt to remove moisture and possible contamination. Sn(Oct)2 was added to the flask. ε-caprolactone monomer (5.3 ml) was added to the flask and the flask was then sealed and the reaction was kept to proceed for 24 h. The termination phase was performed using 5 ml solution of distilled water and a few droplets of 37% HCl. The final copolymer was precipitated in cold ether solvent. The efficiency of the polymerization was 94.6% and the resulted of Gel Permeation Chromatography showed that the final block copolymer possessed a number-averaged molecular weight of 4127 D with a PDI of 1.22.
Thermoset polyurethane synthesis
Table 1 provides the formulation recipe for synthesis of the thermoplastic PU and the thermoset PU/CNW nanocomposites. The molar ratio of HDI to each polyol was kept at 3:1. For synthesis of the control specimen of thermoplastic PU, BDO was used and for the thermoset PU nanocomposites the CNW content was 1.0 wt% relative to the prepolymer.
The synthesis procedure is as follows: (1) Weighting the polyol(s) and adding them into the 3-necked bottom round flask; (2) heating and stirring the polyol(s) at 80 °C and 250 rpm; (3) using vacuum for 15 min to eliminate the possible moisture and then, applying a continuous dry nitrogen purge; (4) adding the PU catalysis solution, Dibutyl tin dilaurate in DMF, to the polyol(s) using a syringe; (5) dropwise addition of HDI to the reaction media and allowing the reaction to proceed for 2 h to ensure complete conversion of diols into prepolymers; (6) adding BDO or the pre-sonicated CNW in DMF suspension to the flask; (7) allowing for the reaction to proceed for 30 min and then pouring the highly viscose solution into Teflon mold; finally, (8) putting the Teflon mold in oven at 70 °C for 24 h. For synthesis of the PCL-based thermoplastic PU (PCL-TPPU), the molar ratio of PCL:HDI:BDO was set to 1:3:2.
Cross-link density and degree measurement
The equilibrium swelling test was used for measuring the cross-link density (CLD) of the thermoset PU/CNW nanocomposites. Three samples of each specimen with dimensions of 1 cm \(\times\) 1 cm \(\times\) 1 mm were cut and then immersed into a glass containing toluene and the result for each specimen was averaged over three. The samples were kept in the solvent for 72 h at room conditions. The sealed glass containers were placed on a shaker to ensure dissolution of unreacted polyols into the solvent. The final stable weight of the swollen samples was selected as the equilibrium swelling weight (ESW). The thermodynamically developed Flory–Rehner equation, Eq. 1, was applied to measure the CLD.
The parameters of \(\upsilon\), \({V}_{0}\), \(\chi\), and \({V}_{r}\) represent the CLD, solvent molar-volume, polymer–solvent interaction parameter, volume fraction of the swollen thermoset PU/CNW nanocomposite. The cross-link degree or gel content of the specimens is measured using a Soxhlet extractor. Again, three samples of each specimen with dimensions of 1 cm \(\times\) 1 cm \(\times\) 1 mm were cut and placed into the Soxhlet and Toluene was used as the solvent. The siphon tube would create a flow to remove the dissolved polyols and procedure proceeded for 24 h. The final dry-weight of the specimens was measured and divided by the original weight to get the gel fraction.
Shape memory procedure
The shape memory performance analysis of the thermoset PU specimens was performed through two different procedures. In the first procedure, the specimens with 2.5 \(\times\) 1 \(\times\) 0.1 cm3 dimensions were bend over and fixed using a metal clamp. Then, the specimens were immediately put into liquid nitrogen for 5 min. Allowing for more freezing process time leads to brittleness and fracture of the specimens. In the next step, the specimens were put at room temperature and the clamp was removed. The camera was set to take pictures of the specimens every 15 s to follow the shape recovery process. The second procedure was to use a DMA instrument working in tension mode. The specimens’ dimension was 2 \(\times\) 0.5 \(\times\) 0.1 cm3. The thermal programming procedure was as follows: (A) increasing temperature to 37.5 °C; (B) applying a 30 N dynamic force to stretch the specimens; (C) decreasing temperature to − 25 °C; (D) Removing the force; (E) Increasing the temperature to 37.5 °C. The parameters of the shape memory performance, recovery ratio (Rr) and fixity ratio (Rf), are defined as Eqs. 2 and 3, respectively.
L1, L2 and L3 represent the stretched length at the elevated temperature, the length after unloading the applied force and the specimen length at the end of the recovery process.
Cell culture procedure
The Human Foreskin Fibroblast (HFF) cells were seeded onto the thermoset PU/CNW nanocomposites according to the procedure described in detail in our previous paper (Jafari et al. 2020). Here, we describe the procedure in summary: (1) washing the PU films with distilled water and after drying, they were UV-sterilized; (2) Tissue-culture grade polystyrene (TCPS, 48 wells) was used for cell seeding onto the tPU/CNW nanocomposite films; (3) HFF cells suspended in DMEM, with 10% FBS and 1% penicillin/ streptomycin, with a cell density of 2 \(\times\) 104 cells/specimen were seeded on the tPUs; (4) Using the standard condition of 37 °C with 96% humidity and 5% CO2 atmosphere in the incubator for maintaining the seeded cells; (5) The cell fixing was performed by 2.5% glutaraldehyde. The cell viability of the HFF cells seeded onto the tPU/CNW nanocomposites was measured through MTT assay. After 3 and 5 days of culture, 50 ml of MTT solution (5 mg/ml) was added to each one of the wells. The plates then were incubated for three hours. At the end, the supernatant was collected and a micro-plate reader (BioTek) was used for analysis of optical density. The cell viability is measured using Eq. 4.
OD and OD0 represent the optical density of suspensions related to cell-seeded plates and the blank control, respectively.
Characterization
The chemical structure analysis was performed using ATR-FTIR (MOD SRG 1100G, BOMEM) and Raman spectroscopy (Confocal Raman Microscope, equipped with two laser wavelengths of 532 and 785 nm). Scanning electron microscopy (SEM), Phenom Pharos, used for surface and cross-section imaging. Dynamic mechanical analysis (DMA, Triton, TA) was performed for phase behavior and shape memory performance analyses. Differential scanning calorimetry (DSC, Mettler/Toledo) was used for analysis of crystallization with a heating and cooling rate of 10 °C/min. The stress–strain analysis was performed using the tensile instrument (BONGSHIN, model: DBBP-2 t) with a jaw speed of 20 mm/min. The surface topography of the thermoset PU/CNW nanocomposites was analyzed using AFM (NT-MDT co.) instrument operating at room temperature in tapping mode. Inverted fluorescent microscopy was used to analyze the state of the seeded HFF cells after three and five days. For this analysis, the cells were fixed and treated with DAPI for cell staining.
Results and discussion
Raman spectroscopy
According to Fig. 1a, the characteristic Raman bands of crystalline and amorphous PCL segments are summarized as follow: 917 cm−1 (\(\nu\)–C–C (= O)–O–) crystalline, 1030–1120 cm−1 (skeletal stretching, 1071 cm−1 \(\nu\) –C–O–C–crystalline), 1276–1352 cm−1 (\(\omega\) CH2), 1410–1487 cm−1 (\(\delta\) CH2, 1445 crystalline), 2835–3000 cm−1 (\(\nu\) CH, crystalline)(Parnell et al. 2003); regarding PTMG, the Raman bands are as follow: 2939, 2887, 2840 cm−1 (alkyl chains), 1478, 1442 cm−1 (bending mode of C–H group), 1278, 1226 cm−1 (CH twisting vibrations), 1123–1146 cm−1 (C–O, C–OH, C–C), 846, 860 cm−1 (skeletal vibrations), and 533, 362 cm−1 C–O–C bending (Tian et al. 2019; Wu et al. 2021). The Raman spectrum of the blend, block copolymer and their mixture indicate presence of both PCL and PTMG segments.

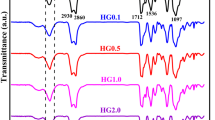
Raman spectroscopy of a the polyols and b the thermoset PU nanocomposites. c, d ATR-FTIR spectrum of the synthesized thermoset PU nanocomposites
Figure 1b shows the Raman spectrum of the synthesized PCL-TPPU and the thermoset PU/CNW nanocomposites. The characteristic isocyanate bands, which are –N=C=O asymmetric stretching band at 2272 cm−1 and symmetric stretching at 1443 cm−1 are absent in the spectrum as an indication of complete consumption of the functionality. Based on the images, the polyurethane bands are as follows: –NH– (amide I 1732 cm−1, amide II 1530 cm−1, amide III 1303 cm−1), Ester \(\upsilon\)(C=O) (urethane amide I \(\upsilon\)(C=O) 1732 cm−1, \(\delta\)(CH2): 1443 cm−1 and \(\delta\)(CH) (urethane amide III 1249) (Ha et al. 2019; Qu et al. 2021). The band related to the ester carbonyl group is not available in the Raman spectrum of the PTMG-tPU and its intensity for the other three tPU specimens is lower than that of PCL-tPU.
FTIR analysis
Figure 1c represents the full FTIR spectrum of the synthesized PUs. The absorption peaks in the wavenumber range of 900–1800 cm−1, which is represented in Fig. 1d, provides the majority of the structural information on the soft segments and the urethane functionality. Based on Fig. 1c, the absorption peak around 3332 cm−1 corresponds to the stretching vibrations of –NH– groups in the urethane group formed by the condensation reaction between the –N=C=O group in the prepolymers’ structure and the –CH2–OH groups available on the CNW surface. The –NH– group in the urethane group usually shows two absorption peaks at 3330 and 3340 cm−1, which are related to hydrogen-bonded and free –NH– groups, which is seen in the spectrum of the PCL-TPPU specimen. The single absorption peak at around 3332 cm−1 is due to the lack of free –NH– vibrations (Goodarzi et al. 2020), which is specific of thermoset PU nanocomposites (Behrouz et al. 2022). The double peak available in the range of 2800–3000 cm−1 corresponds to the stretching vibrations of –CH– groups in the organic polymer structure. Absence of the peak at 2200 cm−1 for all the specimens indicates that the –N=C=O reaction conversion has been completed (Noormohammadi et al. 2021). Regarding the highlighted region, Fig. 1d, the PTMG20000 polyol is distinguished from PCL2000 and other soft segments by the absence of the carbonyl (–C(=O)–) peak at 1734 cm−1 (Sarkhosh et al. 2021). The carbonyl peak in the range of 1700–1750 cm−1 is characteristic of crystalline or amorphous polyesters. Moving towards lower wavenumbers, the two peaks available at 1624 cm−1 and 1575 cm−1 correspond to the stretching vibrations of –C(=O)– and bending vibrations of –NH– functionalities in the urethane structure (–NH–C(= O)–), respectively (Jafari et al. 2020). The intense peak at 1105 cm−1 available for all the specimens corresponds to stretching vibrations of alkoxy group (Goodarzi and Rao 2021). The peak at 1170 cm−1, which is present only for polyols with PCL segments, is specific of ester functionalities (Nourany et al. 2021).
Morphological analysis
Figure 2 shows the SEM cross-sectional and surface images of PCL-tPU, Block 1:1-tPU and PTMG-tPU nanocomposites of CNW. The first and second row images show the cross-sectional images of the cryo-fractured specimens. Based on these images, the CNW nanoparticles with 1.0 wt% content are uniformly distributed and dispersed and there is not any sign of clustering even at 5 μm scale. The reason for absence of any CNW aggregation or clustering is the covalent attachment of the nanoparticles to the matrix chains (Jafari et al. 2020; Noormohammadi et al. 2021). The third row shows the SEM surface images of the specimens with 1 μm scale. The images show that the CNWs are aligned on the surface, parallel to the normal line of the cross-sectional surface, and they seem to have integrated to the matrix.

SEM images of PCL-tPU, Block 1:1-tPU, PTMG-tPU: (first row) cross-section images with 5 μm scale, (second row) cross-section images with 1 μm scale and (third row) surface images of the tPUs with 1 μm scale-bar
DMTA analysis
The viscoelastic properties of thermoset PUs can be evaluated by DMTA instrument. The results of this test provides insights into the changes in the microstructure of the specimens and the results consist of storage (E′) and loss (E′′) modulus. The ratio of E′′ and E′ is called tanδ, which is used to identify the second order transitions (Nourany et al. 2021). In a common logE′ vs. temperature, there are three main regions and two transitions. The regions are glassy, rubbery plateau and melt states. The two main transitions are glass transition temperature (Tg) and flow (or melting) temperature, which are second and first order transitions, respectively (Sarkhosh et al. 2021). For thermoset polymers, the flow of the specimens are absent and for a semi-crystalline polyol, the melting process of the crystallites can be identified as a one-order of magnitude fall in the storage modulus (Ranjbar et al. 2021). The PUs with linear morphology, also called thermoplastic polyurethanes, show another second order transition for the hard segments in a higher temperature range compared to the soft segments (Jafari et al. 2020). Based on Fig. 3a, it can be seen that the transition for the hard segments is available at 39 °C for the thermoplastic PCL (PCL-TPPU) with peak span of 10–93 °C. However, this transition peak is absent for the tPUs as they are immobilized through covalent bonding to the CNWs and cannot go through a long-range segmental movement. Lack of an upturn in tanδ curves in the temperature range of 50–80 °C, Fig. 3b, confirms absence of this transition (Noormohammadi et al. 2021). As it can be seen from Fig. 3a, the Tg transition of the soft domain segments of all the thermoset specimens are very broad and extends from − 85 °C to + 15 °C. This feature is specific of semi-crystalline soft segments while amorphous soft segments show an abrupt fall in this region. This feature has also manifested itself in the wide temperature range of tanδ peak for this transition. Amorphous soft segments usually have a tanδ peak height of about 0.5 or higher with a relatively narrow width (Jafari et al. 2020). The PCL-TPPU specimen show a relatively narrower tanδ transition in with a Tg of − 62 °C and the tanδ peak height is 0.36, which is very higher than the thermoset PU specimens. The reasons for the increased tanδ peak height are the amorphous nature of PCL2000 polyol, as seen by abrupt fall in storage modulus in this region, and absence of CNWs for this specimen. However, the thermoset specimens possess a very low peak height (below 0.14) confirming their semi-crystalline nature. The tanδ peak heights of each PU specimen is shown in Fig. 3c alongside their Tg.

a Storage modulus and b tanδ vs. temperature of the synthesized tPUs; c Tg and tanδ values of the tPUs
Regarding the storage modulus (E′) of the thermoset specimens, which is critical in determining the shape memory parameters, it remains high and constant at elevated temperatures. This is due to the thermoset nature of the PU nanocomposites. However, for the PCL-TPPU specimen, E′ experiences multiple abrupt fall and its value falls in the range of 400–200 kPa for the temperature range of − 23 to 25 °C. Presence of a second fall in the E′ of PCL-TPPU specimen at − 16 °C is related to the phase miscibility of hard/soft segments, which is confirmed by presence of a tanδ peak at this temperature. A high E′, well above 10 MPa even at 100 °C, creates a large driving force for shape recovery process (Zhang et al. 2019a). Another distinct feature of the PU/CNW thermosets is the strong temperature dependency of E′ in the temperature range of − 85 to + 15 °C, which acts as a tool for tuning the shape fixity of the specimens (Kolesov et al. 2015; Argun et al. 2019; Zeng et al. 2021).
The soft domain Tg transition of each specimen is also shown in Fig. 3c. A significant difference in this figure is the sharp increase in PCL segments’ Tg value for the PCL-tPU compared to PCL-TPPU, which confirms the fact that chemical confinement of polyol between CNW reduces their mobility. Another interesting phenomenon that has occurred is that the Tg values of the thermoset PU specimens composed of both PCL and PTMG segments are lower than the Tg values of the soft segments composed of only PCL2000 or PTMG2000. They have also shown higher tanδ peak height. Neat PCL’s Tg is around – 61 ± 1 °C, while for PTMG the Tg value is − 65 °C (Bershtein et al. 2005; Zhuravlev et al. 2011). Compared to PCL2000 and PTMG2000 segments, both polymers have experienced an increase of about + 14 °C in their Tgs once they are used in the thermoset PU/CNW structure. Chemical confinement of the polyols restricts the soft segments’ motion and reducing the ease of soft segment rotation. The declined segmental motion of the PCL2000 and PTMG2000 segments in the PCL-tPU and PTMG-tPU specimens is responsible for the + 14 °C increase in their Tg. The interesting fact is that both the PCL2000 and PTMG2000 soft segments have experienced almost the same extent of increase. The negligible variation can be due to the small difference in their chain flexibility as indicated by the 4 degrees difference in their Tg. The reason behind the lower values of Tgs for soft domain chains with different combinations of PCL and PTMG segments can be explained by their increased chain dynamics (Jafari et al. 2020). The lower Tg values indicate higher segmental mobility as it manifests itself in the slight increase in tanδ peak heights related to the specimens. For the thermoset PUs, the lowest Tg value corresponds to the Blend-tPU specimen, with soft domains composed of PCL2000 and PTMG2000, and the highest tanδ peak height is also corresponds to this specimen. The soft domain of the Blend-tPU specimen is composed of incompatible segments and the single tanδ peak behavior of the soft domain indicates their increased miscibility. The increase in phase miscibility of two semi-crystalline polymers results in a decrease in order and consequently a decline in their crystallization ability. The same concept is applicable for Block 1:1-tPU and mixture-tPU specimens. The impact of morphological and compositional complexity on crystallization potential of the soft domain chains will be discussed later in the DSC and XRD results.
Soft domain crystallization
DSC analysis
For a system of two incompatible semi-crystalline segments, the characteristic crystallization parameters provide insights into the phase organization of the segments. The results of crystallization analysis for the used polyols are shown in Fig. 4a, b, and the characteristic parameters are summarized in Table 2. Based on the results, PCL2000 has the highest crystallization (Tc) and melting (Tm) temperatures of 26.6 and 51.7 °C, respectively. PTMG2000 has a Tc and Tm of 12.8 and 25.7 °C, respectively. Block copolymerization of these two polymers with PCL1000-PTMG2000-PCL1000 architecture has led to two distinct crystallization peaks at 18.5, 8.3 °C and three melting peaks at 39.3, 33.7, and 22.8 °C. Besides the depression of crystallization and melting peak points for both polymer segments, the crystallization enthalpies has also experienced a decline compared to PCL2000 and PTMG2000 homopolymers. Regarding the Tcs, the peak points of + 18.5 and + 8.3 °C correspond to PCL1000 and PTMG2000blocks, respectively. As a general fact, block copolymerization of two semi-crystalline polymers reduces their thermal stability through changing their crystallites’ morphology, which is dependent on their block lengths (Nojima et al. 1995; Müller et al. 2002). The melting temperatures of 39.3 and 33.7 °C correspond to PCL segments and the peak at 22.8 °C is related to the PTMG2000 block. The higher Tc value of the PCL indicate that its crystallites have a higher thermal stability compared to the PTMG crystallites, which leads to its higher crystallization kinetics. Therefore, due to its higher Tc value, the PCL segments would first undergo crystallization, as their crystallites nucleation starts at higher temperatures, leading to increased phase segregation. Early crystallization of PCL segments would affect the PTMG crystallization and increase the imperfectness of its crystallites. The covalent bonding of the PCL1000 and PTMG2000 blocks in an ABA architecture limits their phase separation and increases disorder in the block copolymer chain. These structural features affect the crystallites’ morphology and thermal stability of both crystallizable blocks. The limited phase separation of the PCL-PTMG blocks alongside with early crystallization of PCL segments reduces the conformational entropy of PTMG segments and their ability to fold and form stable crystallites.

The DSC thermograms of the polyols and the thermoset PUs: a second heating and b cooling regime of the polyols; c second heating and d cooling regime of the tPUs. The X-ray diffraction spectrum of e the polyols and f the thermoset PU/CNW nanocomposites
The depression in the characteristic temperatures are lesser in the Blend sample, which comprises of 1:1 w:w PCL2000:PTMG2000 homopolymers. The PCL2000 and PTMG2000 chains are not covalently connected to each other and upon cooling they can easily separate and form relatively stable crystallites and the characteristic crystallization would not be impacted greatly. The reason for the decline in the characteristic temperatures is a combination of kinetics and thermodynamics effects (Zakizadeh et al. 2021). The kinetics works in the course of crystallization and impacts the lamella thickness (L), which is related to Tm through Eq. 5. This equation indicates that the lower L results in lower Tm (Castillo and Müller 2009; He and Xu 2012).
Here, \({v}_{e}\) is the free energy of chain folding at the lamella’s surface and \(\Delta {H}_{f}\) is the lamella heat fusion. As mentioned earlier, covalent bonding of the two incompatible polymers limits their phase separation and this structural feature affects the crystallites’ morphology and predominantly reduces L and a reduction in Tm of each block would be a direct result of this change (Chen et al. 2001). From thermodynamic point of view, the controlling parameter of the changes in thermal stability of the crystallites is the product of the interaction parameter (\({\chi }_{AB}\)) and the block lengths (N), i.e. \({\chi }_{AB}\).N. For higher block lengths the impact of covalent bonding on the characteristic temperatures would be lower (Vilgis and Halperin 1991).
For the mixture specimen, the depression in Tc and Tm of PCL segments are not very large compared to the Block 1:1 specimen. The reason for the relatively higher Tc and Tm of PCL segments can be defined as follows: higher crystallization temperature of PCL segments compared to PTMG segments results in their early crystallization, which results in crystallization-assisted phase separation of PCL segments. Presence of the block copolymer reduces the interfacial tension between the incompatible PCL and PTMG domains, hence, reducing the extent of phase separation and resulting in smaller domains of PCL and PTMG (Müller et al. 2002; He and Xu 2012).
Regarding the PCL-TPPU and the thermoset PU-CNW specimens, as their crystallization and melting spectrum are represented in Fig. 4c, d, the polyols as soft segments have experienced an enormous decline in their crystallization potential. As a general fact, incorporation of semi-crystalline polyols into polyurethane structure leads to a decline in their crystallization ability, however, as reported in our previous paper (Jafari et al. 2020), thermoset PUs of PEG-PCL polyols with CNWs had a larger impact compared to their thermoplastic counterparts. Based on Fig. 4c, d, the PCL-TPPU had a higher Tc and Tm values compared to its thermoset PU counterpart, which is in agreement with our previous results. The reasons behind the sharp declines are: (1) chemical attachment of the polyol chains to CNWs, which highly limits their mobility and kinetics of crystallization, and (2) the repulsive interaction between the polyols and the urethane functionalities (Saralegi et al. 2013; Shirole et al. 2018; Khadivi et al. 2019).
According to the summarized data in Table 2, the Blend-tPU, Block 1:1-tPU and mixture-tPU specimens have negative Tm values and their Tc values are the lowest compared to tPUs of PCL2000 and PTMG2000. This result is in accordance with the results shown for the polyols as block copolymerization and blending reduces the thermal stability of the crystallites. Therefore, the adverse impact of thermosetting on these soft segments would be higher.
X-Ray diffraction analysis
The XRD analysis provides information on the structure of the crystallites’ unit cells and the impact of crystallites’ perfection or defects can be seen as the changes in their 2θ positions, the width and the intensity of the peaks. Based on Bragg’s law, Eq. 6, the change in 2θ positions directly changes the d-spacing (Nourany et al. 2021; Noormohammadi et al. 2021).
According to Fig. 4e, the PCL2000 polyol showed diffraction peaks at 2θ = 21.65°, 22.22°, and 23.95°, which are attributed to the 110, 111 and 200 planes (Hoidy et al. 2010). PTMG2000 showed two diffraction peaks at 2θ = 20.10° and 24.52° corresponding to d-spacing of 0.42 and 0.38 nm. According to the literature, block copolymerization of semi-crystalline polymers reduces their crystallization extent and as a direct result Tc and Tm decreases, which is an indication of lowered thermal stability of the crystallites. For the PCL1000-PTMG2000-PCL1000 block copolymer with hydroxyl end-groups (Block 1:1), the intensity of the diffraction peaks related to the PCL1000 blocks (2θ = 21.40°, 22.03° and 23.73°) have declined by about half compared to PCL2000 homopolymer and the peaks related to PTMG2000 have almost disappeared except for the peak available at 2θ = 20.18°, with extremely low intensity. The results indicate that the block copolymerization had a higher impact on the middle block (PTMG2000). The reason behind this result can be the higher Tg of PTMG compared to PCL and consequently lower chain dynamics, which affects its crystallization kinetics. The changes in 2θ degrees show the structural changes of crystallites’ unit cells as the blocks are covalently attached and affect the nature of the crystallites’ morphology and their development (Jiang et al. 2001; Luyt and Gasmi 2016).
For the Blend polyol (1:1 w:w PCL2000/PTMG2000 blend), the peak positions have not experienced any significant change and only the diffraction peaks’ intensities have declined. This is due to the fact that the two incompatible semi-crystalline polymers have not affected the crystallization thermodynamics and only crystallization kinetics has been impacted (Luyt and Gasmi 2016). Compared to the homopolymers, for the mixture (the relative 1:1:1 weight ratios of PTMG2000, PCL2000 and the block copolymer), the diffraction peak positions have shifted towards lower diffraction peaks (2\(\theta\) = 21.46, 22.10, and 23.73 with a broad peaks at 20.18°). Addition of the block copolymer into the blend reduces the interfacial tension between the two phases. This change improves the phase miscibility of the PCL and PTMG segments at the interface and limiting the extent of phase separation.
As seen in Fig. 4c, d, the PCL-TPPU specimen showed higher Tc and Tm values compared to the thermoset PU/CNW nanocomposites, which is attribute to higher mobility restriction imposed on soft segments through chemical cross-linking by CNWs with numerous active structural –OH groups. The thermoplastic PU showed 2θ peaks at 19.09, 19.96, 21.66 and 23.07, which shows the availability of the PCL crystallites at room temperature. The first two peaks are related to formation of hard segment crystallite and the latter two peaks corresponds to PCL crystallites. Regarding the PU/CNW thermosets, the crystallization ability and the thermal stability of the crystallites has reduced to a large degree. Based on Fig. 4f, the overall shape of the spectrums are broad indicating the amorphous nature of the soft segments at room temperature. Presence of a distinguishable peak at 2\(\theta\) of 23.4° is related to the characteristic diffraction peak of CNW (Noormohammadi et al. 2021). The 1.0 wt% content of the CNW allows for detection of their crystallites’ structure.
Mechanical properties
Since the thermoset PU/CNW nanocomposites do not contain any stable soft segment crystallites at room temperature, they can be considered as rubbers and their deformation under a uniaxial stretching should follow the thermodynamics of rubbers (Vorov et al. 2008).
Based on Eq. 7, the force \(f\) applied to the PUs comprises of two factors, energetic and entropic contributions. Since the nanocomposites have amorphous state at room temperature, \(f_{E} \cong 0\) and the entropic contribution is more dominant. The energetic contribution is more dominant in highly crystalline specimens in which stretching distorts the lattice spacing. At constant temperature, upon stretching the specimens, the soft segments face a decline in their conformational entropy and according to Eq. 8, the required force for further stretching increases (Stribeck et al. 2015). According to Fig. 5a, this behavior is seen for the majority of the specimens, especially for the PCL-TPPU which shows strain-hardening behavior at higher extensions.

The stress–strain behavior of the thermoplastic PCL-based PU and the thermoset PU/CNW nanocomposites a in full-scale and b low-strain scale. c Digital photographs of the PCL- and mixture-tPUs recovering at room temperature, d shape fixity ratio as a function of temperature, and e the shape memory process of the thermoset PU nanocomposites fixed at − 25 °C with the recovery temperature of + 37.5 °C
Overall, at low strains, λ < 1, the energetic contribution is negligible and the entropic contribution dominates. Based on Fig. 5a and Table 3, among the thermoset PU nanocomposites, PCL-tPU had the highest elongation at break (\({\upvarepsilon }_{{\text{u}}}\), ultimate strain) of 289.0% and ultimate stress (\({\upsigma }_{{\text{u}}}\)) of 11.67 MPa, while PTMG-tPU had an \({\upvarepsilon }_{{\text{u}}}\) and \({\upsigma }_{{\text{u}}}\) of 140.4% and 9.27 MPa, respectively. The \({\upvarepsilon }_{{\text{u}}}\) of the block 1:1-tPU declines sharply with a value of 66.2% despite of having a higher soft segment length or higher molecular length between cross-link points (Mc). The Blend-tPU and mixture-tPU specimens have also experienced an intense decline in their \({\upvarepsilon }_{{\text{u}}}\) and besides this change, their \({\upsigma }_{{\text{u}}}\) have also declined with a \({\upsigma }_{{\text{u}}}\) of 4.85 MPa for the Blend-tPU. Regarding the Young’s modulus (E), as summarized in Table 3, Block 1:1-tPU specimen has the highest E value of 166.9 MPa. The E values are measured from the low-strain linear elastic regions of the stress–strain curves (Fig. 5b, below 2% strain). For a uniaxial deformation of a cross-linked system with the assumption of \(\Delta V \cong 0\), no change in volume upon stretching, we have Eq. 9:
Based on this general equation for a network of a homopolymer, the modulus of the specimen increases as a linear function of number density of the strands (an increase in strand length decreases the number of strands per unit volume) (Vorov et al. 2008; Scetta et al. 2021). However, by looking at cross-link density (CLD, \(\nu\)) of the specimens summarized in Table 3, it can be seen that the Block 1:1-tPU has the lowest CLD value but showed the highest modulus of 166.9 MPa. As the chemistry and configuration of soft domain chains differ, the Eq. 8 that has been developed for simple networks in uniaxial deformations fails to predict the overall trend of the mechanical behavior.
One possible explanation for the peculiar behavior can be found in the elastic modulus of the specimens at room temperature. According to Fig. 4a, the E′ of PTMG-tPU and mixture-tPU are around 30 and 60 MPa, respectively. However, the E′ values of the remaining specimens are in the range of 100–200 MPa, which are roughly in correlation with the Young’s modulus of the specimens. The rod-like morphology of the highly crystalline CNW, which creates an asymmetry in the system, and its chemical bonding to the prepolymer chains would have also played a key role in controlling the mechanical properties of the thermoset PU/CNW nanocomposites.
The Young’s modulus of the thermoplastic PUs, depending on the curing process and the nature of the Diisocyanate, is usually below 10 MPa (Tito et al. 2019; Xiang et al. 2021). The high E values of the thermoset PU/CNW nanocomposites compared to PCL-TPPU and other thermoplastic PUs reported in the literature suggests that the CNWs, as high-modulus rod-like nanoparticles, have played a key role in increasing the strength and elasticity of the thermoset PUs. As shown in SEM images, the CNWs are dispersed in the matrix in a relatively uniform way, which is due to the fact that the chemical bonding of the nanoparticles to the prepolymer prohibits their self-assembly and further clustering.
Shape memory performance
According to Fig. 5c, the digital photographs of the PCL-tPU and mixture-tPU specimens that are fixed in liquid N2 and their recovery process was performed at room temperature. The images show that the folded specimens have unfolded to a large degree. In a more systematic study, the glass transition phenomena is usually considered as the base for selection of the fixity temperature (Garle et al. 2012; Zeng et al. 2021). In this research, the three different temperatures of − 50, − 25 and 0 °C were selected for evaluation of the fixity ratio (Rf). Figure 5d shows the change in Rf as a function of temperature for each specimen. The results indicate that as the temperature increases, the ability of the specimens to fix their temporary shape declines. The reason for this change is the gradual decline of elastic modulus after the Tg transition and an increase in soft segment entropy, which tends to regain the relaxed state (Gao et al. 2018; Nessi et al. 2019; Zare et al. 2019; Du et al. 2021). For amorphous polymers, the decline in elastic modulus after Tg is sharp and the temperature dependency of Rf is not very strong. However, for the tPUs with semi-crystalline nature, the temperature dependency of Rf is strong due to the broad Tg transition and the specimens are considered to consist of an infinite number of nano-domains with corresponding Tgs (Zhang et al. 2019a; Gupta and Mekonnen 2022). The broad Tg transition allows for tuning the fixity ratio as the nano-domains with higher Tg values are able to freeze the stored entropic energy (Liu et al. 2015; Mi et al. 2018; Khadivi et al. 2019; Nourany et al. 2021; Ranjbar et al. 2021). Based on the results, the highest shape fixity of 100% corresponds to PCL-tPU at − 50 °C, which is due to the fact that this temperature is below its Tg peak value and the soft segments lack any elastic recovery (Chen et al. 2010; Yu et al. 2014; Wu et al. 2014). According to the literature, elastic modulus is responsible for controlling shape fixity and crystallization of the soft segments, as a key factor, also increase the elasticity of the specimens in the plateau region (Kumar Patel and Purohit 2018; Argun et al. 2019). At the temperature of − 25 °C, the Rf is still high for all the specimens, which is due to the fact that melting temperatures of the existing crystallites are well above this temperature. The temperature of 0 °C is very close to Tm of the crystallites, slightly below or above Tm for each specimen, which in turn cause a sharp decline in Rf, as shown in Fig. 5d. Figure 5e shows the DMA results of the shape memory performance analysis for the shape fixity temperature of − 25 °C and the recovery temperature of + 37.5 °C. According to the DMTA results, the thermoset specimens possessed a high elastic modulus (above 30 MPa) even at higher temperatures, which is due to the thermoset nature of the specimens and the presence of the high modulus CNW nanoparticles. However, the PCL-TPPU specimen with thermoplastic nature showed an intense decline in its elastic modulus, which leads to an increased potential of the specimen to dissipate the stored elastic energy. According to Table 4, the PCL-TPPU showed a low recovery ratio of 75%, which is due to the high entropic energy dissipation caused by higher mobility of the chains. Based on Table 4 and Fig. 5e, regarding to the thermoset PUs, the PTMG-tPU and mixture-tPU specimens showed relatively low recovery ratios. The remaining three thermoset specimens showed very high recovery ratios falling in the range of 95–98%. The reason for the high Rr of the three specimens can be their high and stable elastic modulus compared to the other two specimens. The advantage of thermoset PUs over thermoplastic PUs is their constant elastic modulus at elevated temperatures and over a wide temperature range, which is a sign of a constant entropic driving force for recovering the original shape (Sarkhosh et al. 2021).
In a PU structure, the soft domain is the active phase for recovering the original shape and upon stretching the specimens at elevated temperatures, the chains in the active phase experience a decline in their conformational entropy (Jiu et al. 2016; Zhang et al. 2019b). By keeping the applied force and decreasing temperature, parts of the stretched chain segments are aligned in the crystalline structures (Barot et al. 2008; Bouaziz et al. 2017). Upon removing the force at − 25 °C, since the temperature is well above the Tg transition peak, the specimens experience a partial spontaneous shape recovery. However, as the temperature increases, the segments tend to increase their conformational entropy and shrink in size (Zhou et al. 2014; Zhang et al. 2019a), which in our case it is expressed as a decrease in longitudinal length. For a thermoset system, the most important parameter in determining the shape recovery ratio is the cross-link degree and density, which provides a network point for reducing the chain slippage and energy dissipation ability of the soft segments (Li et al. 2018; Gao et al. 2018). In reality, achieving an ideal chemical network is practically impossible. However, the CNWs with abundant hydroxyl groups were able to create a high cross-linking degree being in the range of 96–98%. The reason for the high and close to each other can be the abundance of hydroxyl groups, which serve as the cross-linking point for the prepolymers (Nourany et al. 2021; Ranjbar et al. 2021). Since the cross-linking degrees of the thermoset PUs were high and close to each other, the elastic modulus was considered to be the main parameter in determining the final shape recovery at the experimental time period.
Surface topography analysis
The surface physics of biomaterials, including surface roughness and uniformity, is a key feature that affects adhesion of seeded cells. For the thermoset PU/CNW nanocomposites, the physical features of the surface can also provide information on the possible clustering of the CNWs. Since CNWs are highly hydrophilic and due to the hydrogen bonding between the nanorods, formation of CNW clusters is a common phenomenon in hydrophobic matrixes. Prohibiting clustering of nanorods can highly increase their reinforcement effect (Noormohammadi et al. 2021). Regarding the surface features, Average roughness, \(R_{a}\) (Eq. 10), and Root Mean Square (RMS) roughness, (\(R_{q}\) Eq. 11), are the statistical representations of deviation and standard deviation in the surface height of each substrate. Skewness, \(R_{Sk}\) (Eq. 12), which is statistically expressed as the third moment of profile amplitude probability density function (\(z_{i}\) − \(\overline{z}\)), is an indication of the deviation in the surface height symmetry from normal distribution. The normal distribution is mathematically defined as \(R_{Sk} = 0\), and a positive Skewness physically indicates presence of more peaks than valleys and planar structures. The surface sharpness, expressed by Kurtosis (\(R_{Ku}\), Eq. 13), is obtained by taking the fourth moment of (\(z_{i}\) − \(\overline{z}\)) and the value of 3 represents a surface with Gaussian amplitude distribution. The higher values represent the predominant peaks and as the value increases the sharpness of the peaks also increases (Kumar et al. 2011; Landa et al. 2014).
Figure 6 shows the topographical 3D images of the substrates’ surface in two different scales of 10 and 2 μm. The statistical information of the surface topography is summarized in Table 5. The results indicate that all the PU specimens show a positive skewness, which is an indication of a surface with predominant peaks. The average roughness (\(R_{a}\)) falls in the range of 16.21–26.0 nm, which is a relatively narrow range. The AFM topographical 3D images with the scale of 2 μm (right column) provide a better visual perspective into the morphology of the peaks. As it can be seen, for the PU/CNW thermosets, the morphology of the peaks have changed compared to PCL-TPPU. The peaks have an elongated morphology, which might be due to the existence of CNWs close to the surface, as seen in the SEM surface images (third row).

The 3D topographical surface images of the thermoset PUs at room temperature
According to Table 5, the Block 1:1-tPU had the highest RMS roughness of 33.45 nm and the PCL-TPPU had the lowest RMS roughness being equal to 19.35 nm. The \(R_{sk}\) values are not very high and are close to the normal surface height distribution. The low \(R_{Ku}\) values of the thermoset PU/CNW nanocomposites indicate that the surface is inclined towards flatness and the surface peaks are not very sharp (Behrouz et al. 2022).
The reason for the relatively higher roughness and predominant peaks in the thermoset PU/CNWs is related to the presence of CNWs on the surface, which are seen in the topographical images presented in Fig. 6.
Cell culture analysis
MTT assay is performed on the specimens using HFF cells to evaluate their cytotoxicity. The viability of the cells on the specimens relative to the control TCPS culture plate is considered as a basis for evaluating the cytotoxicity of the specimens (Nourany et al. 2021). Based on the statistical results of the MTT assay presented in Fig. 7 (top image), in the first three days of the culturing, the cell growth on the specimens, being around 76%, compared to the control is low. The lowest cell growth is for Block 1:1-tPU with a relative cell growth of 68%. The reason for the lower relative cell growth can be the induction time for cell attachment (Ranjbar et al. 2021). After 5 days of cell culture, the seeded HFF cells tend to grow with an average relative cell growth of 102%. The lowest cell growth was again corresponded to the Block 1:1-tPU specimen with a relative cell growth of 93%. The low cell viability of the Block 1:1-tPU can be related to the presence of free CNWs as this specimen has the lowest cross-link density.

(Top) MTT assay using HFF cells cultured on the thermoplastic PCL-based PU and the thermoset PU/CNW nanocomposites for 3 and 5 days. (Bottom) Fluorescent images of the cell-seeded films after 3 and 5 days of culturing HFF cells
Overall, the cell viability of 90% or higher for the materials is considered non-toxic and based on the results of 5 days of culture, the specimens are considered to be highly biocompatible (Nourany et al. 2021). The fluorescent images of PCL-tPU, Block 1:1-tPU and PTMG-tPU are shown in Fig. 7, which indicate that the cell density and growth is higher for PCL-tPU and PTMG-tPU. Based on the images, the cell density of the Block 1:1-tPU is lower compared to other specimens.
Conclusion
The results of shape memory analysis indicated that the thermoset PUs possessed high Rr values falling in the range of 86–98%, while the PCL-based thermoplastic PU showed a low Rr value. The higher Rr values of the thermoset PUs is related to their thermoset nature and lack of chain slippage. There was a direct correlation between the storage modulus (E′) and Rr values as the thermoset PU/CNW nanocomposites showed stable and high E′ values at temperatures above 20 °C. The tanδ peak height of the thermoset specimens was below 0.14, while the high tanδ value of 0.35 for the TPPU is an indication of high chain mobility, which favors loss of stored elastic energy. The results of shape fixity (Rf) analysis indicated the temperature-dependency of this parameter, which was due to the broad Tg transition caused by chemical cross-linking and crystallinity of the soft segments. The higher E′ values of tPUs compared to PCL-TPPU at low temperatures showed their improved ability in freezing the stored elastic energy. The high biocompatibility and HFF cell growth proved the successful development of new biomaterials with multi-functional role.
Data availability
The experimental data will be provided by the corresponding author upon reasonable request.
References
Argun A, Gulyuz U, Okay O (2019) Semi-crystalline, three-segmented hybrid gels with multiple shape-memory effect. Macromol Symp 385:1800164. https://doi.org/10.1002/masy.201800164
Barot G, Rao IJ, Rajagopal KR (2008) A thermodynamic framework for the modeling of crystallizable shape memory polymers. Int J Eng Sci 46:325–351. https://doi.org/10.1016/j.ijengsci.2007.11.008
Behrouz T, Behrooz S, Sarkhosh H, Nourany M (2022) A novel multi-functional model thermoset and PDA-coated PU nanocomposite based on graphene and an amphiphilic block copolymer. Polym Adv Technol. https://doi.org/10.1002/pat.5703
Bershtein VA, David L, Egorov VM et al (2005) Structural/compositional nanoheterogeneity and glass-transition plurality in amorphous polycyanurate–poly(tetramethylene glycol) hybrid networks. J Polym Sci Part B Polym Phys 43:3261–3272. https://doi.org/10.1002/polb.20614
Bouaziz R, Roger F, Prashantha K (2017) Thermo-mechanical modeling of semi-crystalline thermoplastic shape memory polymer under large strain. Smart Mater Struct 26:55009. https://doi.org/10.1088/1361-665x/aa6690
Cao F, Jana SC (2007) Nanoclay-tethered shape memory polyurethane nanocomposites. Polymer (Guildf) 48:3790–3800. https://doi.org/10.1016/j.polymer.2007.04.027
Castillo RV, Müller AJ (2009) Crystallization and morphology of biodegradable or biostable single and double crystalline block copolymers. Prog Polym Sci 34:516–560. https://doi.org/10.1016/j.progpolymsci.2009.03.002
Chen H-L, Hsiao S-C, Lin T-L et al (2001) Microdomain-tailored crystallization kinetics of block copolymers. Macromolecules 34:671–674. https://doi.org/10.1021/ma001485e
Chen S, Hu J, Zhuo H et al (2010) Study on the thermal-induced shape memory effect of pyridine containing supramolecular polyurethane. Polymer (Guildf) 51:240–248. https://doi.org/10.1016/j.polymer.2009.11.034
Choi JT, Dao TD, Oh KM et al (2012) Shape memory polyurethane nanocomposites with functionalized graphene. Smart Mater Struct 21:75017. https://doi.org/10.1088/0964-1726/21/7/075017
Domingues RMA, Gomes ME, Reis RL (2014) The potential of cellulose nanocrystals in tissue engineering strategies. Biomacromolecules 15:2327–2346. https://doi.org/10.1021/bm500524s
Du H, Liu W, Zhang M et al (2019) Cellulose nanocrystals and cellulose nanofibrils based hydrogels for biomedical applications. Carbohydr Polym 209:130–144. https://doi.org/10.1016/j.carbpol.2019.01.020
Du W, Zhang Z, Yin C et al (2021) Preparation of shape memory polyurethane/modified cellulose nanocrystals composites with balanced comprehensive performances. Polym Adv Technol 32:4710–4720. https://doi.org/10.1002/pat.5464
Fortunati E, Luzi F, Janke A et al (2017) Reinforcement effect of cellulose nanocrystals in thermoplastic polyurethane matrices characterized by different soft/hard segment ratio. Polym Eng Sci 57:521–530. https://doi.org/10.1002/pen.24532
Gao Y, Liu W, Zhu S (2018) Reversible shape memory polymer from semicrystalline poly(ethylene-co-vinyl acetate) with dynamic covalent polymer networks. Macromolecules 51:8956–8963. https://doi.org/10.1021/acs.macromol.8b01724
Garle A, Kong S, Ojha U, Budhlall BM (2012) Thermoresponsive semicrystalline poly(ε-caprolactone) networks: exploiting cross-linking with cinnamoyl moieties to design polymers with tunable shape memory. ACS Appl Mater Interfaces 4:645
George J, Sabapathi SN (2015) Cellulose nanocrystals: synthesis, functional properties, and applications. Nanotechnol Sci Appl 8:45–54. https://doi.org/10.2147/NSA.S64386
Goodarzi K, Rao SS (2021) Hyaluronic acid-based hydrogels to study cancer cell behaviors. J Mater Chem B 9:6103–6115. https://doi.org/10.1039/D1TB00963J
Goodarzi K, Jonidi Shariatzadeh F, Solouk A et al (2020) Injectable drug loaded gelatin based scaffolds as minimally invasive approach for drug delivery system: CNC/PAMAM nanoparticles. Eur Polym J 139:109992. https://doi.org/10.1016/j.eurpolymj.2020.109992
Gupta A, Mekonnen TH (2022) Cellulose nanocrystals enabled sustainable polycaprolactone based shape memory polyurethane bionanocomposites. J Colloid Interface Sci 611:726–738. https://doi.org/10.1016/j.jcis.2021.11.174
Ha Y, Kim Y-O, Ahn S et al (2019) Robust and stretchable self-healing polyurethane based on polycarbonate diol with different soft-segment molecular weight for flexible devices. Eur Polym J 118:36–44. https://doi.org/10.1016/j.eurpolymj.2019.05.031
He W-N, Xu J-T (2012) Crystallization assisted self-assembly of semicrystalline block copolymers. Prog Polym Sci 37:1350–1400. https://doi.org/10.1016/j.progpolymsci.2012.05.002
Hoidy WH, Al-Mulla EAJ, Al-Janabi KW (2010) Mechanical and thermal properties of PLLA/PCL modified clay nanocomposites. J Polym Environ 18:608–616. https://doi.org/10.1007/s10924-010-0240-x
Jafari S, Nourany M, Zakizadeh M et al (2020) The effect of controlled phase separation of PEG/PCL-2000 homopolymer polyols using their PCL500-PEG1000-PCL500 tri-block copolymer and CNCs in the final polyurethane hydrogels on their shape memory behavior. Compos Commun 19:194–202. https://doi.org/10.1016/j.coco.2020.03.016
Jiang S, Ji X, An L, Jiang B (2001) Crystallization behavior of PCL in hybrid confined environment. Polymer (Guildf) 42:3901–3907. https://doi.org/10.1016/S0032-3861(00)00565-6
Jiu H, Jiao H, Zhang L et al (2016) Graphene-crosslinked two-way reversible shape memory polyurethane nanocomposites with enhanced mechanical and electrical properties. J Mater Sci Mater Electron 27:10720–10728. https://doi.org/10.1007/s10854-016-5173-2
Joo Y-S, Cha J-R, Gong M-S (2018) Biodegradable shape-memory polymers using polycaprolactone and isosorbide based polyurethane blends. Mater Sci Eng C 91:426–435. https://doi.org/10.1016/j.msec.2018.05.063
Kausar A (2019) Shape memory polyurethane/graphene nanocomposites: Structures, properties, and applications. J Plast Film Sheeting 36:151–166. https://doi.org/10.1177/8756087919865296
Khadivi P, Salami-Kalajahi M, Roghani-Mamaqani H, Lotfi Mayan Sofla R (2019) Fabrication of microphase-separated polyurethane/cellulose nanocrystal nanocomposites with irregular mechanical and shape memory properties. Appl Phys A 125:779. https://doi.org/10.1007/s00339-019-3082-y
Kolesov I, Dolynchuk O, Radusch HJ (2015) Shape-memory behavior of cross-linked semi-crystalline polymers and their blends. Express Polym Lett 9:255–276. https://doi.org/10.3144/expresspolymlett.2015.24
Kumar TP, Saravanakumar S, Sankaranarayanan K (2011) Effect of annealing on the surface and band gap alignment of CdZnS thin films. Appl Surf Sci 257:1923–1927. https://doi.org/10.1016/j.apsusc.2010.09.027
Kumar Patel K, Purohit R (2018) Future Prospects of shape memory polymer nano-composite and epoxy based shape memory polymer—a review. Mater Today Proc 5:20193–20200. https://doi.org/10.1016/j.matpr.2018.06.389
Lahiji RR, Xu X, Reifenberger R et al (2010) Atomic force microscopy characterization of cellulose nanocrystals. Langmuir 26:4480–4488. https://doi.org/10.1021/la903111j
Landa M, Canales J, Fernández M et al (2014) Effect of MWCNTs and graphene on the crystallization of polyurethane based nanocomposites, analyzed via calorimetry, rheology and AFM microscopy. Polym Test 35:101–108. https://doi.org/10.1016/j.polymertesting.2014.03.008
Li M, Guan Q, Dingemans TJ (2018) High-temperature shape memory behavior of semicrystalline polyamide thermosets. ACS Appl Mater Interfaces 10:19106–19115. https://doi.org/10.1021/acsami.8b03658
Liu Y, Li Y, Yang G et al (2015) Multi-stimulus-responsive shape-memory polymer nanocomposite network cross-linked by cellulose nanocrystals. ACS Appl Mater Interfaces. https://doi.org/10.1021/am5081056
Lu P, Hsieh Y-L (2010) Preparation and properties of cellulose nanocrystals: rods, spheres, and network. Carbohydr Polym 82:329–336. https://doi.org/10.1016/j.carbpol.2010.04.073
Luyt AS, Gasmi S (2016) Influence of blending and blend morphology on the thermal properties and crystallization behaviour of PLA and PCL in PLA/PCL blends. J Mater Sci 51:4670–4681. https://doi.org/10.1007/s10853-016-9784-z
Mariano M, El Kissi N, Dufresne A (2014) Cellulose nanocrystals and related nanocomposites: review of some properties and challenges. J Polym Sci Part B Polym Phys 52:791–806. https://doi.org/10.1002/polb.23490
Mi H-Y, Jing X, Yilmaz G et al (2018) In situ synthesis of polyurethane scaffolds with tunable properties by controlled crosslinking of tri-block copolymer and polycaprolactone triol for tissue regeneration. Chem Eng J 348:786–798. https://doi.org/10.1016/j.cej.2018.04.198
Mu R, Hong X, Ni Y et al (2019) Recent trends and applications of cellulose nanocrystals in food industry. Trends Food Sci Technol 93:136–144. https://doi.org/10.1016/j.tifs.2019.09.013
Müller AJ, Balsamo V, Arnal ML et al (2002) Homogeneous nucleation and fractionated crystallization in block copolymers. Macromolecules 35:3048–3058. https://doi.org/10.1021/ma012026w
Nessi V, Falourd X, Maigret J-E et al (2019) Cellulose nanocrystals-starch nanocomposites produced by extrusion: structure and behavior in physiological conditions. Carbohydr Polym 225:115123. https://doi.org/10.1016/j.carbpol.2019.115123
Nojima S, Yamamoto S, Ashida T (1995) Crystallization of block copolymers IV. Molecular weight dependence of the morphology formed in ε-caprolactone–butadiene diblock copolymers. Polym J 27:673–682. https://doi.org/10.1295/polymj.27.673
Noormohammadi F, Nourany M, Mir Mohamad Sadeghi G et al (2021b) The role of cellulose nanowhiskers in controlling phase segregation, crystallization and thermal stimuli responsiveness in PCL-PEGx-PCL block copolymer-based PU for human tissue engineering applications. Carbohydr Polym 252:117219. https://doi.org/10.1016/j.carbpol.2020.117219
Nourany M, Ghelichkhani S, Sarkhosh H et al (2021) The effect of PCL/PEG ABA block lengths on the crystallization of homo/block-based polyurethane/CNW nanocomposites. J Polym Res. https://doi.org/10.1007/s10965-020-02376-y
Park SH, Oh KW, Kim SH (2013) Reinforcement effect of cellulose nanowhisker on bio-based polyurethane. Compos Sci Technol 86:82–88. https://doi.org/10.1016/j.compscitech.2013.07.006
Parnell S, Min K, Cakmak M (2003) Kinetic studies of polyurethane polymerization with Raman spectroscopy. Polymer (Guildf) 44:5137–5144. https://doi.org/10.1016/S0032-3861(03)00468-3
Pei Z, Yu Z, Li M et al (2021) Self-healing and toughness cellulose nanocrystals nanocomposite hydrogels for strain-sensitive wearable flexible sensor. Int J Biol Macromol 179:324–332. https://doi.org/10.1016/j.ijbiomac.2021.03.023
Prataviera R, Pollet E, Bretas RES et al (2018) Nanocomposites based on renewable thermoplastic polyurethane and chemically modified cellulose nanocrystals with improved mechanical properties. J Appl Polym Sci 135:46736. https://doi.org/10.1002/app.46736
Qu Q, He J, Da Y et al (2021) High toughness polyurethane toward artificial muscles, tuned by mixing dynamic hard domains. Macromolecules 54:8243–8254. https://doi.org/10.1021/acs.macromol.1c01098
Ranjbar HA, Nourany M, Mollavali M et al (2021) Stimuli-responsive polyurethane bionanocomposites of poly(ethylene glycol)/poly(ε-caprolactone) and [poly(ε-caprolactone)-grafted-] cellulose nanocrystals. Polym Adv Technol 32:76–86. https://doi.org/10.1002/pat.5062
Reid MS, Villalobos M, Cranston ED (2017) Benchmarking cellulose nanocrystals: from the laboratory to industrial production. Langmuir 33:1583–1598. https://doi.org/10.1021/acs.langmuir.6b03765
Roman M, Dong S, Hirani A, Lee YW (2009) Cellulose nanocrystals for drug delivery. In: Edgar KJ, Heinze T, Buchanan CM (eds) Polysaccharide materials: performance by design. American Chemical Society, Washington, DC, pp 4–81
Rueda L, Saralegui A, Fernández d’Arlas B et al (2013) Cellulose nanocrystals/polyurethane nanocomposites. Study from the viewpoint of microphase separated structure. Carbohydr Polym 92:751–757. https://doi.org/10.1016/j.carbpol.2012.09.093
SaifulAzry SOA, Chuah TG, Paridah MT et al (2021) Influence of cellulose II polymorph nanowhiskers on bio-based nanocomposite film from Jatropha oil polyurethane. Mater Res Express 8:15003. https://doi.org/10.1088/2053-1591/abc6ce
Saralegi A, Fernandes SCM, Alonso-Varona A et al (2013) Shape-memory bionanocomposites based on chitin nanocrystals and thermoplastic polyurethane with a highly crystalline soft segment. Biomacromol 14:4475–4482. https://doi.org/10.1021/bm401385c
Sarkhosh H, Nourany M, Noormohammadi F et al (2021) Development of a semi-crystalline hybrid polyurethane nanocomposites for hMSCs cell culture and evaluation of body-temperature shape memory performance and isothermal crystallization kinetics. J Polym Res. https://doi.org/10.1007/s10965-021-02522-0
Scetta G, Ju J, Selles N et al (2021) Strain induced strengthening of soft thermoplastic polyurethanes under cyclic deformation. J Polym Sci 59:685–696. https://doi.org/10.1002/pol.20210060
Shirole A, Nicharat A, Perotto CU, Weder C (2018) Tailoring the properties of a shape-memory polyurethane via nanocomposite formation and nucleation. Macromolecules 51:1841–1849. https://doi.org/10.1021/acs.macromol.7b01728
Stribeck N, Li X, Kogut I et al (2015) Morphological failure mechanisms in tensile tests of crosslinked polyurethanes with poorly developed domain structure. Macromol Mater Eng 300:699–711. https://doi.org/10.1002/mame.201500007
Tang J, Sisler J, Grishkewich N, Tam KC (2017) Functionalization of cellulose nanocrystals for advanced applications. J Colloid Interface Sci 494:397–409. https://doi.org/10.1016/j.jcis.2017.01.077
Tian D, Wang F, Yang Z et al (2019) High-performance polyurethane nanocomposites based on UPy-modified cellulose nanocrystals. Carbohydr Polym 219:191–200. https://doi.org/10.1016/j.carbpol.2019.05.029
Tingaut P, Zimmermann T, Sèbe G (2012) Cellulose nanocrystals and microfibrillated cellulose as building blocks for the design of hierarchical functional materials. J Mater Chem 22:20105–20111. https://doi.org/10.1039/C2JM32956E
Tito NB, Creton C, Storm C, Ellenbroek WG (2019) Harnessing entropy to enhance toughness in reversibly crosslinked polymer networks. Soft Matter 15:2190–2203. https://doi.org/10.1039/C8SM02577K
Trache D, Hussin MH, Haafiz MKM, Thakur VK (2017) Recent progress in cellulose nanocrystals: sources and production. Nanoscale 9:1763–1786. https://doi.org/10.1039/C6NR09494E
Urbina L, Alonso-Varona A, Saralegi A et al (2019) Hybrid and biocompatible cellulose/polyurethane nanocomposites with water-activated shape memory properties. Carbohydr Polym 216:86–96. https://doi.org/10.1016/j.carbpol.2019.04.010
Vanderfleet OM, Cranston ED (2021) Production routes to tailor the performance of cellulose nanocrystals. Nat Rev Mater 6:124–144. https://doi.org/10.1038/s41578-020-00239-y
Vilgis T, Halperin A (1991) Aggregation of coil-crystalline block copolymers: equilibrium crystallization. Macromolecules 24:2090–2095. https://doi.org/10.1021/ma00008a058
Vorov OK, Livesay DR, Jacobs DJ (2008) Conformational entropy of an ideal cross-linking polymer chain. Entropy. https://doi.org/10.3390/e10030285
Wang Y-J, Jeng U-S, Hsu S (2018a) Biodegradable water-based polyurethane shape memory elastomers for bone tissue engineering. ACS Biomater Sci Eng 4:1397–1406. https://doi.org/10.1021/acsbiomaterials.8b00091
Wang Y, Cheng Z, Liu Z et al (2018b) Cellulose nanofibers/polyurethane shape memory composites with fast water-responsivity. J Mater Chem B 6:1668–1677. https://doi.org/10.1039/C7TB03069J
Wu XL, Kang SF, Xu XJ et al (2014) Effect of the crosslinking density and programming temperature on the shape fixity and shape recovery in epoxy–anhydride shape-memory polymers. J Appl Polym Sci. https://doi.org/10.1002/app.40559
Wu H, Xie H, Tian X et al (2021) Hard, tough and fast self-healing thermoplastic polyurethane. Prog Org Coat 159:106409. https://doi.org/10.1016/j.porgcoat.2021.106409
Xiang N, Zhang X, Zheng M et al (2021) Investigation of tensile behavior and molecular structure of the thermoplastic polyurethane sheets injection molded at different mold temperatures. J Appl Polym Sci 138:50959. https://doi.org/10.1002/app.50959
Xie H, Du H, Yang X, Si C (2018) Recent strategies in preparation of cellulose nanocrystals and cellulose nanofibrils derived from raw cellulose materials. Int J Polym Sci 2018:7923068. https://doi.org/10.1155/2018/7923068
Yu K, Ge Q, Qi HJ (2014) Reduced time as a unified parameter determining fixity and free recovery of shape memory polymers. Nat Commun 5:1–9. https://doi.org/10.1038/ncomms4066
Zakizadeh M, Nourany M, Javadzadeh M et al (2021) Analysis of crystallization kinetics and shape memory performance of peg-pcl/mwcnt based pu nanocomposite for tissue engineering applications. Express Polym Lett 15:418–432. https://doi.org/10.3144/expresspolymlett.2021.36
Zare M, Prabhakaran MP, Parvin N, Ramakrishna S (2019) Thermally-induced two-way shape memory polymers: mechanisms, structures, and applications. Chem Eng J 374:706–720. https://doi.org/10.1016/j.cej.2019.05.167
Zeng H, Sun H, Gu J (2021) Modeling the one-way and two-way shape memory effects of semi-crystalline polymers. Smart Mater Struct 30:95020. https://doi.org/10.1088/1361-665x/ac179e
Zhang K, Zhao Z, Huang J et al (2019a) Self-recoverable semi-crystalline hydrogels with thermomechanics and shape memory performance. Sci China Mater 62:586–596. https://doi.org/10.1007/s40843-018-9347-5
Zhang Y, Hu J, Zhao X et al (2019b) Mechanically robust shape memory polyurethane nanocomposites for minimally invasive bone repair. ACS Appl Bio Mater 2:1056–1065. https://doi.org/10.1021/acsabm.8b00655
Zhou J, Turner SA, Brosnan SM et al (2014) Shapeshifting: reversible shape memory in semicrystalline elastomers. Macromolecules 47:1768–1776. https://doi.org/10.1021/ma4023185
Zhuravlev E, Schmelzer JWP, Wunderlich B, Schick C (2011) Kinetics of nucleation and crystallization in poly(ɛ-caprolactone) (PCL). Polymer (Guildf) 52:1983–1997. https://doi.org/10.1016/j.polymer.2011.03.013
Acknowledgments
We would like to thank Seyed Sadi Hosseini for his insightful comments on the cytotoxicity analysis.
Funding
This work was partially funded by Amirkabir University of Technology and the grant number is not applicable.
Author information
Authors and Affiliations
Contributions
EA: Data acquisition, data validation, formal analysis, writing the original draft; MAM, SB, FN: Data validation, formal analysis, editing the original draft; YRD: Data acquisition, formal analysis; MN: Conceptualization, data validation, writing the original draft, funding and grant acquisition.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Aboueimehrizi, E., Makaremy, M.A., Bazrpash, S. et al. Synthesis of high-modulus thermoset PUs of PCL-PTMG/CNW biomaterials with different soft domain architecture and composition for high shape memory performance. Cellulose 29, 8651–8674 (2022). https://doi.org/10.1007/s10570-022-04796-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-022-04796-z