
Abstract
Synthesis of maleated pimaric acid (MPA) cellulose esters is first reported in this work. Cellulose esterification was performed by reacting microcrystalline cellulose with monoacid chloride of MPA (MPA-Cl) in presence of pyridine as catalyst and reaction medium. The syntheses were started in a heterogeneous solid–liquid reaction medium, but as the reaction advanced, the reaction mass turned into a homogeneous solution. The effects of MPA-Cl/anhydroglucose unit molar ratio, reaction temperature, and reaction duration on the yield and degree of substitution (DS) of cellulose esters (CEs) were investigated. CEs with DS ranging from 2.6 to 2.8 were achieved at molar ratios of 5.5–6.0 after 12–16 h at 118 °C. The purified products were characterized by elemental analysis, IR and 13C-NMR spectroscopy, and thermogravimetric analysis. CEs are soluble or partially soluble in usual organic solvents, depending on DS. Transparent films were prepared using CE-cyclohexanone solutions.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cellulose has been used as a chemical raw material since the mid-nineteenth century, when cellulose esters (CEs) were prepared for the first time (Schönbein 1846; Schützenberger 1865). Since then, CEs were the vanguard compounds of cellulose chemistry, and they remain the most important technical derivatives of cellulose (Klemm et al. 2005). The short-chain carboxylic acid CEs, such as cellulose acetate, cellulose acetate propionate and cellulose acetate butyrate proved to be of great importance for a whole range of advanced industries like textiles, films and membranes, biodegradable materials, modern coatings, or additives (Edgar et al. 2001; Klemm et al. 2005; Vaca-Garcia et al. 1998).
More recently, cellulose has become a basic component in the manufacturing of fiber-reinforced polymer composites. Long-chain fatty acid cellulose esters (FACE) proved to be able to solve the problems arising from the incompatibility between the hydrophilic cellulose and the hydrophobic synthetic polymers (El Seoud and Heinze 2005). Maim et al. (1951) synthesized for the first time FACE in 1951, but the esterification of cellulose with fatty acid has been widely studied in last few decades (Freire et al. 2006; Hon 1996; Vaca-Garcia et al. 2003; Wang and Tao 1995; Wei et al. 2007; Zhou et al. 2014).
Other important CEs are produced by the esterification of cellulose with aromatic carboxylic acids, such as benzoic (Cross and Bevan 1907; Ost and Klein 1913), naphthoic (Chen et al. 2013), phthalic (Levey 1920; Kawamoto et al. 2005), cinnamic (Arai and Satoh 1992; Minsk et al. 1959), or with the aromatic sulfonic acids (Bernoulli and Stauffer 1940; Heinze and Petzold-Welcke 2012; Rahn et al. 1999; Schmidt et al. 2014). CEs of aromatic acids have performed well as modern coatings, polymer supports for controlled release formulations, plastics and biodegradable plastics, composites and laminates, separation media in high performance chromatography, support or protective films for optical media with good optical isotropy, water resistance, thermal and dimensional stability, liquid crystalline materials (Arai and Satoh 1992; Edgar et al. 2001; Kawamoto et al. 2005).
But very little research has been devoted to CEs with the resin acids, accounted as pseudoaromatic carboxylic acids, and especially with their Diels–Alder addition derivatives. Indeed, literature cites only the few researches undertaken by Heinze et al., those who investigated esterification of abietic acid with some simple polysaccharides like pullulan or dextran but not with cellulose itself, or with some cellulose derivatives as hydroxypropylcellulose (Hussain 2004, 2008; Hussain and Heinze 2008). In these studies some unconventional esterification methods based on the in situ activation of abietic acid with tosyl chloride, N,N′-carbonyldiimidazole, or iminium chloride have been used. The abietic acid esters of cellulose derivatives were recommended for applications as amphyphilic polymeric materials, self-assembling materials, or as materials with potential uses in pharmaceutical industry as macromolecular prodrugs. Furthermore, if the situation in the case of the abietic acid cellulose esters is as above, that regarding cellulose esters with the most representative Diels–Alder derivatives of abietic acid (with acrylic acid or maleic anhydride) is even more precarious, literature to date making no reference in this respect.
Rosin is a natural product, composed mostly (about 90%) of resin acids, which in turn consist of a mixture of abietic type (abietic, levopimaric, dehydrobietic, neoabietic, pallustric) and pimaric type (pimaric and isopimaric) organic acids. The major uses of rosin are in the production of the derivatives for papermaking, adhesives, coatings, and inks. However, in the past few decades resin acids have been extensively used as intermediate in the organic synthesis because they are abundant, renewable, inexpensive, biocompatible, biodegradable, rich in carbon and hydrogen, and capable of chemical transformations owing to their particular chemical structure. The chemical structure of abietic type resin acids comprises a bulky and rigid hydrophenanthrene moiety, similar in some respects to that of the aromatic compounds, and two chemically reactive centers, the conjugated double bonds and the carboxyl group (Maity et al. 1989). Of these three functionalities, the hydrophenanthrene moiety confers to the resin acids thermal stability and hydrophobicity, whereas the reactive centers impart to them a high chemical reactivity. However, the chemical transformation of abietic type resin acids is difficult because their molecules are bulky and the carboxylic groups are some tertiary and hindered. As such, the chemical transformation of abietic acid can only be carried out at high temperatures and prolonged reaction durations (Bicu and Mustata 2007). More easily develop the chemical transformations in which the conjugated double bonds of resin acids are involved. In this regard one of the most interesting routes to modify resin acids proved to be the Diels–Alder cycloaddition. This consists of coupling of resin acids with dienophiles, e.g. acrylic acid, fumaric acid, maleic anhydride, acrylonitrile, to be transformed into di- or polyfunctional derivatives. The various cycloaddition adducts of rosin acids have been widely used as monomers for polymer synthesis. New polymers with specific chemical structures and special properties were prepared. Among resin acids derivatives maleopimaric acid anhydride is of special importance because the most resin acids used for technical purposes are that modified with maleic anhydride and the resulting polymers exhibit excellent phisico-chemical properties (Atta et al. 2005; Liu et al. 2010; Abo-Elenin et al. 2014; El-Ghazawy et al. 2014, 2015).
In this context particularly attractive appears the coupling of MPA with cellulose to produce novel fully bio-based polymers with many properties (thermal stability, crystallinity, hydrophobicity, chemical reactivity or biodegradability) superior to those of the forerunners. The esterification reaction between the hydroxyl groups of cellulose and the carboxyl groups of MPA may be a simple route for achieving this goal. Moreover, as will be seen later in this paper, MPA-Cl, the actual agent for the esterification of cellulose with MPA, although a difunctional compound, behaves like a monofunctional one in this regard, leading to valuable, uncrosslinked, soluble and processable cellulose esters.
That is why, the objective of this work is to initiate the research in the field of CEs derived from the chemically modified resin acids by investigating preparation and properties of CEs with the Diels–Alder adduct of abietic acid (in its isomeric form of levopimaric acid) with maleic anhydride (MA).
Experimental
Materials
Microcrystalline cellulose (MCC, Avicel PH-101, ~50 μm, DP 235, Fluka), was used as starting material in the experiments after drying under reduced pressure at 50 °C for 6 h. Thionyl chloride (Fluka) was purified before use by distillation under reduced pressure to give a clear colorless liquid. Pyridine (Py, puriss, Fluka) was dried over KOH and distilled before using. Maleic anhydride (purum) was purchased from Fluka. Methanol (reagent grade, Chimopar, Romania) was used as a precipitation agent. The last two chemicals were used without further purification.
Resin acids (acid number = 184 mg of KOH· g−1, 99%) were separated by crystallization from the cooled and concentrated acetone solutions of a commercial rosin (Wuzhou Forestry & Chemicals Co., Guangxi, China) and purified by recrystallization from the same solvent. The main components of resin acids were abietic acid (85.9 wt%) and levopimaric acid (8.3 wt%). This composition was estimated from the UV spectrum. A mention must be made about the isomerization of abietic acid to levopimaric acid by its heating at elevated temperatures (e.g. 150 °C and over), as indeed happens during the Diels–Alder addition reaction. The 13C-NMR spectrum of levopimaric acid can be accessed on line from the Spectral Database for Organic Compounds (SDBS), (SDBS Catalogue).
Synthesis of MPA
MPA was synthesized by heating resin acids (230.0 g, 0.75 mol) and maleic anhydride (75.0 g, 0.75 mol) under nitrogen at 150 °C for 0.5 h, at 170 °C for 0.5 h, and at 190 °C for 4 h. Crude MPA (293 g) was separated from the cooled reaction mass by a treatment with petroleum ether (b.p. 28–60 °C). The purification of the crude product was carried out by its dissolution into warm acetone, filtering, removal of the solvent, and crystallization of MPA as a tetrahydrate. The purification was continued by the recrystallization, twice, from acetone and drying at 110 °C for 8 h, to give 264 g (86.5%, related to the weight of the reactants) of purified product. The purified MPA, as a white granular material, exhibited a melting point of 232 °C, and an acid number of 418 mg KOH· g−1 (99.5% purity).
FT-IR (KBr, cm−1) 3554–3496 ν(OH in –COOH), 2959–2871 ν(CH), 1843 ν(C=Oasym in anhydride ring), 1772 ν(C=Osym in anhydride ring), 1689 ν(C=O in carboxylic group), 1634 ν(C=C), 1461 δ[(CH3)asym in isopropyl group], 1388 δ[(CH3)sym in isopropyl group], 1286–1226 ν(C–O in carboxylic group, C–Oasym in anhydride ring), 1164 and 1141 ν(C–C in isopropyl group), 1080 ν(C–Osym in anhydride ring), 945–927 δ(C–C in isopropyl group), 853, 671, 580 (Bacher 2002; Vogt et al. 2011). 1H-NMR (CDCl3, δ ppm) 5.51 (s, 1H, H-5, CH=C), 3.11–3.08 (d, 1H, H-15, CHC=C), 2.74–2.70 (d, 1H, H-10, CHC=O), 2.52–2.47 (d, 1H, H-7, CHC=O), 2.18–2.29 (m, 1H, H-17,CH(CH3)2), 1.88–1.85 (d, 2H, H-16, CH2), 1.78–1.24 (m, 12H; H-6, CH; H-8, CH; H-12 CH 2; H-14, H-14 CH 2; H-18 CH 2; H-19 CH 2; H-22 CH 2), 1.15 (s, 3H, H-23, CH 3), 1.00–0.98 (d, 6H, H-20, CH 3; H-21, CH 3), 0.59 (s, 3H, H-24, CH 3). 13C-NMR (CDCl3, δ ppm) 184.67 (C-1, COOH), 172.56, 170.86 (C-2, C-3, O=C OC=O), 148.08 (C-4, C = CH), 125.16 (C-5, C = C H), 53.51(C-6), 53.07 (C-7), 49.15 (C-8), 48.21 (C-9), 45.97 (C-10), 40.58 (C-11), 37.91 (C-12), 37.59 (C-13), 36.72 (C-14), 35.68 (C-15), 35.00 (C-16), 32.69 (C-17), 27.19 (C-18), 21.65 (C-19), 20.59 (C-20), 19.97 (C-21), 17.08 (C-22), 16.65 (C-23), 15.76(C-24). The signals were assigned according to the 13C-NMR spectrum of levopimaric acid (SDBS Catalogue, Kruk et al. 1990).
Synthesis of MPA-Cl
The esterifying agent, MPA-Cl was prepared by gently refluxing a mixture of MPA and thionyl chloride. A mixture consisting of 250.0 g (0.62 mol) of MPA and 489 g (300 mL, 4.1 mol) of thionyl chloride was stirred at room temperature until all solid particles dissolved. Shortly after mixing, a vigorous evolvement of gas was observed. As a result, temperature of the reaction mass decreased by itself below 10 °C. After a reaction duration of about 2 h the reaction mass temperature returned to the initial value. Next, the reaction mass was heated at 30 °C for 4 h to finish up synthesis, diluted with 150 mL of carbon tetrachloride, cooled at low temperatures (near 0 °C) overnight, filtered over a fiberglass cloth, and washed three times with 100 mL portions of diethyl ether, to give 222.5 g of dried MPA-Cl (85.8% yield) as white (yellowish) fine grains.
FT-IR (KBr, cm−1) 2959–2871 ν(CH), 1844 ν(C=Oasym in anhydride ring), 1775 ν(C=Osym in anhydride ring; C=O in acyl chlorides), 1636 ν(C=C), 1458 δ[(CH3)asym in isopropyl group], 1390 δ[(CH3)sym in isopropyl group], 1229–1211 ν(C–O in acyl chloride group, C–Oasym in anhydride ring), 1161 and 1149 ν(C–C in isopropyl group), 1086 ν(C–Osym in anhydride ring), 948–908 δ(C–C in isopropyl group), 853, 702 ν(C–Cl), 672, 582). 1H-NMR (CDCl3, δ ppm) 5.50 (s, 1H, H-5, CH=C), 3.10 (s, 1H, H-15, CHC=C), 2.77–2.74 (d, 1H, H-10, CHC=O), 2.53–2.49 (d, 1H, H-7, CHC = O), 2.19–2.26 (m, 1H, H-17,CH(CH3)2), 1.88–1.85 (d, 2H, H-16, CH 2), 1.86–1.21 (m, 12H; H-6, CH; H-8, CH; H-12 CH 2; H-14, H-14 CH 2; H-18 CH 2; H-19 CH 2; H-22 CH 2), 1.14 (s, 3H, H-23, CH 3), 1.00–0.98 (d, 6H, H-20, CH 3; H-21, CH 3), 0.58 (s, 3H, H-24, CH 3). 13C-NMR (CDCl3, δ ppm) 181.23 (C-1, COOH), 172.56, 170.86 (C-2, C-3, O=C OC=O), 148.24 (C-4, C=CH), 124.79 (C-5, C=C H), 57.14 (C-6), 52.78 (C-7), 52.51 (C-8), 49.21 (C-9), 45.47 (C-10), 40.28 (C-11), 37.61 (C-12), 37.49 (C-13), 35.72 (C-14), 35.48 (C-15), 34.40 (C-16), 32.63 (C-17), 27.09 (C-18), 21.50 (C-19), 20.43 (C-20), 19.87 (C-21), 17.90 (C-22), 16.62 (C-23), 15.40(C-24). The signals were assigned according to the spectra of MPA.
-
Anal. Calcd. for C24 H31 O4 Cl: C, 68.81; H, 7.41; Cl, 8.48; a.n. 535.9 mg KOH g−1.
-
Found: C, 69.22; H, 7.36; Cl, 8.39; a.n. 530.3 mg KOH g−1.
Synthesis of cellulose esters
In a typical esterification procedure cellulose (4 g) was suspended into 200 mL of anhydrous pyridine and the mixture was stirred at room temperature for 2 h under nitrogen. Then, definite amounts of MPA-Cl were added (as specified in Table 1) at once to the heterogeneous mixture. After an initial preheating under mechanical stirring at 85 °C for 1 h, reaction mass was refluxed at 118–125 °C for 12–16 h (Table 1), cooled at 40 °C, and treated under vigorous stirring with 250 mL of 80% (v/v) aqueous methanol to stop the reaction and consume the remaining MPA-Cl. The reaction product was recovered by filtration over a fine glass fiber fabric and then repeatedly (2 × 100 mL) washed with methanol. Crude cellulose ester was purified by Soxhlet extraction with methanol for 16 h, and finally dried to constant weight, at 50 °C in vacuum. An optional additional purification involved the dissolution of CE in dimethylformamide or cyclohexanone, filtration, precipitation with methyl alcohol, repeated (twice) washing on the filter with the same alcohol, and drying under vacuum at 50 °C. By this route some colored derivatives were removed, and as a result CEs get lighter colors.
Peracetylation of MPA cellulose esters for DS determination (Hasani and Westman 2007; Einfeldt et al. 2005).
In a typical peracetylation procedure 0.3 g of cellulose ester CE-4 (Table 1) was suspended into a mixture of pyridine (7 mL), acetic anhydride (6 mL) and N,N-dimethylaminopyridine (20 mg) and allowed to react at 70 °C for 24 h. The crude product was collected by precipitation into ethanol (150 mL), washed several (three) times with the same solvent and dried at 40 °C for 24 h, under vacuum.
-
Yield: 0. 35 g.
1H-NMR (CDCl3): δ = 5.04, 4.77, 4.38, 4.04, 3.70, 3.51 (anhydroglucose protons), 2.20–1.92 (CH3 acetate protons), 1.78–0.59 (pimarate protons). The complete acetylation of the free OH groups was confirmed by the absence of the characteristic absorption band at about 3500 cm−1 in the FTIR spectra of the respective peracetylated CEs.
Measurements
Acid number was determined with 0.1 N aqueous KOH in the presence of phenolphthalein with acetone as solvent. Density measurements were performed with a pycnometer at 25 °C, using water as reference liquid.
Elemental analysis (C and H) was performed with a Perkin-Elmer CHNS-2420 Series II analyzer. Cellulose ester yields were calculated using the following equation:
where M1 is the mass of cellulose, 382 is the mass of the acyl moiety, and M2 is the mass of cellulose ester. The endo/exo ratio was determined by column chromatography on silica gel, using ethyl acetate: hexane (2:1) as mobile phase. The degree of substitution (DS) of cellulose-MPA esters in Scheme 1 was determined by three different methods: elemental analysis, traditional saponification, and 1H-NMR spectroscopy after peracetylation. DS was calculated from the elemental analysis data by using CEs general molecular formula:
where DS is the degree of substitution. This formula allows for establishing equations for carbon or hydrogen contents (Cc, Hc) as a function of DS (Vaca-Garcia et al. 2001).
These two equations can be solved to give DS. For example, if the first equation is solved, DS can be expressed as:
and when the second equation is solved, it can be expressed as:
where, Cc and Hc represent carbon content and hydrogen content expressed as decimal fractions.

Schematic representation of cellulose esterification with MPA-Cl
For reasons of accuracy, DS value was determined by saponification method, too. The method of Genung and Mallatt (1941), slightly modified, was employed to determine the percent maleated pimaryl content (MPC %) of cellulose esters. Cellulose ester (1 g) was added to 50 mL of 75% v/v aqueous ethanol in a 250-mL conical flask, and the mixture was heated under stirring for 30 min at 50 °C. After cooling to room temperature, 50 mL of 0.5 N KOH solution were added and the novel mixture was heated at 60 °C for 30 min and then kept for 48 h at 45 °C with occasional shaking. The mixture was titrated in presence of phenolphthalein with 0.5 N HCl up to the disappearance of the pink color and retitrated after standing for 3 h to account for alkali from the swollen regenerated cellulose. The same procedure was repeated in absence of cellulose ester for the determination of the blank value. MPC % (g/100 g of cellulose ester) was calculated by the following equation:
where VB is the volume in mL of HCl used for the titratation of blank, VS is the volume in mL of HCl used for the titratation of cellulose ester sample, N is the normality of HCl used, 383/3 is the equivalent weight of the maleated pimaryl rest, 0.1 is a conversion factor, and M is the sample mass in grams. DS can be calculated from the MPC % data, according to the equation:
where 162 is the molar weight of glucose unit, 383 is the molar weight of the maleated pimaryl group, 382 is the molar mass of the maleated pimaryl group minus 1, and MPC % is percent maleated pimaryl content.
DS value was also determined by 1H-NMR spectroscopy after the peracetylation of the nonesterified hydroxyl groups in cellulose esters. Peracetylation, but not propionylation, proved to be useful in the case of the MPA cellulose esters, because the signals of the acetate methyl protons are not overlapping with those of the pymaroyl moiety. DS value for the peracetylated CEs was determined by using the following equation (Hasani and Westman 2007):
IH,acetyl = spectroscopic integral of acetate methyl protons; IH,AGU = spectroscopic integral of anhydroglucose unit protons.
The FTIR spectra were recorded on a Bruker Vertex 70 spectrophotometer with the KBr discs. 1H- and 13C-NMR spectra were recorded on a Bruker Avance DRX 400 spectrometer in CDCl3. Molecular weight was determined by gel permeation chromatography using a WGE SEC/GPC multidetection instrument consisting of a pump, two PLgel columns (PLgel 5micro Mixed C Agilent and PLgel 5micro Mixed D Agilent), dual detector RI/VI WGE SEC-3010, with dichloromethane, flow rate of 1.0 mL/min−1 at 25 °C. The RI/VI detector was calibrated with polystyrene standards. The system was also equipped with a Bi-MwA Brookhaven multi-angle SLS detector. Data were analyzed using PARSEC Chromatography software. Wide angle X-ray diffraction (WAXD) was performed with a Bruker AD8 Advance diffractometer. WAXD patterns were obtained using CuKα radiation (λ = 1.541 Å) and a diffraction angle 2θ ranging from 4° to 40°. Differential scanning calorimetry (DSC) was carried out by using a NETZSCH DSC 200 F3 MAIA instrument operated at a 10 °C/min heating rate, under nitrogen atmosphere. Sample (10 mg) was subjected to heating/cooling cycles between 20 and 350 °C to obtain reproducible glass transition temperature (Tg) value. Tg value was recorded as the midpoint of heat capacity change in glass transition region. Melting temperatures were determined by means of a microscope with a heated plate and verified by DSC measurements. Thermogravimetric measurements were performed on a STA JUPITER 409 PC device. Measurements were carried out in the temperature range of 50–700 °C, under dry nitrogen flow, at a heating rate of 10 °C/min.
Water retention value (WRV) was determined in the following way: Approximately one gram of powdered cellulose ester was suspended in 100 mL of distilled water. The suspension was homogenized by stirring for 0.5 h, shaken for another 4 h at 25 °C, transferred into a G4 sintered-glass funnel to remove excess water under vacuum, and centrifuged at 1000 g for 30 min. Then, the sample was transferred into a preweighed glas vial and the weight of the wet sample was determined. Subsequently, the sample was dried at 103 °C to constant weight, and WRV was calculated according to the formula:
where W1 is the weight of wet sample after centrifuging; W2 is the weight of dry sample.
Water contact angle (WCA). Samples, the transparent CE films, were prepared by drying CE cyclohexanone solutions. Cellulose esters were dissolved in cyclohexanone to reach a minimum concentration of 4.0 wt%. The solutions, after a preliminary filtration, were cast onto a glass plate and dried in open air at room temperature. The complete removal of solvent was carried out by drying at 50 °C, under vacuum. Uniform drops of test liquid (5 μL) were deposited onto the film surface, and the contact angles were measured with a home-made instrument which allowed determination of contact angle by a goniometric method, with a precision of ±1%. Each value was the average of five determinations. As probe liquid, double-distilled water was used.
The room temperature solubility of CEs was tested by direct visual examination at 1% (w/v) concentration in different aprotic polar and nonpolar organic solvents.
Results and discussion
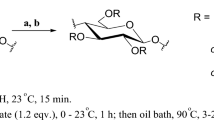
CEs with the pseudoaromatic acyl substituents were prepared in heterogeneous solid–liquid reaction medium using a novel synthetic method based on the use of MPA-Cl as an esterification reagent, and of pyridine as catalyst and solvent, Scheme 1. MCC was used as polymeric starting material in the esterification process. This pure cellulose material was chosen to eliminate the possible influences exerted by lignin on the reaction yield and especially on the cellulose ester color.
Synthesis of cellulose ester precursors
Two precursors, maleated pimaric acid (MPA) and monoacid chloride of maleated pimaric acid (MPA-Cl) were synthesized by the Diels–Alder addition of maleic anhydride to levopimaric acid, and by the chlorination of the above adduct by the treatment with thionyl chloride, respectively. Some specifications are noted here in relation to MPA and MPA-Cl). According to the rule of the Diels–Alder addition reaction, the majoritar product (55–85 wt%, depending on the reaction conditions) of the reaction between levopimaric acid and maleic anhydride was endo-maleated pimaric acid (Wiyono et al. 2007; Li et al. 2012). In the present experiments, performed with purified resin acids but not with rosin, the actual ratio endo/exo was found to be 89/11. The purity of endo MPA used to get endo-MPA-Cl and finally CEs must be as high as possible (e.g. 99.5%) so that the resulting derivatives to exhibit the same very high purities, and implicitly colors as close to white. Another prerequisite for obtaining intermediates and finished products of white color, or as close to it, consists in the use of a thionyl chloride purified by distillation under reduced pressure until the clear colorless liquid was achieved.
As regards MPA-Cl it should be also noted that, unlike acid chlorides of resin acids or of the Diels–Alder adducts of levopimaric acid with some organic acids such as acrylic acid, this compound proved to be a very stable one, even in the presence of the atmospheric water traces. This lower reactivity might be due to the steric hindrance exerted by the bulky hydrophenanthrene group in the structure of MPA on the acyl and maleic moieties (Wang et al. 2013).
Therefore, if necessary, MPA-Cl may be easily purified by recrystallization from suitable solvents, such as carbon tetrachloride. And yet MPA-Cl exhibits an enough high reactivity towards the hydroxyl groups of cellulose. In such reactions MPA-Cl acts only as a monofunctional acylating agent, although in its chemical structure also exist other functional groups capable of reacting with OH groups of cellulose, such as anhydride functional group. Indeed, experimentally we found that anhydride functional group did not participate in the reaction of esterification of cellulose, and that it has not even undergone the possible ring-opening transformation. This observation is of great importance for cellulose ester synthesis since it is basically a guarantee that the esterification reaction will end in non-crosslinked CEs, with a good solubility in the various specific organic solvents. Again mention must be made for the fact that to achieve CEs with light colors, possibly white, the starting MPA-Cl should have the same white color, and a minimum purity of 99.5%.
Synthesis of cellulose esters
As a rule, the synthesis started in a heterogeneous solid–liquid reaction medium consisting of pyridine, cellulose, and MPA-Cl. But after a few hours of heating, depending on the used amount of acyl chloride, an almost total dissolution of the solid particles in the reaction mass occurred. This was possible because cellulose esters almost always have had high degrees of substitution and, consequently, they became soluble in the used organic solvent, pyridine.
The optimum conditions for the esterification reaction of cellulose with MPA-Cl were investigated in terms of molar ratio, reaction time, and reaction temperature. The last ones were experimentally varied as summarized in Table 1. The data in Table 1 show that reaction yield, or rather the degree of substitution (DS), of CEs constantly increase with increasing molar ratios, reaction time and reaction temperature. At low reaction temperatures, for example at about 65 °C (Table 1, Exp. CE-4), the DS increases are very small, because the reaction rates are small, too. But, at 118 °C, which is the reflux temperature of the reaction mass containing pyridine as a solvent, the reaction rate, and consequently DS of CEs, significantly increases. It can also be seen that similarly act both the reaction time and molar ratio. The data entered in Table 1 also show that, degrees of substitution near the maximum were registered for reaction times around 16 h and molar ratios of reactants around 6:1. At higher reaction durations (Table 1, Exp. CE-6) the additional yield increases become practically insignificant. Generally speaking, the enhancement of the esterification reaction by increasing reaction temperature can be ascribed to the fact that high temperatures can facilitate swelling of cellulose and implicitly diffusion of bulky acid chloride inside the swollen particles. In addition, a long reaction time could allow the esterifying agent to react and substitute the majority of the free hydroxyl groups of cellulose, resulting in the higher DS values. It also should be observed that the molar ratios higher than those specified above, for example 9:1, become unjustified and unnecessary because these can no longer contribute to accelerating the esterification reaction or to moving the reaction equilibrium to the right. The small decreases in cellulose ester yield recorded in Table 1, rows 9, 10 and 11, can be explained by the fact that the chosen cosolvents, DMF and DMAc, as very good solvents for the highly substituted CEs, disturbed to some extent the high efficiency of the precipitating agent (aqueous methanol), recorded for example in presence of alone pyridine. Experiments CE-10 and CE-11 prove also that, in fact, syntheses performed in the solvent mixtures Py/DMAc (DMF) ended with the same results (or even better) compared with that carried out in presence of sole pyridine, and that in such cases minor problems occur only in connection with the polymer recovery efficiency, which this time decreases somewhat.
Thus, we can conclude that the esterification of cellulose with MPA-Cl should be conducted at molar ratios equal or up to 6:1 mol MPA-Cl to mol AGU (2:1 equiv. acyl to OH group), at 118 °C for 16 h to achieve values close to maximum for the reaction product yield (92.9%) and DS (2.78).
Structural characterization
CEs were characterized by FTIR, 1H-NMR and 13C-NMR spectroscopy to confirm cellulose esterification, their chemical structure (rendered in Scheme 1) and purity, to determine DS value of some of them, and to analyze the regioselectivity aspects of the esterification process. The existence of CEs is evidenced by the presence in their IR spectra (Fig. 1b, c) of the characteristic absorption bands located at 1726 cm−1 [ν(C=O) in esters], 1232 and 1087 cm−1 [ν(C–O–C) in esters], and 1469 [δ(CH3)asim in isopropyl group] and 1390 cm−1 [δ(CH3)sim in isopropyl group of hydrophenanthrene moiety] (Pandiarajan et al. 2005; Bicu and Mustata 2007; Liu et al. 2009; Liu et al. 2010). The presence of the bands at 1469 and 1390 cm−1 in the spectra in Fig. 1b–d proves the existence in the chemical structure of CEs of the hydrophenanthrene moieties coming from the starting levopimaric (resin) acid, whereas the presence of bands at 1781 and at 1850 cm−1 gives evidence for the presence in the chemical structure of CEs of the maleic anhydride rests. An additional strong absorption band at 2870–2990 cm−1 represent methylene and methyne groups in the same hydrophenanthrene moieties. Other absorption bands characteristic of CEs are that at 3473 cm−1 assigned to stretching of O–H groups, and at 1637 cm−1 attributed to the O–H bending of the adsorbed water. From the spectra in Fig. 1b, c it can also be seen that, as expected, the intensity of the two above absorption bands decreases with increasing DS. Furthermore, the decrease in intensity of the band at 1637 cm−1, due to the bend vibrations of the hydrogen bonded water in cellulose and cellulose derivatives, should be correlated with a predictable increase in cellulose ester hydrophobicity as a result of the insertion of hydrophenanthrene moieties in their chemical structure. On the other hand, the significant decrease in the intensity of the absorption band of OH groups at 3473 cm−1, correlated with the appearance of the strong band at 1726 cm−1 suggest the high efficiency of the esterification reaction. And finally, a last remark: the good purification by a Soxhlet extraction of CEs is reflected by the fact that no absorption bands characteristic of free MPA or MPA-Cl were registered by IR spectroscopy. The absence of the absorption bands at 1690 cm−1, characteristic of the (MPA)-COOH group (Liu et al. 2009), and at 702 cm−1 (C–Cl stretch in MPA-Cl), indicates that CEs are free of regenerated MPA, or unreacted MPA-Cl, respectively.

FTIR spectra of a MCC, b cellulose ester sample CE-1 (DS = 0.53), c cellulose ester sample CE-7 (DS = 2.78) and d MPA
The 13C-NMR spectrum in Fig. 2 also supports the existence of CEs in Scheme 1. Thus, the appearance of the characteristic signals at 176.6–179.0 ppm (C-6 at 179.0 ppm, C-2 at 177.5 ppm and C-3 at 176.6 ppm) due to the carbonyl carbon of ester linkage, constitutes a first evidence of the ester formation. Further, the presence of the carbon atoms belonging to the maleated hydrophenanthrene moieties, indicated in the spectrum by the complex series of signals in the range from 16 to 53 ppm, and of the unsaturated carbons in the same structure, indicated by the signals at 125.2 (C-9) and 148.1 (C-8) ppm, also confirm the attachment by esterification of the maleated pimaroyl groups to the cellulose backbone. The presence of the maleic anhydride moiety in the chemical structure of MPA, and ultimately in the chemical structure of CEs, is evidenced by the peaks at 171.2 (C-11) and 172.7 (C-10) ppm. The signals of the carbon atoms of the modified anhydroglucose units are observed between 62.1 and 100.7 ppm. The high degree of substitution of the analysed CEs (DS = 2.78) is reflected in the spectrum by the appearance of the broad, but not sharp, peaks at 176.6–179.0 ppm (C-7), 100.7 ppm (C-1 atom influenced by the O-2 esterification), 79.3 ppm (C-4 atom influenced by the O-3 functionalization), 62.1 ppm (C-6 atom influenced by the esterification of the primary hydroxyl group). Because of the same esterification of the secondary hydroxyl groups, the peaks of C-2, C-3 and C-5 atoms decreased in intensity and overlapped forming a single broad peak at 71.1–75.2 ppm.

13C-NMR spectrum of cellulose ester with maleated pimaric acid, sample CE-7, DS = 2.78
But this 13C-NMR spectrum is not sufficiently well resolved to allow assignment of the signals of the carbons influenced by the esterification at C-2 and C-6, C-1′ and C-6′, respectively. To obtain well resolved NMR spectra CE-samples were peracylated before being analyzed in order to become soluble in chloroform regardless of their initial DS. Peracylation also allowed us to approach the regioselectivity aspects of the esterification process, or to determine DS values.
Figure 3 shows the 13C-NMR spectra of the samples CE-1, CE-4, and CE-5 peracetylated. In can be seen that at low DSs (CE-1, DS = 0.53) the peaks arising from C-1, C-4, C-6, and C-7 are very sharp and well resolved. Further, the peak of C-6′ (C-6 atom influenced by the esterification of O-6) appears at δ = 62.9 ppm, exhibiting a downfield shift of about 3 ppm as compared to the corresponding carbon of unsubstituted cellulose (60.1 ppm). All these prove that MPA-Cl reacted preferentially with the C-6 primary hydroxyl group, less hindered. In spectrum (b) of the sample C-4 with the DS = 1.25 the signal of C-7 splits into a doublet, which indicates that a part of the hydroxyl groups at C-2 have already participate in the reaction. This finding is also supported by the fact that in spectrum (b) the signal of C-1 at 102.2 ppm was doubled by the signal at 99.2 ppm arising from C-1′ (C-1 atom influenced by an O-2 esterification). In spectrum (c) of the sample C-5 with the DS = 2.47 the signal of C-1 almost disappears and concurrently C-1′ gets stronger. Also a more pronounced splitting of the signals in the carbonyl region can be noticed. Based on the above we can conclude that in the reaction of esterification of cellulose with MPA-Cl in pyridine medium the distribution of MPA moieties among the three hydroxyl groups of anhydroglucose unit is as follows: C6–OH > C2–OH > C3–OH. Otherwise, the same order of reactivity has also been identified when cellulose was esterified with other bulky acyl chlorides such as pivaloyl, adamantoyl, benzoyl chlorides (Zhang et al. 2009; Xu et al. 2011).

13C-NMR spectra of peracetylated cellulose esters: a sample CE-1 (DS = 0.53), b sample CE-4 (DS = 1.25), and c sample CE-5 (DS = 2.47)
1H-NMR spectroscopy was used mainly to determine the DS value of CEs. When cellulose esters proved to be insoluble in the solvents routinely used in the NMR analysis, CEs were peracylated. Usually peracylatyion means peracetylation or perpropionylation (Liebert and Heinze 2005). But this time perpropionylation could not be used for this purpose due to the overlapping of the propionate methyl proton signals with those of the pimaroyl moiety. Figure 4 shows the 1H-NMR spectrum of peracetylated CE-1 (Table 1). In this spectrum the peak at 5.56 ppm is attributed to the unsaturated carbon (H-8) formed by the addition between levopimaric acid and maleic anhydride. The peaks between 3.53 and 5.06 ppm are attributed to anhydroglucose unit protons (the signal at 5.04 ppm attributed to H-3, at 4.77 ppm to H-2, at 4.38 ppm to H-1, H-6, at 4.08 ppm to H-6′, at 3.70 ppm to H-4, and at 3.51 ppm to H-5) according to Zheng et al. (2013). The peaks between 1.93 and 2.12 ppm arise from the methyl protons of the acetyl moiety, and the signals between 0.59 and 1.78 ppm originate from the pimaryl moiety. DS value for the peracetylated CEs was determined according to the following equation (Hasani and Westman 2007):

1H-NMR spectra of peracetylated cellulose esters: a sample CE-1 (DS = 0.53), b sample CE-4 (DS = 1.25), and c sample CE-5 (DS = 2.47)
IH,acetyl = spectroscopic integral of acetate methyl protons; IH,AGU = spectroscopic integral of anhydroglucose unit protons.
Results for three representative CEs (CE-1, CE-4, and CE-7, Table 1) are inserted in Table 1. The data in Table 1 show that 1H-NMR method gives results similar to those achieved by saponification or by elemental analysis. It seems that these three methods can substitute for each other.
At the same time the spectra in Fig. 4 once again highlight the preference for the MPA derivatization at position 6 of anhydroglucose unit (AGU). But, as regards the distribution of MPA moieties among the hydroxyl groups at positions 2 and 3, these spectra show that the substitution order is the opposite of that inferred by 13C-NMR spectroscopy, so that the new order becomes C6–OH > C3–OH > C2–OH. Thus, if in these spectra the signals appearing at 2.2–1.9 ppm are assigned to the methyl moieties of the acetate group, the presence of the weaker signal at 2.2 ppm, attributed to the acetylation at position 6 of the AGU unit, denote a preferred derivatization by the MPA moieties at the same position 6. Also the presence of the intense signals at 1.90 ppm and especially at 1.97 ppm indicates an intense acetylation of cellulose at positions 3 and 2, respectively, and implicitly a less intense MPA derivatization at these positions. Obviously, between the two hydroxyls under discussion the one at the position 2 was the least MPA-derivatized. Anyway, it is clear that the bulky MPA group in MPA-Cl preferentially reacts in pyridine medium with the primary hydroxyl groups of cellulose, and that the reaction also takes place at the secondary hydroxyl groups, but with an incomplete regioselectivity. The same conclusion was reached by Xu et al. (2011) when esterified cellulose with other bulky acyl chlorides such as, pivaloyl chloride, a trisubstituted benzoyl chloride, or adamantoyl chloride.
Properties of cellulose esters
The polymers synthesized by the substitution of MPA on cellulose chain are brittle, solid materials having colors varying from white-yellowish to brown, depending primarily on the quality of the starting raw materials and less on the reaction conditions (unnecessarily prolonged reaction durations, local overheating, inadequate purity of the co-solvents if present, a.s.o.).
Experimentally we observed that the produced CEs can be cast as films by spreading their cyclohexanone or trichloromethane solutions over the surface of a flat glass, but the resulting films are brittle because of both the medium-molecular-weight of the synthesized CEs and the bulkiness of the maleated hydrophenanthrene moiety grafted onto the polymer chains. It should be mentioned here that the organic hydrophilic solvents can not be used for the preparation of CEs solutions because finally the films change from transparent to translucent or even opaque. Other properties of CEs are presented in the following.
Thermal properties
The thermal properties of CEs prepared from MCC in pyridine-MPA-Cl system were investigated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). In Fig. 5 are given the TG/DTG thermograms of MCC and CEs resulting from the experiments 5 (CE-5) and 7 (CE-7) included in Table 1. Figure 6 shows the DSC thermograms of the two above CEs. Table 2 summarizes some thermal characterization data calculated from TG/DTG and DSC curves. Taken together, the TGA data in Table 2 reveal that CEs can be considered as fairly thermostable substances. Indeed, these data prove that DT0 increases from underivatized cellulose to CEs and that for the latter DT0 increases as the DS increases. Furthermore, the data regarding the decomposition temperature associated to 10% weight loss (DT10), which often is considered as criterium for thermal stability, fall in the range 335–350 °C, while that for the underivatized cellulose was only 307 °C. The fast decomposition of CEs took place at temperatures placed in the range 350–470 °C, with the maximum degradation rates ranging between 357 and 362 °C, each time the higher values corresponding to the higher DS. Again the underivatized cellulose is characterized by lower values compared to CEs, the corresponding values being 310–370 and 338 °C, respectively. Only in the case of the amount of residue after pyrolysis (WR600) the situation has reversed. This time the char residue of cellulose material reaches the value 11.1%, whereas the CEs samples are characterized by smaller values, namely 7.1–7.6%. This could be explained by an easier formation and release of some volatile products in the case of CEs as compared with the underivatized cellulose, which can be easily accepted. So, it is obvious that the thermal stability of the CEs resulting from the reaction of cellulose with MPA is higher than that of the starting cellulose. This increased thermal stability can be ascribed to the simultaneous action of some factors as follows: the insertion on the cellulose macromolecular chain of the hydrophenanthrene units with a pseudoaromatic character, the disappearance of the more vulnerable conjugated double bonds in the hydrophenanthrene moieties by diene coupling, and the insertion of the maleic anhydride segments into their chemical structure. A final remark, as can be seen in the same Fig. 5, no significant differences exist between the thermal properties of the two analysed polyesters, although their DSs differ significantly from each other.

TG/DTG traces of a sample CE-7, b sample CE-5, and c microcrystalline cellulose

DSC thermograms (first heating, second heating and cooling) of cellulose ester sample CE-7
DSC was performed to characterize the thermal properties of CEs as glass transitions and melting temperatures. The DSC thermograms of a film-forming cellulose ester sample, (CE-7, DS = 2.78, Table 1) are presented in Fig. 6. The first heating scan shows a melting range falling between 270 and 335 °C, with a melting peak at 313 °C. This endothermic peak can be attributed to the melting of crystalline phase in highly substituted CE-7. Moreover, a melting range of 280–310 °C was also determined using a microscope with a heated plate. During the second scan the existence of a glass transition region with a glass transition temperature (Tg) of 111 °C was observed. In a first instance, this finding could be attributed to the fact that MPA acylation produces an internal plastification in cellulose, as a result of the decrease of the intermolecular interactions between its hydroxyl groups. All these findings indicate that the analyzed cellulose ester consists inter alia, of one significant, but not preponderant, amorphous phase in balance with a preponderant crystalline one. Further, we arrived at the same conclusion and by X-ray analysis. Hence, once again one can conclude that, when a polymer incorporates both amorphous and crystalline regions in its inside, it can show a glass transition temperature, together with a melting temperature.
Having such thermal performances, MPA cellulose esters stand at the same level with other cellulose esters, such as e.g. cellulose acetates. Indeed, for the cellulose triacetate, literature cites a transition temperature of 177 °C and a melting temperature of 289 °C (Edgar 2004). On the other hand, the thermal properties of the polymer materials in general, or the thermal properties characteristic of MPA cellulose esters in particular, are particularly important in defining of a possible their thermal processability, or of a specific their application. On another occasion Edgar (Edgar 2004) showed that the decomposition temperature, as it was determined by TGA, provides information about the upper limit of processing, and that the temperature range between the melting temperature determined by DSC and the onset decomposition temperature determined by the same TGA, offers information about the processing range in which the polymer material could be thermally processed without decomposition problems. In the case of cellulose esters in question it can be seen that this temperature range is an enough large one, falling between 270 and 335 °C. Therefore, this study shows that these bulky hydrophenanthrene substituents could improve the thermal processability of cellulose by esterification.
XRD
The crystalline structure of the original MCC and of two CEs with different DSs was studied by WAXD. The XRD patterns of MCC and of CE-2 (DS = 1.59) and CE-7 (DS = 2.78) are shown in Fig. 7. It can be seen that the diffraction pattern of MCC exhibits three strong characteristic diffraction peaks at 2θ = 14.9°, 22.4°, and 34.5° and a shoulder peak at about 16.1°, representing cellulose I crystal planes (101), (002), (040) and (10−1), respectively. The existence of the sharp diffraction peak at 22.4° once again emphasizes the high degree of crystallinity of the used cellulose material, MCC. But, in the WAXD spectra of CEs in Fig. 7 the peaks characteristic of original cellulose decrease in amplitude or even disappear altogether as DS increases. Indeed, in the spectrum (b) of CE-2, with a DS of only 1.59, the peaks at 14.9° and 34.5° disappeared and the peak at 22.4° became a shorter and wider one, whereas in the spectrum (c) of CE-7 all the three peaks disappeared. All these indicate a strong drop in the degree of crystallinity of cellulose material as a result of the disruption of the ordered packing of cellulose chains by the insertion of bulky maleated hydrophenanthrene moieties by esterification, as expected. But Fig. 7 also shows that, as the esterification goes to completion and the peaks characteristic of cellulose crystallinity disappear, other two new strong diffraction peaks at diffraction angles (2θ) of 4.5° and 13.5° appear and grow in intensity, which suggests that new crystalline regions were formed in MPA cellulose esters. Otherwise, the emergence of such new peaks was also reported when other cellulose esters were studied. Thus, for cellulose acetate the corresponding diffraction angle values were found to fall in the range from 5 to 10 degrees (Li et al. 2009). Therefore, the low-substituted CEs are constituted by both crystalline and amorphous regions, whereas the high-substituted CEs exhibit rather a crystalline structure.

X-ray diffraction spectra of a MCC and of cellulose esterified with maleated pimaric acid: b sample CE-2 (DS = 1.59) and c sample CE-7 (DS = 2.78)
Furthermore, this preponderant crystalline structure of the high-substituted esters is also evidenced by the fact that they present melting temperatures, or rather melting ranges, like crystalline crystalline polymers. Indeed, the thermal analysis showed that cellulose ester with a degree of substitution of 2.78 has exhibited a melting temperature placed in the range between 270 and 315 °C (Fig. 6).
On the other hand, the emergence of these new strong crystalline structures (zones) could also suggest that cellulose ester synthesis has not led to a too advanced degradation of cellulose polymer chains, even under the working conditions that may be considered as being harsh, as that associated for instance to the experiment 7 in Table 1. This assumption was confirmed by molecular weight measurements. Indeed, we found that CE-7 (DS = 2.78) showed a DP = 146, whereas the starting MCC had a DP = 235.
Solubility
The solubility of cellulose esters was tested in some aprotic polar and nonpolar solvents such as N,N-dimethylacetamide, N,N,-dimethylformamide, cyclohexanone, pyridine, tetrahydrofuran, trichloromethane, toluene, ethyl acetate, and n-hexane. The results are presented in the first columns of Table 3. It may be noted that, as with other cellulose esters, the solubility of the esters synthesized in the present work largely depends on DS and the polarity of the solvents. CEs with DS 1.82 are soluble in all tested solvents, except for trichloromethane, tetrahydrofuran, cyclohexanone, toluene, and ethyl acetate. CEs with DS value from 2.0 to 2.86 dissolves in cyclohexanone and that with DS above 2.4 dissolves in trichloromethane. It is possible that bulky and hydrophobic maleated hydrophenanthrene moieties to break intermolecular and intramolecular hydrogen bonding at medium and high DSs which results in the dissolution of such polymer products in aprotic polar and nonpolar organic solvents. But CEs synthesized in this work are insoluble in very non-polar organic solvents such as n-hexane, n-heptane, and even toluene.
It is interesting to note here that CEs with DS greater than 2.5 proved to be fully soluble in the cold dilute (around 1%) aqueous alkaline solutions. In our opinion, the solubilization of the highly substituted cellulose esters in the cold dilute aqueous alkaline solutions is the result of the maleic ring opening and of the neutralization of the resulting COOH groups, without the saponification of ester linkage. As we have already mentioned in the work, the saponification of ester linkage took place with a significant rate only at temperatures higher than room temperature, e.g. at 45–60 °C, and also in presence of the (alcoholic) concentrated alkaline solutions (0.5 N). Therefore, these solutions will remain stable (transparent, clear) for a long time at room temperature.
Hydrophobic properties
The hydrophobicity of CEs was indirectly estimated from the wettability (contact angle, CA) and water retention value (WRV). The wettability is an important property of solid substances because it plays a major role in defining their applications. CA is the angle between a liquid drop and a solid surface. When the test liquid is water, the measured contact angle becomes water contact angle (WCA).
The values of WCA according to the DS of CEs are presented in the antepenultimate column of Table 3. One can observe that, initially, the higher the DS values, the higher the WCA values of CEs and that later, after DS exceeds 2.5, WCA values reach a maximum of 64 degrees or even tend to decrease. Certainly, these values exceed those characteristic of chemically unmodified celluloses, which are characterized by the contact angles close to zero degrees, but does not reach the values characteristic of, e.g., long chain fatty acid cellulose esters. The literature cites for such cellulose esters WCA values in the range of 70°–90° (Freire et al. 2006), or even of 105 degrees (Crepy et al. 2009). The increase in the hydrophobicity of the CEs following the esterification of cellulose with MPA-Cl is suggested by the corresponding increase in WCA values. In the first instance, this evolution can be ascribed to the well-known surface activity of the appended hydrophenanthrene moieties. But, MPA moieties also possess numerous maleic anhydride groups with lower water repellency. On the whole, the presence of the pseudoaromatic moiety in the chemical structure of MPA compensates this adverse effect and even significantly increases its specific hydrophobicity. Indeed, we experimentally measured for the starting MPA contact angle values of 65°–67°. On the other hand, as was above mentioned and the data in Table 3 show, hydrophobicity of CEs, constituted of the hydrophilic cellulose polymer backbone and the hydrophobic pendant maleated hydrophenanthrene moieties, is strongly dependent on DS. And conversely, by varying DS different amphiphilic CEs could be synthesized. In brief, hydrophobicity of CEs, expressed as WCA, increased to 62–64 degrees for DSs of about 2.75–2.85.
To get more information about the hydrophobic-hydrophilic properties of CEs WRV measurements were also performed. This time, WRV is a measure of the capacity of CEs to retain water, not only on the solid particle surface, but especially inside them. The values of WRV for two representative CEs are presented in the penultimate column of Table 3. It can be seen that WRV amounts to about 56% for a DS of 0.53 and that it decreases to about 37% when DS increases to 2.78. As expected, WRV data in Table 3 shows once more that wettability of CEs is strongly dependent on DS. At lower DS the hydrophobic maleated pimaryl groups are in the minority as compared with the cellulose OH groups and as a consequence WRV reaches higher values. Such CEs behave as hydrophile materials. On the contrary, at higher DS the hydrophobic groups becomes dominant, WRV decreases, and CEs behave as fairly hydrophobic materials. The low WRV registered at the high value of DS indicates that the esterification process was not a surfacial one.
Finally, we can conclude that hydrophobicity of CEs expressed either as WCA or as WRV is dependent on the degree of acylation and that the magnitude of their hydrophilic-hydrophobic character can be adjusted by varying DS.
Density of CEs
Generally, the density of cellulose esters is mainly affected by DS and the acyl group size (Edgar 2004). In the particular case of MPA-cellulose esters the density was determined on the solvent-cast polymer films prepared at high DS values, e.g. of 2.78. The resulting value is given in the last column of Table 3. One can observe that the inserted density value (1.212 g/cm3) is lower than that of the starting cellulose (1.5 g/cm3) and higher than that of, e.g., abietic acid (1.06 g/cm3). This value can be ascribed to the simultaneous action of some already mentioned factors, namely the bulky conformation of the maleinized hydrophenanthrene group in the chemical structure of CE and its high DS.
Conclusion
Direct acylation of cellulose in pyridine proved to be an effective way to prepare esterification products with high degrees of substitution, even when maleated pimaryl chloride, a bulky pseudoaromatic acyl chloride, was used as an acylating agent.
Highly substituted cellulose esters were synthesized with MPA-Cl in heterogeneous media, consisting of microcrystalline cellulose, acyl chloride, and pyridine. Both reaction yield and DS of cellulose esters increased with increasing molar ratio between the reactants, reaction duration, and reaction temperature. Various degrees of substitution ranging from 0.56 to 2.86 were obtained. Cellulose esters with high DSs (around 2.8) were synthesized after 16 h at 118 °C with 6–6.5 molar ratios expressed as MPA-Cl/AGU. Under these reaction conditions a strong degradation of cellulose chain had not occurred. Fairly hydrophobic fully transparent films were prepared using cellulose ester cyclohexanone solutions. The contact angle of cellulose ester films was found to be 64 degrees when DS was 2.78. Thermogravimetric analysis and differential scanning calorimetry showed improved thermal stability and high glass transition temperature for the synthesized materials. Also, these high-substituted CEs exhibited a melting temperature placed in the range 270–315 °C, which reveals that the bulky maleated hydrophenanthrene moieties could improve the thermal processability of cellulose by esterification.
The properties of the maleated pimaric acid cellulose esters, as these are seen at the moment, recommend them as thermoplastic materials, thermoadhesives, superhydrophobization and consolidation agents for paper and paperlike products, or as simply biodegradable polymers. The future research will provide additional information about the best uses of these cellulose esters.
These results also demonstrate that rosin acids and cellulose are important feedstock for the preparation of fully biobased cellulose esters, which could be valuable substitutes for those cellulose esters prepared, e.g., using aromatic organic substituents.
In order to extend applications of the MPA cellulose esters, in the next research step, more experiments should be carried out to investigate use of the high molecular-weight cellulose materials as starting materials in an in solution esterification reaction, to optimize the work parameters according to the desired degree of substitution, or to deep the investigations regarding some their properties, such as the mechanical, thermal and thermo-adhesive properties, hydrophobic properties, or the biodegradability.
References
Abo-Elenin OM, El-Saeed AM, El-Sockari MA (2014) Synthesis of new polyurethane coating based on rosin for corrosion protection of petroleum industries equipment. Int J Eng Res Appl 4:148–155
Arai K, Satoh H (1992) Liquid crystalline phase-formation behavior of cellulose cinnamate. J Appl Polym Sci 45:387–390. doi:10.1002/app.1992.070450302
Atta AM, Mansour R, Abdou MI, El-Sayed AM (2005) Synthesis and characterization of tetra-functional epoxy resins from rosin. J Polym Res 12:127–138. doi:10.1007/s10965-004-2936-x
Bacher A (2002) IR handout-UCLA chemistry and biochemistry-infrared spectroscopy. www.chem.edu/~bacher/spectroscopy/IR1.html
Bernoulli AL, Stauffer H (1940) Darstellung und einige physikalische Eigenschaften der 1-Chlor-2,3,4,6,-tetra-p-toluolsulfonyl-glucose. Helv Chim Acta 23:615–626. doi:10.1002/hlca.19400230182
Bicu I, Mustata F (2007) Polymers from a levopimaric acid–acrylic acid Diels–Alder adduct: synthesis and characterization. J Polym Sci A Polym Chem 45:5979–5990. doi:10.1002/pola.22352
Chen J, Zhang JM, Chen WW, Feng Y, Zhang J (2013) Homogeneous synthesis of cellulose naphthoate in an ionic liquid. Acta Polym Sin 10:1235–1240. doi:10.3724/SP.J.1105.2013.13168
Crepy L, Chaveriat L, Banoub J, Martin P, Joly N (2009) Synthesis of cellulose fatty esters as plastics—influence of the degree of substitution and the fatty chain length on mechanical properties. ChemSusChem 2:165–170. doi:10.1002/cssc.200800171
Cross CF, Bevan EJ (1907) Researches on cellulose, 1895–1900. Chapt. Cellulose benzoates, 2nd edn. Longmans, Green & Co, London, pp 35–41
Edgar KJ (2004) Cellulose esters, organic. In: Kroschwitz JI (ed) Encyclopedia of polymer science and technology, vol 9, 3rd edn. Wiley, New York, pp 131–133. doi:10.1002/0471440264.pst045
Edgar KJ, Buchanan CM, Debenham JS, Rundquist PA, Seiler BD, Shelton MC, Tindall D (2001) Advances in cellulose ester performance and application. Prog Polym Sci 26:1605–1688. doi:10.1016/S0079-6700(01)00027-2
Einfeldt L, Gunther W, Klemm D, Heublein B (2005) Peracetylated cellulose: end group modification and structural analysis by means of 1H-NMR spectroscopy. Cellulose 12:15–24
El Seoud OA, Heinze T (2005) Organic esters of cellulose: new perspectives for old polymers. Adv Polym Sci 186:103–149. doi:10.1007/b136818
El-Ghazawy RA, El-Shafey HI, El-Saeed MA, Abdel-Raheim A-RM, El-Sockhary MA (2014) Liquid crystal bio-based epoxy coating with enhanced performance. Int J Eng Res Appl 4:90–96
El-Ghazawy RA, El-Saeed MA, El-Shafey HI, Abdel-Raheim A-RM, El-Sockhary MA (2015) Rosin based epoxy coatings: Synthesis, identification, and characterization. Eur Polym J 69:403–415. doi:10.1016/j.eurpolymj.2015.06.025
Freire CSR, Silvestre AJD, Pascoal Neto C, Belgacem MN, Gandini A (2006) Controlled heterogeneous modification of cellulose fibers with fatty acids: effect of reaction conditions on the extent of esterification and fiber properties. J Appl Polym Sci 100:1093–1102. doi:10.1002/app.23454
Genung LB, Mallatt RC (1941) Analysis of cellulose derivatives. Determination of total combined acyl in cellulose organic esters. Industrial and Engineering Chemistry
Hasani MM, Westman G (2007) New coupling reagents for homogeneous esterification of cellulose. Cellulose 14:347–356. doi:10.1007/s10570-007-9107-2
Heinze T, Petzold-Welcke K (2012) Recent advances in cellulose chemistry. In: Habibi Y, Lucia LA (eds) Polysaccharide building blocks: a sustainable approach to the development of renewable biomaterials. Wiley, Hoboken, pp 2–6
Hon DNS (1996) Chemical modification of lignocellulosic materials. Marcel Dekker, New York, pp 97–129
Hussain MA (2004) Alternative routes of polysaccharide acylation: synthesis, structural analysis, properties. Dissertation, Friedrich Schiller University of Jena
Hussain MA (2008) Unconventional synthesis and characterization of novel abietic acid esters of hydroxipropylcellulose as potential macromolecular prodrugs. J Polym Sci Pol Chem 46:747–752. doi:10.1002/pola.22461
Hussain MA, Heinze T (2008) Unconventional synthesis of pullulan abietates. Polym Bull 60:775–783. doi:10.1007/s00289-008-0918-6
Kawamoto H, Kawanishi H, Okazaki M, Sata H (2005) Cellulose ester of aromatic carboxylic acid. EP 1,215,216 B1
Klemm D, Heublein B, Fink HP, Bohn A (2005) Cellulose: fascinating biopolymer and sustainable raw material. Angew Chem Int Ed 44:3358–3393. doi:10.1002/anie.200460587
Kruk C, de Vries NK, van der Velden G (1990) Two-dimensional INADEQUATE 13CNMR studies of maleopimaric acid, the Diels-Alder adduct of levopimaric acid and maleic anhydride, and of abietic acid. Magn Reson Chem 28:443–447
Levey HA (1920) Cellulose phthalate: Its preparation and properties. J Ind Eng Chem 12:743–744. doi:10.1021/ie50128a012
Li J, Zhang L-P, Peng F, Bian J, Yuan T-Q, Xu F, Sun R-C (2009) Microwave-assisted solvent-free acetylation of cellulose with acetic anhydride in the presence of iodine as a catalyst. Molecules 14:3551–3566. doi:10.3390/molecules14093551
Li J, Rao X, Shao S, Gao Y, Song J (2012) Synthesis and antibacterial activity of amide derivatives from acrylopimaric acid. Bioresources 7:1961–1971. doi:10.15376/biores.7.2.1951-1971
Liebert TF, Heinze T (2005) Tailored cellulose esters: synthesis and structure determination. Biomacromolecules 6:333–340. doi:10.1021/bm049532o
Liu X, Xin W, Zhang J (2009) Rosin-based acid anhydrides as alternatives to petrochemical curing agents. Green Chem 11:1018–1025. doi:10.1039/b903955d
Liu X, Xin W, Zhang J (2010) Rosin-derived imide-diacids as epoxy curing agents for enhanced performance. Bioresour Technol 101:2520–2524. doi:10.1016/j.biortech.2009.11.028
Maim CJ, Mench JW, Kendall DL, Hiatt GD (1951) Aliphatic acid esters of cellulose. Preparation by acid-chloride-pyridine procedure. Ind Eng Chem 43:684–688. doi:10.1021/ie50495a033
Maity S, Ray SS, Kundu AK (1989) Rosin: a renewable resource for polymers and polymer chemicals. Prog Polym Sci 14:297–338. doi:10.1016/0079-6700(89)90005-1
Minsk LM, Smith JG, Van Densen WP, Wright JF (1959) Photosensitive polymers. I. Cinnamate estes of poly (vinyl alcohol) and cellulose. J Appl Polym Sci 2:302–307. doi:10.1002/app.1959.070020607
Ost H, Klein F (1913) Die Benzoylester der Cellulose. Angew Chem 26:437–440. doi:10.1002/ange.19130266102
Pandiarajan S, Umadevi M, Rajaram RK, Ramakrishnan V (2005) Infrared and Raman spectroscopic studies of l-valine L-valinium perchlorate monohydrate. Spectrochim Acta A 62:630–636. doi:10.1016/j.saa.2005.02.008
Rahn K, Diamantoglou M, Klemm D, Berghmans H, Heinze T (1999) Homogeneous synthesis of cellulose p-toluenesulfonates in N,N-dimethylacetamide/LiCl solvent system. Angew Makromol Chem 238:143–163. doi:10.1002/apmc.1996.052380113
Schmidt S, Liebert T, Heinze T (2014) Synthesis of soluble cellulose tosylates in an eco-friendly medium. Green Chem 16:1941–1946. doi:10.1039/C3GC41994K
Schönbein CF (1846) Lettre de M. Schoenbein a M. Dumas sur le cotton fulminant. Comptes Rendus 23:678–679
Schützenberger P (1865) Action de l’acide acétique anhydre sur la cellulose, l’amidon, les sucres, la mannite et ses congeners, les glucosides et certaines matieres colorants vegetales. Comptes Rendus 61:485–486
SDBS (Integrated Spectral Data Base System for Organic Compounds) Catalogue. http://www.aist.go.jp/RIODB/SDBS/cgi-index.cgi. Accessed 26 Oct 2016
Vaca-Garcia C, Thiebaud S, Borredon ME, Gozzelino G (1998) Cellulose esterification with fatty acids and acetic anhydride in lithium chloride/N,N-dimethylacetamide. J Am Oil Chem Soc 75:315–319. doi:10.1007/s11746-998-0047-2
Vaca-Garcia C, Borredon ME, Gaseta A (2001) Determination of the degree of substitution (DS) of mixed cellulose esters by elemental analysis. Cellulose 8:225–231. doi:10.1023/A:1013133921626
Vaca-Garcia C, Gozzelino G, Glasser WG, Borredon ME (2003) Dynamic mechanical thermal analysis transitions of partially and fully substituted cellulose fatty esters. J Polym Sci Pol Phys 41:281–288. doi:10.1002/polb.10378
Vogt N, Demaison J, Rudolph HD (2011) Equilibrium structure and spectroscopic constants of maleic anhydride. Struct Chem 22:337–343. doi:10.1007/s11224-010-9714-7
Wang P, Tao BY (1995) Synthesis of cellulose-fatty acid esters for use as biodegradable plastics. J Environ Polym Degrad 3:115–119. doi:10.1007/BF02067487
Wang J, Yu J, Liu Y, Chen Y, Wang C, Tang C, Chu F (2013) Synthesis and characterization of a novel rosin-based monomer: free radical polymerization and epoxy curing. Green Mater 1:105–113. doi:10.1680/gmat.12.00013
Wei Y, Cheng F, Hou G (2007) Synthesis and properties of fatty acid esters of cellulose. J Sci Ind Res 66:1019–1024
Wiyono S, Tachibana S, Tinambunan D (2007) Reaction of abietic acid with maleic anhydride and attempts to find the fundamental component of fortified rosin. Pak J Biol Sci 10:1588–1595
Xu D, Li B, Tate C, Edgar KJ (2011) Studies on regioselective acylation of cellulose with bulky acid chlorides. Cellulose 18:405–419. doi:10.1007/s10570-010-9476-9
Zhang J, Wu J, Cao Y, Sang S, Zhang J, He J (2009) Synthesis of cellulose benzoates under homogeneous conditions in an ionic liquid. Cellulose 16:299–308. doi:10.1007/s10570-008-9260-2
Zheng X, Gandour RD, Edgar KJ (2013) TBAF-catalyzed deacylation of cellulose esters: reaction scope and influence of reaction parameters. Carbohydr Polym 98:692–698. doi:10.1016/j.carbpol.2013.06.010
Zhou Y, Min D-Y, Wang Z, Yang Y, Kuga S (2014) Cellulose esterification with octanoyl chloride and its application to films and aerogels. Bioresources 9:3901–3908
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Bicu, I., Mustata, F. Novel cellulose esters derived from the levopimaric acid-maleic anhydride adduct: synthesis, characterization, and properties. Cellulose 24, 2029–2048 (2017). https://doi.org/10.1007/s10570-017-1243-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-017-1243-8