
Abstract
Ultra-lightweight cellulose foams were prepared by regeneration of sodium dodecyl sulfate (SDS)/cellulose/NaOH/urea blend solution via mechanical agitation and then freeze-drying. The morphology and properties of the blend solutions and foams were investigated via optical microscope, rheometer, BET and SEM. As a result, it was found that the inclusion complex structure between cellulose macromolecules and the solvent molecules was not destroyed. Moreover, the bubbles were about 20–50 μm in the solutions and larger (>100 μm) in the foams. Not only the micropores (bubbles) but also the nanopores could be observed in the wet and dried foams. The cellulose foams possessed ultra-low density of about 30 mg/cm3 and high specific surface area. The result of X-ray diffraction and Fourier transform infrared spectroscopy indicated that the cellulose foams were transited from cellulose I to cellulose II after dissolution and gelation. Bubbles inside the wet foams weakened the mechanical properties, but inversely increased the mechanical properties in the dried foams. Typical “J”-shaped curves were observed during the mechanical test, which revealed good compressive strength of dried foams. In this work, cellulose foams with ultra-lightweight and good mechanical properties were obtained, which exhibited great potentials for further development and comprehensive utilization of cellulose.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cheap, lightweight, disposable packaging materials for the industrial demand have led to an extensive use of petroleum-based polymeric foams such as expanded polystyrene (EPS) and polyurethane (PU) foams (An et al. 2015; Bernardini et al. 2015; Karlsson et al. 2015). In 2010, the global production of PU was about 14 million tons and about three-quarters of which was consumed in the form of foams (Quadrini et al. 2013). PS and PU are petroleum based and undegradable. Therefore, the preparation and the foam forming technique of biodegradable foams are of great interest (Ahmadzadeh et al. 2015).
Foam forming process of papermaking fibers has been developed in the early 1960s (Radvan 1964). In this process, papermaking fibers were rapidly agitated in a surfactant solution to create a suspension having an average bubble size between 20 and 200 μm. Subsequently, the material was pressed on a traditional or slightly modified paper machine (Dwiggins and Bhat 2002; Skaugen 1981) under significant load before evaporative drying to create paper sheet of reduced density. In certain applications, the foam forming technique was used in multilayer operations (Pounder et al. 1993; Rokman et al. 2001; Blomqvist et al. 2008). In their work, paper-based foams with low density were obtained. However, to increase strength, this paper-based foam should be mechanically compressed, and therefore, it was very difficult to obtain ultra-low density (Madani et al. 2014).
The novel porous materials, aerogels (Dorcheh and Abbasi 2008) and nanofoams (Vukovic et al. 2011), for example, have been rapidly developed by utilizing nanotechnology to obtain the materials with similar or even better properties. Recently, aerogel-like materials have gained great attention due to their unique properties via the so-called sol–gel method. During this process, solid matter in the solution is aggregated and then transformed to gel state. Afterward, the supercritical drying method is utilized to replace the solvent such as water and some organic reagents in the gel with gas (Fujii et al. 2001). Most of aerogel-like materials, which are mainly based on inorganic materials and PU, have been used to manufacture thermal insulation material (Cai et al. 2012; Carlsson et al. 2012; Hua et al. 2011). Cellulose, as the most abundant natural polymer and an inexhaustible source of raw material, is environmentally friendly and biocompatible. However, the utilization of cellulose is limited because it is insoluble and unmeltable due to the strong intra- and inter-molecular hydrogen bonding and van der Waals attractions (Fengel and Wegener 1989).
Recently, NaOH/urea aqueous solution as a novel cellulose solvent system at low temperature has been developed to prepare various functional materials (Cai et al. 2007; Lue and Zhang 2008). After being precooled to −12.5 °C, cellulose could be dissolved rapidly at ambient temperature in 2 min (Qi et al. 2009). The cellulose dissolution at low temperature arises as a result of a fast dynamic self-assembly process among solvent molecules (NaOH, urea and water) and cellulose macromolecules, leading to formation of inclusion complexes that are relatively stable at low temperature (Cai et al. 2008; Shi et al. 2015). As a result, regenerated cellulose materials and functional material have been prepared, as well as the industrialized trial of regenerated cellulose multifilaments (Li et al. 2010). In this work, ultra-lightweight cellulose foams were obtained by combination of the foam forming technique and the novel cellulose solvent system of cellulose by adding sodium dodecyl sulfate (SDS) to the NaOH/urea aqueous solution with a further mechanical stir. The mechanism of foam forming process and the effect of SDS as well as the formed bubbles on the properties of cellulose foams were investigated in detail. We hope to provide useful information of the new cellulose foams to promote the further application of cellulose and this green solvent.
Experimental
Materials
The cellulose (cotton linter pulp) was provided by Hubei Chemical Fiber Group Ltd. (Xiangfan, China), in which the α-cellulose content was more than 95%. The cellulose was washed by distilled water and then oven-dried for 24 h before using. The viscosity–average molecular weight (M η ) of cellulose in cadoxen was determined using an Ubbelohde viscometry at 25 °C to be 9.6 × 104 (degree of polymerization, DP = 600) according to Mark–Houwink equation [η] (mL/g) = 3.85 × 10−2 (M w )0.76 (Brown and Wikström 1965). All other chemical reagents, such as sodium dodecyl sulfate (SDS) and sulfuric acid, were purchased from Shanghai Chemical Reagent Co., Ltd., China, and were of analytical grade.
Preparation of cellulose foams
Cellulose solution was prepared according the previous method (Zhang et al. 2005). A desired amount of NaOH, urea and distilled water (7:12:81 by weight) was added into a 250-mL beaker, and then, the mixed aqueous NaOH/urea solution was placed in a refrigerator until the temperature reached −12.5 °C. After that, desired amount of cotton linter pulp was immediately added into the precooled solution with stirring vigorously for 5 min at room temperature; as a result, 2–4 wt% transparent cellulose solution was obtained. The cellulose solution was centrifuged at 7200 rpm for 15 min at 10 °C to remove the slightly remaining undissolved part, impurities and bubbles. A certain amount of sodium dodecyl sulfate (SDS) was mixed to the transparent NaOH/urea/cellulose aqueous solution and then vigorously agitated at room temperature for 30 min. Then, a milky bubble solution was obtained. The bubble solution was poured into cylindrical tube and heated at 60 °C for 4 h to form cellulose foams. The resulting foams were washed with running water and then distilled water until neutral, and finally dried through lyophilization. Samples were coded as x/y, which x and y% was the concentration of cellulose and SDS in cellulose solution, respectively. In this paper, cellulose gels with bubble inside were called cellulose foams, while cellulose gels without bubble were called cellulose gels.
Characterization
The bubbles in the solution were observed and photographed by optical microscope at magnification of 10–40. Scanning electron micrographs (SEM) were taken on a Hitachi S-570 scanning electron microscope with 20 kV accelerating voltage and at magnification of 50 and 5000, respectively. The cellulose foams at wet state were treated in liquid nitrogen, immediately snapped and then freeze-dried. The samples were sputtered with gold, then observed and photographed. X-ray diffraction (XRD) measurement was taken on an XRD diffract meter (D8-Advance, Bruker). The patterns with CuKα radiation (λ = 0.15406 nm) at 40 kV and 30 mA were recorded in the region of 2θ from 5 to 45°. Samples were ground into powders and dried in a vacuum oven at 60 °C for 48 h. The N2 adsorption and desorption were tested on an instrument (BET, ASAP 2020, Micromeritics, USA). The specific surface area of the cellulose foams was calculated according to the Brunauer–Emmett–Teller equation from the adsorption data in the relative pressure P/P 0 from 0.35 to 0.5. The dried cellulose foams were cut into cubes to measure the volume (V) and the mass (m), and then, the density (ρ) was calculated through the equation: ρ = m/V.
As previous works suggested (Shi et al. 2012), the dynamic rheology experiment was carried out on an ARES-RFS III rheometer (TA Instruments, USA). A double concentric cylinder geometry with a gap of 2 mm was used to measure dynamic viscoelastic parameters such as the shear storage modulus (G′) and loss modulus (G′′) as functions of temperature (T). The rheometer was equipped with two force transducers allowing the torque measurement in the range from 0.004 to 1000 gm. The values of the strain amplitude were checked to ensure that all measurements were set as 10%, which is within a linear viscoelastic regime. Fresh NaOH/urea/cellulose solution and cellulose bubble solutions were prepared for each measurement. Then, cellulose bubble solutions and degassed NaOH/urea/cellulose solution were poured into the couette geometry instrument, which had been kept at each measurement temperature without preshearing or oscillating. Temperature control was set up a Julabo FS18 cooling/heating bath kept within ±0.2 °C over an extended time. The dynamic temperature sweep measurements were taken at an angular frequency of 1 rad/s and with heating or cooling rates of 0.5 °C per min from room temperature to 70 °C (Cai and Zhang 2006).
Results and discussion
Bubbles in the cellulose solutions
Cellulose viscose dope is formed by dissolution of cellulose in NaOH/urea aqueous solution at low temperature (Lue and Zhang 2008). During the dissolution procedure, bubbles are appeared because of mechanical agitation. These bubbles are unfavorable for the mechanical properties, and thus, they are usually removed by centrifugation, vacuum degasification or storing. However, in the present paper, more and more bubbles were expected in order to produce cellulose foam with very low density.
Bubbles in the cellulose solutions were observed through optical microscope. The images are shown in Fig. 1. In general, the bubbles sizes (D bubble) were of 20–50 μm and decreased with an increase in cellulose solution (Table 1). As a result, higher cellulose concentration possesses higher viscosity, leading to smaller bubble size. Also few bubbles were found to break open after the bubble solution was stored for 1 h at 10 °C, indicating that bubbles were stable.

Optical morphology of cellulose solutions. a 4/0, b 4/2, c 3.5/2, d 3/2. Scale bar is 150 μm
Sol–gel transition of the cellulose bubble solutions
The rheological properties are sensitive to variations in the polymer structure, and they are used to study structure–function relationships for polymer solutions (Lue and Zhang 2008). The effect of temperature on sol–gel transition of the resultant cellulose bubble solutions was measured by using dynamic viscoelastic method. Figure 2 shows the storage modulus (G′) and loss modulus (G′′) values of the different bubble solutions, and Table 2 summarizes the gelation temperatures. The G’ values of the cellulose bubble solutions were smaller than G′′ at lower temperature, indicating a viscose behavior. However, both G′ and G′′ increased with the increase in the temperature and showed a crossover representing the critical gel point, and afterward G′ became larger than G′′ at further higher temperature. The results suggested that the temporary networks composed of cellulose chains were enhanced with increasing temperature and finally transformed to a well-established 3D network with elastic property. The sol–gel transition could be explained that the inclusion complex structure composed of solvent small molecules and cellulose macromolecules was broken at elevated temperature, and the cellulose hydrogen bonds were reconstructed to form 3D network structure (Li et al. 2015). The gelation temperatures of the bubble solutions decreased when the SDS content increased, because the inclusion complex structure in cellulose solution was unstable and easier to be destroyed with fluctuations at higher temperature. Obviously, the gelation temperature also decreased when the cellulose content increased, which was consistent with previous work (Lue and Zhang 2008). The results also proved that SDS did not affect the dissolution procedure of cellulose in NaOH/urea aqueous solution.

Temperature dependence of the storage modulus G′ and loss modulus G′′ for cellulose bubble solutions. The data are shifted along the vertical axis by 10a to avoid overlapping
Morphology and structure of cellulose foams
According to the rheological research, after heating in a drying oven at 60 °C for 4 h, cellulose solution was transformed to regenerated cellulose foam, which is shown in Fig. 3a. The wet cellulose foam can float on water, while the wet cellulose gel sinks to the bottom, which reveals that the density is decreased obviously after introducing SDS into the NaOH/urea aqueous solution of cellulose. The density of the dried cellulose foams listed in Table 2 also proved the ultra-lightweight feature, which was 0.02–0.05 g/cm3. The density of the cellulose gels (control, dry state) is over 0.1 g/cm3, and that of some cellulose fiber foams is about 0.05 g/cm3 (Madani et al. 2014).

Digital photographs of the wet cellulose foam 4/2 (up, a), wet cellulose gel 4/0 (bottom, a) and dried cellulose foam 4/2 (b)
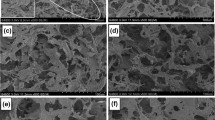
The SEM images of the resultant cellulose gel are illustrated in Fig. 4. Microholes appeared on cellulose foams, while there was no similar observation on cellulose gels. The microholes in the foams (>100 μm) were bigger than those in the solutions (as given in Table 1), which indicated the enlargement of the bubbles. The structure of the bubble lamella is shown in Fig. 5. SDS is composed of hydrophobic chain (tail) and hydrophilic group (head). The tail of SDS extends out of the solution, into the air due to the hydrophobicity of air, while the head remains in the solution or the gel. Lamella with thickness of several micrometers separates the bubbles, which is composed of cellulose channel inclusion complex in solution and cellulose chains in gel. During heating, water evaporation might make it larger. On the other hand, bubbles were coalesced in solution. From the rheology test (Fig. 6), the viscosity of cellulose bubble solution firstly decreased and then increased with temperature increment. The rising of the viscosity revealed the gelation process of the cellulose bubble solution. According to Figs. 6 Fig. 2, when we got lower cellulose concentration, the gelation temperature was higher. After gelation, bubbles will keep their size because the bubble lamellas are transformed from liquid to solid. So, after heating, the bubbles grew much bigger in the cellulose bubble solution with lower cellulose concentration, leading to the larger micropores of dried cellulose foams.

SEM images of the cellulose foams. a, b Foam 3/2; c, d foam 3.5/2; e, f foam 4/2; g, h cellulose gel 4/0. a, c, e, g Overview of cellulose foams; b, d, f, h lamellas

Schematic diagram of the structure of the lamellas between the bubbles

Temperature dependence of viscosity η* for cellulose bubble solutions
Otherwise, there were nanoscale pores inside the lamellas as a result of hydrophily of the cellulose molecules and lyophilization (Fig. 4b, d, e), which further increased the porosity and also leaded to a high surface area of over 75 m2/g (shown in Table 3). The nitrogen adsorption of cellulose foams changes gradually with an increase in cellulose concentration (Fig. 7a). The mesopore distribution of the foams is shown in Fig. 7b. The main size of nanoscale pores was about 30–200 nm, which were consistent with the result in SEM. Thus, the results revealed the existence of multilevel pores and high surface area, leading to potential application in filtration, catalyst loading and biology application (Kim et al. 2013).

Nitrogen adsorption and desorption isotherms (a) and Barrett–Joyner–Halenda (BJH) pore-size distribution (b) of ultra-lightweight cellulose foams calculated from the desorption branch of the isotherm
From FTIR spectrum shown in Fig. 1S, the classic structure of cellulose was observed, which revealed no reaction between the surfactant molecules and the cellulose chains. Figure 2 shows the powder X-ray diffraction of the cellulose foams. All the results of FTIR and XRD indicated that the native cellulose I of cellulose pulp has been transferred to cellulose II after dissolution and regeneration, as we mentioned in our previous work.
Mechanical properties
Compressive strength of the cellulose gel and cellulose foams is listed in Table 2. Obviously, the mechanical properties of materials will be extremely weakened by bubbles inside, because bubbles shape is deformed and then makes the breakage of the bubble lamella under exogenous force. Thus, wet cellulose foams exhibited lower compressive strength than that of wet cellulose gel. The compressive strength of the wet foams increased when SDS content increased, and the sample containing 2% SDS revealed the best mechanical properties, due to the smallest bubble size.
Interestingly, the bubbles inside the dried foams played a different role. The compressive strength–strain curves of the cellulose foams (dry state) are shown in Fig. 8. All the cellulose foams reached a constant strength (σ c ) firstly and then displayed typical “J”-shaped curves. The cellulose foams were not broken during the test, and at last the force reached the highest level (1 kN) of the machine. Although the compressive strength of dried cellulose gel was higher than σ c of cellulose foams, the dried cellulose gel was broken during the test. From SEM images shown in Fig. 4, multilevel pore structure was observed, and micropores were connected through nanoscale pores, leading to the “open-cell” structure of cellulose foams. Thus, we believed that air in micropores could be vented through the nanoscale pores, so that cellulose foam lamella was not broken under exogenous force, as shown in the inside SEM image of Fig. 8. Micropores almost disappeared after the tests, leaving superposed foam lamella. In one word, the results indicated perfect compressive strength of cellulose foams.

Compressive stress–strain curves of the cellulose foams. Digital photographs inside the figure: left—cellulose foam 4/2 before test, right—cellulose foam 4/2 after test, middle—SEM image of the cellulose foam 4/2 after test
Conclusion
Ultra-lightweight cellulose foams were successfully prepared by adding SDS to NaOH/urea aqueous solution with rapid agitation. After adding SDS, the inclusion complex structure composed of cellulose macromolecules and the solvent was not destroyed. Moreover, the bubbles are about 20–50 μm in the solutions and larger (>100 μm) in the foams, where both micropores and nanopores can be observed. As a result, the cellulose foams also exhibit ultra-low density (about 30 mg/cm3) and high specific surface area, which are useful in catalysis, sensing, separation and filtration. The cellulose foams were transited from cellulose I to cellulose II after dissolution and regeneration. Bubbles inside the wet foams lead to lower mechanical properties but inside the dry foams played an opposite role. Typical “J”-shaped curves were observed, which revealed good compressive strength. The present paper provided a simple pathway to fabricate novel cellulose foams with ultra-lightweight, high specific surface area and great compressive strength, exhibiting great potentials for the further utilization of cellulose.
References
Ahmadzadeh S, Nasirpour A, Keramat J, Hamdami N, Behzad T, Desobry S (2015) Nanoporous cellulose nanocomposite foams as high insulated food packaging materials. Colloids Surf, A 468:201–210
An W, Jiang L, Sun J, Liew KM (2015) Correlation analysis of sample thickness, heat flux, and cone calorimetry test data of polystyrene foam. J Therm Anal Calorim 119:229–238
Bernardini J, Cinelli P, Anguillesi I, Coltelli MB, Lazzeri A (2015) Flexible polyurethane foams green production employing lignin or oxypropylated lignin. Eur Polymer J 64:147–156
Blomqvist R, Kostamo R, Rokman K (2008) Method and apparatus for foam forming, US Patent 7416636
Brown W, Wikström R (1965) A viscosity-molecular weight relationship for cellulose in cadoxen and a hydrodynamic interpretation. Eur Polymer J 1:1–10
Cai J, Zhang L (2006) Unique gelation behavior of cellulose in NaOH/Urea aqueous solution. Biomacromolecules 7:183–189
Cai J, Zhang L, Zhou J, Qi H, Chen H, Kondo T, Chen X, Chu B (2007) Multifilament fibers based on dissolution of cellulose in NaOH/urea aqueous solution: structure and properties. Adv Mater 19:821–825
Cai J, Zhang L, Liu S, Liu Y, Xu X, Chen X, Chu B, Guo X, Xu J, Cheng H (2008) Dynamic self-assembly induced rapid dissolution of cellulose at low temperatures. Macromolecules 41:9345–9351
Cai J, Liu S, Feng J, Kimura S, Wada M, Kuga P et al (2012) Cellulose–silica nanocomposite aerogels by in situ formation of silica in cellulose gel. Angew Chem Int Ed 124(9):2118–2121
Carlsson DO, Nystrom G, Zhou Q, Berglund LA, Nyholm L, Stromme M (2012) Electroactive nanofibrillated cellulose aerogel composites with tunable structural and electrochemical properties. J Mater Chem 22:19014–19024
Dorcheh AS, Abbasi MH (2008) Silica aerogel; synthesis, properties and characterization. J Mater Process Technol 199:10–26
Dwiggins JH, Bhat DM (2002) Foam forming method and apparatus, US Patent 6413368
Fengel D, Wegener G (1989) Wood: chemistry, ultrastructure, reactions. Walter de Gruyter, New York, pp 66–105
Fujii T, Yano T, Nakamur K, Miyawaki O (2001) The sol–gel preparation and characterization of nanoporous silica membrane with controlled pore size. J Memb Sci 187:171–180
Hua J, Marjo K, Ari L, Hanna PN, Jouni P, Abraham M, Olli I, Ras RHA (2011) Superhydrophobic and superoleophobic nanocellulose aerogel membranes as bioinspired cargo carriers on water and oil. Langmuir 27:1930–1934
Karlsson K, Schuster E, Stading M, Rigdahl M (2015) Foaming behavior of water-soluble cellulose derivatives: hydroxypropyl methylcellulose and ethyl hydroxyethyl cellulose. Cellulose 22:2651–2664
Kim Y, Cha M, Choi Y, Joo H, Lee J (2013) Electrokinetic separation of biomolecules through multiple nano-pores on membrane. Chem Phys Lett 561–562:63–67
Li R, Chang C, Zhou J, Zhang L, Gu W, Li C, Liu S, Kuga S (2010) Primarily industrialized trial of novel fibers spun from cellulose dope in NaOH/urea aqueous solution. Ind Eng Chem Res 49:11380–11384
Li R, Wang S, Lu A, Zhang L (2015) Dissolution of cellulose from different sources in an NaOH/urea aqueous system at low temperature. Cellulose 22:339–349
Lue A, Zhang L (2008) Investigation of the scaling law on cellulose solution prepared at low temperature. J Phys Chem B 112:4488–4495
Madani A, Zeinoddini S, Varahmi S, Turnbull H, Phillion AB, Olson JA, Martinez DM (2014) Ultra-lightweight paper foams: processing and properties. Cellulose 21:2023–2031
Pounder JR, Ahrens FW, Kershaw TN (1993) Multi-layer papers and tissues, US Patent 5227023
Qi H, Chang C, Zhang L (2009) Properties and applications of biodegradable transparent and photoluminescent cellulose films prepared via a green process. Green Chem 11:177–184
Quadrini F, Bellisario D, Santo L (2013) Recycling of thermoset polyurethane foams. Polym Eng Sci 53:1357–1363
Radvan B (1964) Basic Radfoam process, British Patent 1329409
Rokman K, Jansson J, Laine E (2001) Foam process for producing multi-layered webs, US Patent 6238518 B1
Shi X, Lu A, Jie C, Zhang L, Zhang H, Ji L, Wang X (2012) Rheological behaviors and miscibility of mixture solution of polyaniline and cellulose dissolved in an aqueous system. Biomacromolecules 13:2370–2378
Shi Z, Yang Q, Kuga S, Matsumoto Y (2015) Dissolution of wood pulps in aqueous NaOH/urea solution via dilute acid pretreatment. J Agric Food Chem 63(27):6113–6119
Skaugen B (1981) Foam generator for papermaking machine, US Patent 4299655
Vukovic I, Punzhin S, Vukovic Z, Onck P, De Hosson JTM, ten Brinke G, Loos K (2011) Supramolecular route to well-ordered metal nanofoams. ACS Nano 5:6339–6348
Zhang, L., Cai, J., Zhou, J. (2005). Manufacture of regenerated cellulose films and fibers. Chinese Patent ZL200310111566.3
Acknowledgment
This work was supported by Natural Science Foundation of Fujian Province (2016H6005) and Research Foundation of Education Bureau of Fujian Province (JB13037, JAT160148).
Author information
Authors and Affiliations
Corresponding author
Additional information
Joint first author: Jinyan Du.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, R., Du, J., Zheng, Y. et al. Ultra-lightweight cellulose foam material: preparation and properties. Cellulose 24, 1417–1426 (2017). https://doi.org/10.1007/s10570-017-1196-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-017-1196-y