
Abstract
Despite effective anticancer effects, the use of doxorubicin (Dox) is limited due to its side effects as cardiotoxicity. Corosolic acid (CRA) is a pentacyclic triterpene acid isolated from Lagerstroemia speciosa L. (Banaba) leaves, and it has also been shown to improve myocardial hypertrophy and myocardial infarction which expected to be used in clinical pharmaceuticals. The purpose of this study was to explore whether CRA can improve myocardial injury caused by Dox and to clarify potential mechanisms. C57 BL/6J mice and AMPKα2 knockout mice were given a single intraperitoneal (i.p.) injection of Dox (5 mg/kg) every week for 4 weeks, while normal saline (NS) was used as control. Mice were given CRA (10 mg/kg or 20 mg/kg) or equal volumes of normal saline daily after the first time i.p. injection of Dox. After 4 weeks, echocardiography, gravimetric, hemodynamic, histological, and biochemical analyses were conducted. After Dox injury, compared with the control group, CRA increased the survival rate of mice, improved the cardiac function, decreased the oxidative stress, and reduced the apoptosis. CRA may function by promoting transcription factor EB (TFEB) nuclear translocation and thus restoring autophagic flux. We also observed that CRA protected mitochondrial structure and function, which may benefit from oxidative stress reduction or TFEB activation. In vitro, the protective effect of CRA is reversed by TFEB deletion. Then, we evaluated the expression of AMPKα2/mTOR C1 signaling pathway, the main pathway of TFEB activation. In vivo and in vitro, CRA promoted TFEB nuclear translocation by activating AMPKα2/mTOR C1 signaling, while ablating AMPKα2 reversed these results and accompanied with a decrease in the ability of CRA to resist Dox-induced cardiotoxicity. Thus, we suggested that CRA activated TFEB in an AMPKα2-dependent manner to protect against Dox cardiotoxicity.
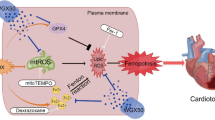
Graphical abstract

This study confirms the role and mechanism of CRA in the treatment of Dox-induced cardiac injury. Dox-induced damage to autophagy includes autophagosomes maturation disorders and autophagolysosomes acidification defects, CRA restored autophagic flux, and promoted lysosomal degradation by activating TFEB in an AMPKα2-depended manner, stabilized mitochondrial function, ultimately protected against Dox-induced cardiotoxicity
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Doxorubicin is a nonselective class I anthracycline antibiotic and widely used in the treatment of cancers. However, Dox can cause many serious side effects, especially cardiotoxicity. Dox-induced cardiotoxicity includes acute and chronic damage, single intravenous injection of Dox can immediately cause cardiac arrhythmia (Singal et al. 1987), and long-term Dox therapy can lead to dilated cardiomyopathy and eventually heart failure (Koleini and Kardami 2017). Recent studies have elucidated many mechanisms that regulate the process of Dox-induced cardiac damage, such as oxidative stress (Alshabanah et al. 2010), autophagy (Govender et al. 2014), apoptosis (Ghosh et al. 2011), and inflammation (Zhang et al. 2017). The clinical application of Dox has been greatly limited due to its cardiotoxicity; thus, understanding Dox-induced cardiotoxicity is critical.
Autophagy is an important regulatory process that maintains cellular homeostasis under normal and stress conditions. By promoting the formation, combination, and clearance of autophagosomes and lysosomes, autophagy removes damaged organelles and misfolded proteins to ensure cell survival (Terman and Brunk 2005). Autophagy is a dynamic process that requires the continuous generation of autophagosomes and stable lysosomal degradation to maintain autophagic flux. Problems in any link will lead to abnormal autophagy and affect cellular homeostasis (Kim and Lee 2014). Previous studies have shown that Dox-induced oxidative stress is related to autophagy deficiency, and enhanced autophagy can reduce the production of reactive oxygen species (ROS) and improve the cardiotoxicity caused by Dox (Zhao et al. 2020; Zhang et al. 2020a, b). Recent studies have confirmed that Dox-induced cardiotoxicity is associated with impaired lysosomal function. Dox destroys lysosomal acidification and blocks autophagic flux, which is regarded as the main cause of reactive oxygen species production in cardiomyocytes (Li et al. 2016). This impairment in lysosomal function involves the lysosomal receptor lysosomal-associated protein 2a (Lamp2A) and transcription factor EB (TFEB) (Cuervo and Dice 1996). Studies have confirmed that long-term Dox exposure reduces the expression of TFEB and Lamp2A in cardiomyocytes (Bartlett et al. 2016). TFEB is a member of the microphthalmia/transcription factor E (MIT/TFE) family of transcription factors and plays an essential role in stabilizing lysosomal function and promoting autophagy (Inpanathan and Botelho 2019). Previous studies have shown that long-term Dox exposure exacerbated TFEB depletion. This change leads to a variety of intracellular dysfunctions, such as decreased autophagolysosomes degradation, blocked autophagic flux, and mitochondrial damage. Overexpression of TFEB promotes the expression of LAMP 2A, initiates lysosomal autophagy, and restores lysosomal acidification and autophagic flux, which are beneficial to cardiomyocyte survival after Dox exposure (Bartlett et al. 2016).
Corosolic acid is a pentacyclic triterpene acid isolated from Lagerstroemia speciosa L. (Banaba) leaves, possesses effective hypoglycemic effects, and is expected to be used in clinical pharmaceuticals (Fukushima et al. 2006). Recent studies have proven that corosolic acid has excellent performance in the treatment of gastric cancer (Cheng et al. 2017), colon cancer (Sung et al. 2014), osteosarcoma cells (Cai et al. 2011), renal cancer (Woo et al. 2018), and others. In this study, we investigated whether CRA could improve Dox-induced changes in cardiac function to provide new strategies for clinical treatments.
Methods and materials
Animals and animal model
All animal experiments followed the Guidelines for the Care and Use of Laboratory Animals approved by the Animal Care and Use Committee of Renmin Hospital of Wuhan University. Male C57BL/6 background mice, aged 8–10 weeks, 23.5–27.5 g weight were purchased from the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences (Beijing, China). AMPKα2 knockout mice were provided by Professor Lianfeng Zhang of the Chinese Academy of Medical Sciences. Animal living in a room with controlled temperature (22 ± 3 °C) and humidity (50 ± 5%) on a 12-h light-dark cycle with free access to standard animal food and water. Mice were randomly allocated to different experimental groups and processed, each group of 30. Dox group received intraperitoneal (i.p.) injection of Dox (5 mg/kg, Sigma-Aldrich) every week for 4 weeks. Dox+CRA group was given CRA (10 mg/kg or 20 mg/kg) daily after the first time i.p. injection of Dox. Normal saline is used as control.
Chemicals
CRA (purity > 98%) was purchased from Baoji Herbest Bio-Tech Co., Ltd.
Echocardiography and hemodynamic analysis
Echocardiography was performed to obtain the index for cardiac function; 1.5% isoflurane was used to anesthetize mice, thereby using a Mylab30CV ultrasound (Biosound Esaote Inc.) to obtain data. Millar pressure-volume system (MPVS-400; Millar, Inc.) was used to record hemodynamic measurement, and the data were analyzed by PVAN data analysis software (version 3.5; Millar, Inc.).
Cardiac morphology and histological examinations
After euthanasia, the hearts were collected and fixed in 10% formaldehyde for over 24 h with gentle shake. Subsequently, the samples in paraffin were dehydrated and embedded and cut into 5-μm-thick sections. Myocyte injury was evaluated by hematoxylin and eosin (H&E) stained.
Biochemical determination
Collect blood and separate serum by centrifugalization at 4000 rpm for 30 min. The creatine kinase isoenzyme (CK-MB) and lactate dehydrogenase (LDH) levels were measured by an automatic biochemical analyzer (ADVIA® 2400, Siemens Ltd., China).
Real-time quantitative PCR
Firstly, total RNAs were extracted using TRIzol reagent (Invitrogen Life Technologies, Carlsbad). Then, the cDNAs were synthesized using oligo (DT) primers and the cDNA synthesis kit (4897030001, Roche, Switzerland). Finally, absolute QPCR SYBR mix (Thermo Fisher Scientific, UK) and Mx3000P real-time PCR system were used to measure qPCR. All PCR primers are listed in Supplementary Table.
Western blotting
By homogenizing the left ventricular samples, cardiac tissue extracts were collected from mice in lysis buffer (50 mM Tris HCl, 5 mM EDTA, 0.25 M NaCl, 50 mM NaF, 0.5 mM Na3VO4, and 0.5% NP-40). After adjusting the protein concentration, protein samples were collected and stored at – 40 °C. Mitochondria were isolated from myocardial tissue by tissue mitochondria isolation kit (Beyotime Institute of Biotechnology, Beijing, China; cat. no. C3006), and then the mitochondrial protein and beyond it were extracted in the above way.
Electrophoresis was performed using SDS-PAGE gels and then transferred onto immobilon-PL transfer membranes (Millipore). After blocked with 5% skim milk and incubated with indicated antibodies (P-AMPKα2, CST, 2535; T-AMPKα2, CST, 2603P; Bax, CST, 2772; C-caspase3, CST, 9661; Bcl2, CST, 2870; T-ULK1, CST, 8054; P-ULK1, CST, 14202T; Beclin1, Abcam, ab55878; P-mTOR, CST, 2971; TmTOR, CST, 2983; p62, CST, 13121s; TFEB, proteintech, 13372-1-AP; LC3I/II, CST, 3868; Lamp 2A, ABCAM, ab18528; Lamin, ABCAM, ab16048; GAPDH, CST, 2118) at 4°C overnight, the membranes were washed 3 times with Tris-buffered saline (TBS) and 0.5% Tween and incubated with secondary antibody (Thermo Fisher Scientific) for 1 h in room temperature. The membranes were covered by enhanced chemiluminescence (ECL), and blots were scanned by ChemiDoc Imaging System (BIO-RAD, 733BR2234, CA, USA) and analyzed using Image J software.
Cell culture and cell viability
H9C2 cells were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and cultured in DMEM containing 10 % fetal bovine serum (FBS) with streptomycin (100 mg/ml) and penicillin (100 U/ml) at standard conditions (37 °C, 5 % CO2). For signal pathway experiments, H9C2 cells were cultured in six-well plates with 2 × 105 cells/well and were exposed to PBS or Dox (1μmol/L), and meantime add CRA (dissolved in 0.1% DMSO PBS solution, 0.1 μM, 0.5 μM, 1 μM, 3 μM, 5 μM for final concentration) or DMSO for 24 h. Cell viability was detected by CCK-8 (Dojindo Molecular Technologies, Rockville, MD, USA). To evaluate the role of autophagy in Dox-induced cardiotoxicity of CRA treatment, Baf A1 (100 nmol; Millipore, cat. no. B1793) was used to inhibit autophagy.
Transfection experiment
In order to knockdown LC3, TFEB, or AMPKα2, according to the manufacturer’s protocol, LC3 siRNA (100 nmol/L, Guangzhou RiboBio Co. Ltd.), TFEB siRNA (100 nmol/L, Guangzhou RiboBio co. Ltd.), AMPKα2 siRNA (100nmol/L, Guangzhou RiboBio Co. Ltd.), or scramble siRNA were mixed with lipo 6000TM (Beyotime Institute of Biotechnology) and transfected into H9C2 cells. After 48 h of culture in serum-free DMEM, the cells were exposed to Dox for 6 h.
Detection of DHE
For the detection of ROS, the cells were incubated with dihydroethidium (DHE, 5 μmol/L). The production of DHE was observed using Olympus IX51 fluorescence microscope (Olympus Corporation).
TUNEL assay
Cell apoptosis was evaluated by TUNEL staining. For heart tissue sections, they need to firstly dewaxing and hydration. For cells, they need to firstly fix in 4% paraformaldehyde for about 60 min at room temperature and then treated with 0.2% TritonX-100 on rotation with gentle shake for 5 min and then washed with PBS. Then TUNEL staining was performed according to the manufacturer’s instructions (S7111; EMD Millipore Crop). Briefly, equilibrium buffer was incubated with tissue sections or cell climbing sheets at room temperature for about 10 s. Then, the TDT enzyme was added and incubated in a worm dark humidified chamber for 1 h. After that, add stop/wash buffer for about 10 min to terminate the reaction and washed by PBS before anti-digoxigenin fluorescein was finally incubated in a dark humidified chamber for 30 min. Nuclei were co-stained with 0.1 g/mL DAPI (Invitrogen; Thermo Fisher Scientific, Inc.). Images were captured under Olympus IX51 fluorescence microscope (Olympus Corporation).
Mitochondrial respiration
Ice-cold KCl-based cardioplegia (110 mM NaCl, 310 mM NaHCO, 16 mM KCl, 216 mM MgCl, 21.2 mM CaCl, pH 7.8) was used to artificially induce cardiac arrest and immediately obtain ventricular tissue samples from ventricle myocardium. Then, the ventricle myocardium were minced into fine fragments whilst placed on ice and CMs were isolated by trypsin digestion method. For H9C2 cells, prepare 2 × 105 freely isolated pretreated H9C2 cells suspended in 2 mL of empty cell culture medium. After preparing the cells, use high-resolution respirometry (Oxygraph-2k, Oroboros, Innsbruck, Austria) to measure mitochondrial respiration function. Oligomycin (1 μM), p-trifluoromethoxy phenylhydrazone (FCCP, 0.5 μM/step until maximal OCR reached), and rotenone/antimycin A (0.5 μM) were used subsequently to perform oxidative consumption rate (OCR). Injection of aucomycin and FCCP, respectively, measured the ATP linked OCR and maximum respiratory.
Autophagic flux analysis
In order to evaluate the autophagic flux of H9C2 cells, we used the mCherry-GFP-LC3 adenovirus (Vigene, MOI = 50) to infect H9C2. Besides, bafilomycin A1 (BafA1, 100 nM, B1793; Millipore) or TFEB siRNA was added with or without CRA (1 μM) existing when Dox (1 μM) is added to detect the influence of CRA to the patency of autophagic flux. H9C2 cells were cultured on cell climbing films in 24-well plates for autophagic flux analysis.
Transmission electron microscopy
Autophagosomes and autolysosomes were observed by transmission electron microscopy. The isolated heart tissue was cut into about 1-mm cubes and fixed with 2.5% glutaraldehyde at 4 °C overnight. After washed by cold PBS (pH 7.4) 3 times for 10 min, tissues were fixed with 1.0% OsO4 at 4 °C for 90 min. Then, the tissues were dehydrated in ascending series of ethanol and sputter coated with silver ions. Finally, image observation was performed using a transmission electron microscope (TM-3000; Hitatchi, Ltd., Tokyo, Japan), and micrographs were captured at different magnifications (5000 and 8000-fold).
Immunofluorescence staining
Immunofluorescence staining was performed in order to determine the localization of TFEB in H9C2 cells. Briefly, cell climbing slices were fixed in 4% paraformaldehyde overnight at 4 °C and then washed by PBS and treated with 0.2% TritonX-100 on rotation with gently shake for 5 min and then washed with PBS. After blocking with 10% goat serum, TFEB, cell climbing slices were incubated in TFEB (Proteintech, 13372-1-AP, 1:100) for 2 h at 37 °C. Washed by PBS, then the cell climbing slices were incubated with respective fluorescent secondary antibodies (Abcam, ab 150077, 1:200) for 60 min at 37 °C. Finally, the cell climbing slices were washed by PBS, and nuclei were stained with 0.1 g/mL DAPI (Invitrogen; Thermo Fisher Scientific, Inc.). Images were captured under Olympus IX51 fluorescence microscope (Olympus Corporation).
Statistical analysis
All Data were using the appropriate statistical analysis methods to analyze. Two-tailed Student’s t test was used for data analysis between two groups. Data of groups for multiple comparisons were analyzed using repeated measures analysis of variance (ANOVA) with SPSS software (version 21.0). p < 0.05 was considered statistically significant.
Results
CRA improved left ventricular contractility, alleviated Dox-induced cardiac injury, and improved survival rate
Compared with the Dox group, CRA treatment improves the heart weight loss caused by Dox (Fig. 1b). The echocardiographic data show that after CRA treatment, the heart rate (HR), ejection fraction (EF%), and fractional shortening (FS%) of mice all increased in varying degrees (Fig. 1a). The left ventricle end-systolic diameter decreases after CRA treatment (Fig. 1a). The results of hemodynamics show that CRA treatment increased pressure decay (± dP/dt) (Fig. 1c). The above results showed that CRA treatment improved the ventricle contraction after Dox exposure. Subsequently, we detected the cardiac injury markers LDH and CK-MB, and the results showed that CRA reduced the content of LDH and CK-MB in the serum, suggesting that CRA reduced Dox-induced damage to myocardial cells (Fig. 1d). HE staining shows that Dox exposure altered the morphology of cardiac myocytes and resulted in disordered cell arrangement, which was improved after CRA treatment (Fig. 1e). Survival statistics show that CRA application improved the survival rate compared with the Dox group (Fig. 1f).

CRA improved left ventricular contractility, alleviated Dox-induced cardiac injury, and improved survival rate. (a) Representative M-mode echocardiographic and the echocardiographic analysis of mice 4 weeks after Dox or CRA: HR (heart rate), EF% (ejection fraction), FS% (fractional shortening), LVEDs (left ventricle end-systolic diameter) (n = 10). (b) Heart weight to tibial length ratio (HW/TL) (n = 10). (c) Hemodynamics analysis of pressure decay (± dP/dt). (d) Results of serum myocardial injury markers creatine kinase isoenzymes (CK-MB) and lactate dehydrogenase (LDH) (n = 6). (e) HE straining results at 4 weeks after Dox (magnification: × 400). (f) Survival statistics (n = 30). *p < 0.05 vs NS; #p < 0.05 vs Dox
CRA treatment reduced DOX-induced oxidative stress and apoptosis in vitro and in vivo
TUNEL staining of cardiac tissues was used to detect cardiomyocyte apoptosis. The results show that the number of cardiomyocyte apoptosis was significantly reduced after CRA treatment compared with Dox group (Fig. 2a). Western blot results showed that the expression of pro-apoptotic protein Bax and C-caspase3 was increased after Dox exposure, while the expression of the antiapoptotic protein Bcl2 was reduced. CRA treatment reversed these results. Besides, CRA treatment also reduces the reactive oxygen species derived from Nox2 (Fig. 2b). QT-PCR results show that the CRA treatment inhibited Nox 2 and Nox 4 transcription in heart tissues (Fig. 2c). Then, we tested the effect of different concentrations of CRA on cardiomyocyte viability with or without Dox to screen the best concentration used in vitro test. The CRA concentration of 1 μm is most conducive to the survival of cardiomyocytes (Fig. 2d), so we used this concentration in the following in vitro experiments. The results of TUNEL and DHE show that CRA treatment inhibited apoptosis caused by Dox and reduced the content of reactive oxygen species in H9C2 cells (Fig. 2e).

CRA treatment reduced oxidative stress and apoptosis in vitro and in vivo. (a) Representative images of TUNEL staining of heart tissue (magnification: × 400). (b) Representative Western blot analyses of Bax, Bcl2, C-caspase3, Nox2 in the left ventricle (n = 5). (c) The relative mRNA levels of Nox2 and Nox4 in the left ventricle (n = 6). (d) The effect of CRA on cell viability with or without Dox via cell counting Kit-8 (CCK8) assay (n = 8). (e) Representative image of TUNEL staining and DHE staining of H9C2 cell (magnification: × 400, n = 30). *p < 0.05 vs NS/PBS; #p < 0.05 vs Dox
CRA protected the structure and function of mitochondria, promoted cardiomyocyte metabolism and ATP generation
As mitochondria is the main source of ROS, mitochondrial dysfunction can lead to insufficient ATP production, thereby reducing the contractility of the heart; we investigated if CRA protected the structure and function of mitochondria. Our results show that CRA enhanced the transcriptional activities of glucose metabolism-related proteins GULT1, GULT4, ATPase4, and PDK which are important for mitochondrial respiration and metabolism (Fig. 3a). To assess basal respiration, maximal respiration, and ATP-production respiration, we measured the oxygen consumption rate (OCR) by using adult ventricular myocytes isolated from mice of each group. These experiments reveal that Dox significantly reduced mitochondrial basal respiration, maximal respiration, and ATP production in cardiomyocytes compared with NS group (Fig. 3b). However, CRA partly rescued the mitochondrial respiration function damaged by Dox, especially CRA20-treated group (Fig. 3b). The results of the transmission electron microscope of the heart tissue showed that Dox damaged the mitochondrial structure, and resulted in swelling of the mitochondria. After CRA treatment, the swelling of the mitochondria is reduced (Fig. 3c). Using JC-1 staining to detect mitochondrial membrane potential, ΔΨm was an important parameter of mitochondrial function; decreased ΔΨm also predicted early cell apoptosis. The red fluorescence represented normal ΔΨm, while the green fluorescence represented decreased ΔΨm which suggested mitochondrial injury. Red fluorescence and green fluorescence ratio was used to calculate the results. The JC-1 results show that Dox exposure caused a decrease inΔΨm, while CRA improved Dox-induced decrease of ΔΨm, suggesting that CRA reduced Dox damage to mitochondria (Fig. 3d). Taken together, CRA protected the mitochondrial structure and function, which promoted cardiomyocyte metabolism and ATP generation.

CRA protected the structure and function of mitochondria, promoted cardiomyocyte metabolism and ATP generation. (a) The relative mRNA levels of GLUT1, GLUT4, ATPase6, and PDK4 in the left ventricle (n = 6). (b) The results of oxygen consumption rate (OCR) in ventricular myocytes. (c) Transmission electron microscope showed mitochondrial structures (× 5000). (d) Changes of mitochondrial membrane potential in H9C2 cells via JC-1 assay. The green fluorescence represents decreased membrane potential, and the red fluorescence represents normal membrane potential (magnification: × 200, n = 20). *p < 0.05 vs PBS; #p < 0.05 vs Dox
CRA restored autophagic flux and promoted protective autophagy
Because autophagy is important for degradation of damaged organelles and mitochondrial quality, we next investigated the effect of CRA on autophagic flux. In vivo experiments showed that Dox reduced the level of LC3II, while the protein expression of p62 increased. CRA treatment promoted the expression of LC3II and reduced the cargo protein P62. Meanwhile, CRA increases the expression of p-ULK1 and Beclin1, suggesting that CRA promoted autophagosomes formation (Fig. 4a). In vitro, the expression levels of LC3II and P62 increased after Dox exposure, which is consistent with previous reports; possibly due to Dox blocked autophagic flux, CRA treatment promoted the expression of LC3II, which may be due to either impaired autophagolysosomes degradation or increased assembly of autophagosomes (Fig. 4b). We used a GFP-mRFP-LC3 adenovirus to transfect H9C2 cells to analyze autophagic flux. The results showed that after Dox stimulation, the yellow puncta representing autophagosomes were decreased, and the red puncta representing autophagolysosomes accumulated in large numbers, suggesting that Dox inhibited the formation of autophagosomes and induced the accumulation of autophagolysosomes. After CRA treatment, the yellow puncta were increased, indicating the enhancement of autophagic flux (Fig. 4c). Then, we used Baf A1 to inhibit the fusion of lysosomes and autophagosomes. After Baf A1 treatment, Dox exposure decreased yellow puncta, indicating impaired autophagosomes formation. Moreover, Dox exposure also increased red puncta, which was consistent with previous reports that Dox affected downstream of lysosome-autophagosome fusion, which was the degradation of autophagolysosomes. Compared with the Dox+Baf A1 group, CRA treatment increased yellow puncta, indicating that CRA promoted the formation of autophagosomes, while CRA treatment also reduced red puncta, indicated CRA reduced accumulation of autophagolysosomes (Fig. 4c). In addition, after Baf A1 blocked the fusion of autophagosomes and lysosomes, the Western blotting results showed that Baf A1 did not significantly change the LC3II expression in the Dox group compared to the control group, suggesting that Dox blocked autophagic flux, while in the CRA intervention group, Baf A1 increased the LC3II level, indicating that CRA ameliorated the Dox-induced blockade of autophagic flux (Fig. 4d). In addition, Western blotting results show that CRA could promote autophagosomes formation (Fig. 4e). Taken together, CRA promoted autophagosomes formation and autophagolysosomes degradation. To confirm the protective effect of autophagy, we used LC3 siRNA to inhibit autophagosomes formation. The Western blot results show that the application of SiLC3 successfully inhibited the expression of LC3II and abrogate the CRA-induced inhibition of apoptosis and oxidative stress (Fig. 4f, g). The DHE results show that after blocking autophagosomes formation, ROS accumulated in CRA-treated cells (Fig. 4h). These results suggested that CRA ameliorated Dox-induced cardiotoxicity by restoring autophagic flux and promoting autophagosomes maturation.

CRA restored autophagic flux and promoted protective autophagy. (a) Representative Western blot analyses in the left ventricle (n = 5). (b) Representative Western blot analyses of LC3I/II and P62 in H9C2 cell after Dox exposure (n = 5). (c) Images of H9C2 cells transfected with Cherry-red fluorescent protein (RFP)-green fluorescent protein (GFP)-LC3 adenovirus with and without Dox treatment. Autophagosome (yellow puncta) and autophagolysosome (red puncta) were calculated (magnification: × 400, n = 20). (d, e) Representative Western blot analyses of LC3I/II in H9C2 cell with or without Baf A1 (100 nmol) after Dox exposure (n = 5). (f) Representative Western blot analyses of LC3I/II after SiLC3 transfected. (g) Representative Western blot analyses of Bcl2, Bax, and Nox2 in in H9C2 cell with or without SiLC3 (n = 5). (h) Representative image of DHE staining of H9C2 cell (magnification: × 400) (n=30). In vivo: *p < 0.05 vs PBS; #p < 0.05 vs Dox; in vitro: *p < 0.05. ns, not significant
By promoting TFEB expression, CRA restored myocardial autophagic flux, reduced Dox-induced oxidative stress and cell apoptosis, and protected mitochondria
Given that TFEB plays an important role in maintaining lysosomal function and autophagic flux, we detected the protein expression of TFEB in heart tissues. The results showed that after Dox treatment, TFEB expression is decreased in the myocardial nucleus, as well as the expression level of LAMP2A, while CRA treatment promotes the expression of TFEB and LAMP 2A (Fig. 5a, b). The transmission electron microscopy results directly showed the abnormal accumulation of autophagolysosomes in the myocardial tissue after Dox exposure. CRA treatment improves the accumulation of autophagolysosomes (Fig. 5c). Immunofluorescence results show that CRA promoted the nuclear translocation of TFEB (Fig. 5d). Subsequently, we used small interfering siRNA to block the transcription of TFEB in H9C2 cells and examined the protective effect of CRA after Dox stimulation. The Western blot results show that TFEB expression was decreased by SiTFEB and that the inhibition of TFEB weakened the protective effect of CRA against apoptosis, as the expression of LAMP 2A and p62 was similar to that in the Dox group treated with CRA due to the inhibition of TFEB (Fig. 5e, f). In addition, TUNEL and DHE staining also show that the inhibition of TFEB weakened the protective effect of CRA by increasing ROS and apoptosis levels (Fig. 5g). Subsequently, we measured autophagic flux. After TFEB inhibition, CRA did not change autophagolysosomes accumulation, and the yellow punta representing autophagosomes also decrease significantly (Fig. 5h). In addition, TFEB inhibition attenuates the expression of LC3II by CRA (Fig. 5i). These results indicated that CRA promoted the autophagosomes formation and autophagolysosomes degradation by promoting TFEB transcription after Dox-induced injury.

By promoting TFEB expression, CRA restored myocardial autophagic flux, reduced Dox-induced oxidative stress and cell apoptosis, and protected mitochondria. (a) Representative Western blot analyses of TFEB and Lamin in nucleus (n = 5). (b) Representative Western blot analyses of Lamp2A in the left ventricle (n = 5). (c) Autophagolysosomes (red arrow) (× 5000) was observed by transmission electron microscope. (d) Localization of TFEB in H9C2 cells shown by immunofluorescence staining (n = 3). (e) Representative Western blot analyses of TFEB in H9C2 cell after SiTFEB transfected. (f) Representative Western blot analyses of Lamp2A, P62, Bax, Bcl2 in H9C2 after Dox exposure with or without SiTFEB transfected. (g) Representative image of TUNEL staining and DHE staining of H9C2 cell (magnification: × 400, n = 30). (h) Images of H9C2 cells transfected with Cherry-red fluorescent protein (RFP)-green fluorescent protein (GFP)-LC3 adenovirus with Baf A1(magnification: × 400, n = 20). (i)Representative Western blot analyses of LC3I/II in H9C2 cell with or without Baf A1 and SiTFEB (100 nmol) after Dox exposure. (j) The results of oxygen consumption rate (OCR) in H9C2 cells. (n = 5). (k) Changes of mitochondrial membrane potential in H9C2 cells via JC-1 assay (magnification: × 200, n = 30). In vivo: *p < 0.05 vs PBS; #p < 0.05 vs Dox; in vitro: *p < 0.05. ns, not significant
To determine whether CRA protects mitochondria by activating TFEB, we examined the function of mitochondria in the absence of TFEB by OCR. The results show that after inhibiting TFEB, the protective effect of CRA on mitochondrial respiratory function disappeared (Fig. 5j). These results are consistent with those of JC-1 staining (Fig. 5k).
CRA activated TFEB in an AMPKα2-dependent manner to protect against DOX-induced cardiotoxicity
To uncover the upstream factors that regulate TFEB, we examined the AMPKα2-mTOR pathway. The Western blot results confirm that AMPKα signaling was suppressed after Dox treatment (Fig. 6a). CRA markedly activates AMPKα2 signaling but reducing mTOR activity (Fig. 6a). In vitro, after using siRNA to inhibit AMPKα2, CRA did not promote the transfer of TFEB into the nucleus in H9C2 cells (Fig. 6b, c), and the levels of apoptosis and oxidative stress are not reversed by CRA due to AMPKα2 inhibition (Fig. 6d). To evaluate whether CRA protects against Dox-induced cardiotoxicity through AMPKα2 in vivo, C57 and AMPKα2 knockout mice were used and treated with 20 mg/kg CRA or equal volumes of normal saline after Dox exposure. Ablating of AMPKα2 reduces survival and exacerbates cardiac injury in mice (Fig. 6e–i). CRA did not promote the survival of AMPKα2 knockout mice (Fig. 6f). In addition, the EF% and FS% are not be improved by CRA treatment due to the ablation of AMPKα2, nor were the LVEDs, indicating that CRA could not rescue the Dox-induced damage to cardiac function in the absence of AMPKα2 (Fig. 6g). After CRA treatment, the myocardial injury markers LDH and CK-MB did not change significantly (Fig. 6h), and the HE staining results also suggested that the cardioprotective effect of CRA disappeared after AMPKα2 knockout (Fig. 6i). These findings indicated that CRA protected against Dox-induced cardiotoxicity by activating TFEB in an AMPKα2-depended manner.

CRA activates the TFEB in an AMPKα2-dependent manner to protect against Dox-induced cardiotoxicity. (a) Representative Western blot analyses of p-AMPKα2, T-AMPKα2, p-mTOR, and T-mTOR in the left ventricle (n = 5). (b) Representative Western blot analyses of T-AMPKα2 in H9C2 cell after SiAMPKα2 transfected. (c) Representative Western blot analyses of TFEB in nucleus or cytoplasm after transfection with SiAMPKα2 (n = 5). (d) After SiAMPKα2 transfected, representative Western blot analyses of Bcl2, Bax, and Nox2 in H9C2 cell. (e) Representative Western blot analyses of T-AMPKα2 in C57 and AMPKα2 knockout mice. (f) Survival statistics. (g) Representative M-mode echocardiographic and the echocardiographic analysis of C57 and AMPKα2-/-mice 4 weeks after Dox or CRA: EF% (ejection fraction), FS% (fractional shortening), HR (heart rate), LVEDs (left ventricle end-systolic diameter) (n = 8). (h) Results of serum myocardial injury markers lactate dehydrogenase (LDH) and creatine kinase isoenzymes (CK-MB) (n = 6). (i) HE straining results at 4 weeks after Dox (magnification: × 400). In vivo: *p < 0.05 vs PBS; #p < 0.05 vs Dox; in vitro: *p < 0.05. ns, not significant
Discussion
Dox is an effective antitumor drug, but its cardiotoxicity limits its widespread use. Several studies have reported the antitumor effect of CRA, and in our previous studies, we verified the cardioprotective effect of CRA (Wang et al. 2019c, 2020b. However, whether CRA can improve Dox-induced cardiomyopathy remains unclear. In our study, we found that CRA improved Dox-induced cardiotoxicity and rescued damaged cardiac function. In this context, CRA promoted mitochondrial biogenesis and improved cellular metabolism. In addition, CRA restored autophagic flux and promoted autophagolysosomes degradation by activating TFEB, thereby reducing ROS production and promoting cardiomyocyte survival. These protective effects of CRA against cardiotoxicity depended on the activation of AMPKα2.
Autophagy has been extensively studied in Dox-induced cardiotoxicity, and previous studies have mostly clarified that Dox-induced cardiotoxicity involves regulating the maturation of autophagosomes (Zhang et al. 2020a; Zhao et al. 2020). Autophagosomes formation is usually initiated by ULK1 kinase complex; AMPKα2 and mTORC 1 are essential to ULK1 kinase complex activation. mTORC 1 negatively regulates autophagy by phosphorylating ULK1 at Ser757 (Zachari and Ganley 2017). Activated ULK1 kinase complex phosphorylates the class III phosphatidylinositol 3-kinase complex composed of beclin1, hVps34, and p150 and cooperates with other ATG proteins to control the assembly of autophagosomes (Gallagher et al. 2016). Activation of ULK1and Beclin1 strongly confirmed that CRA reversed the damage of Dox to autophagosomes formation. The application of LC3siRNA attenuated the protective effect of CRA, suggesting that the protective effect of CRA against Dox-induced cardiotoxicity was dependent on autophagy. However, accelerating autophagosome formation alone is insufficient to protect against Dox damage to the heart. Recent research has shown that Dox-induced damage to autophagy includes autophagosomes maturation disorders and autophagic flux blockade (Zheng et al. 2019) . By causing autophagolysosomes acidification defects, long-term Dox exposure leads to the failure of autophagolysosomes degradation, and promotes the abnormal accumulation of autophagosolysosomes, which are considered to be a major source of ROS (Li et al. 2016). In this context, accelerating autophagosomes maturation alone will not improve autophagy function, but will exacerbate autophagic flux blockade and worsen tissues damage (Li et al. 2016). In our study, we also noticed that CRA reduced the accumulation of autophagolysosomes, suggesting that CRA promoted the degradation of autophagolysosomes and restored autophagic flux. TFEB has been reported to be involved in autophagosomes formation and autophagolysosomes degradation; thus, TFEB has become the focus of research on the impairment of autophagy caused by Dox. TFEB is a member of the MIT/TFE family, maintains the biological activity and function of lysosomes, accelerates lysosome degradation of proteins, and is the master gene in NET CLEAR (Slade et al. 2020). Previous studies have confirmed that TFEB can stabilize the expression of the lysosomal membrane protein LAMP 2A, maintain the acidification function of lysosomes, and promote the degradation of autophagolysosomes (Shen and Mizushima 2014). At the early stage of Dox exposure, defects in lysosomal function led to the excessive accumulation of autophagolysosomes, as indicated by increased LC3II and P62 protein levels. Over time, autophagosomes maturity was further inhibited, and the LC3 esterification cycle was blocked due to the rapid depletion of TFEB, leading to decreases in LC3II protein levels (Bartlett et al. 2016). In addition to regulating of lysosomal autophagy, TFEB has also been demonstrated to activate macroautophagy independently of its regulation of lysosomes (Settembre and Ballabio 2011). Our results indicated that CRA treatment reversed Dox-induced TFEB depletion, restored the blocked autophagic flux, and reduced Dox-induced oxidative stress and mitochondrial damage and maintained cell survival, while TFEB transcription inhibition eliminated the protective effect of CRA.
Under physiological conditions, TFEB remains inactivated in the cytoplasm through phosphorylation-dependent mechanism, and mTORC 1 can bind to its phosphorylation site to inhibit its transcriptional activity. AMPKα2 is an important negative regulator of mTORC 1 and can activate TFEB through the negative regulation of mTORC 1 (Martina et al. 2012). Additionally, AMPKα2 also directly promotes TFEB nuclear translocation (Li and Chen 2019). Abnormal TFEB expression is associated with a variety of diseases. Previous studies have suggested that Dox exposure leads to overall TFEB depletion, including within the nucleus and cytoplasm, accompanied by ROS generation, decreased lysosome function, mitochondrial injury, and apoptosis. Inhibiting mTORC 1 promotes TFEB transcription and the expression of Lamp 2 A, restores lysosome function, maintains autophagic flux, reduces ROS generation, and promotes cell survival (Bartlett et al. 2016; Wang et al. 2019a, b). In addition, TFEB has been reported to be related to the activation of NF-κB in cardiomyocytes after Dox exposure. Interfering with the nuclear translocation of TFEB will activate the phosphorylation of IKKα/β and NF-κB and promote the production of inflammatory factors (Wang et al. 2020a; Brady et al. 2018). In ischemic heart disease, TFEB has been reported to reduce ischemia/reperfusion-induced apoptosis by rescuing autophagic flux and activating lysosomal autophagy. This protective effect is related to the activation of AMPKα2 (Zhang et al. 2020b; Wang et al. 2019b). Similarly, TFEB-mediated autophagy in endothelial cells promotes angiogenesis and improves ventricular remodeling during myocardial infarction (Liu et al. 2020).
In cardiomyopathy caused by protein accumulation, TFEB activates autophagy to remove abnormally accumulated proteins, and promotes the transcription of the chaperone protein HSPB8, restoring the protein’s normal localization in cardiomyocytes (Ma et al. 2019). We observed the activation of AMPKα2 by CRA, and CRA could not activate TFEB in the context of AMPKα2 deficiency. AMPKα2 ablation also attenuated the cardioprotective effects of CRA in vitro and in vivo. This result may be related to the absence of AMPKα2 preventing its inhibition of mTORC1 or the direct regulation of TFEB. In conclusion, we hypothesized that CRA may protect against Dox-induced cardiotoxicity by activating TFEB in an AMPKα2-dependent manner.
Apoptosis caused by oxidative stress and mitochondrial disorder is considered to be the main mechanism of cardiomyocyte death after Dox exposure. Anthracyclines generate many superoxide anion radicals when they degrade, which is one of the mechanisms by which they kill cancer cells; however, anthracyclines also cause damage to the organelles in normal cells and eventually lead to cell death (De Beer et al. 2001). Mitochondria are important energy-producing organelles of cardiomyocytes that provide a large amount of ATP to maintain cardiac contractile function (Zhang et al. 2013); however, mitochondria are also the main sites of Dox damage and ROS production (Damiani et al. 2016). When the heart is injured by Dox, mitochondrial structural damage and excessive ROS accumulation lead to mitochondrial dysfunction and apoptosis. Previous studies have proven that mitochondrial ROS activate NADPH oxidases (Noxs), especially Nox2. Mitochondria are considered to be the largest source of ROS in the heart and promote the production of mitochondrial ROS (Lambeth 2004; Wosniak et al. 2009). Noxs have been reported to be involved in a variety of heart injuries, including cardiomyocyte apoptosis, interstitial fibrosis, and cardiac hypertrophy (Braunersreuther et al. 2013; Jin et al. 2017). Previous studies have suggested that inhibiting Noxs effectively reduced Dox-induced ROS generation and cell apoptosis and improved cardiac function (De Beer et al. 2001). Our study confirmed that CRA reduced oxidative stress through an autophagy-dependent pathway mediated by TFEB and maintained the mitochondrial biological activity. The protective effects of mitochondria may be due to the reduction in oxidative stress. However, in addition to regulating autophagy, TFEB is a novel metabolic regulator associated with mitochondrial biogenesis, and the absence of TFEB leads to mitochondrial damage and impaired glucose metabolism (Markby and Sakamoto 2020). Moreover, TFEB can induce the transcription of the mitochondrial protective protein PGC1α, promote mitochondrial biogenesis, and enhance cardiomyocyte resistance to stress (Ma et al. 2015). Therefore, CRA may also enhance the ability of mitochondria to resist stress and maintain metabolism by activating the mitochondrial protective effect of TFEB.
In previous studies, CRA exhibited excellent antitumor effects (Fujiwara et al. 2011), and in cardiovascular studies, CRA has also been shown to improve myocardial hypertrophy caused by thoracic aortic constriction and myocardial infarction caused by coronary artery ligation (Wang et al. 2019c, 2020b). More importantly, previous studies have confirmed the safety of CRA, and administration of a gel product containing 10 mg corosolic acid could improve the symptoms of diabetes mellitus without side effects. In another study, no adverse events were found in 15 patients who received CRA daily for up to a year (Stohs et al. 2012). These studies provide a reliable basis for the further clinical application of CRA. In this study, our results demonstrated the protective effect of CRA against Dox-induced cardiotoxicity by restoring autophagic flux and promoting mitochondrial biogenesis, ultimately reducing oxidative stress and apoptosis. Whether the combined application of CRA and Dox can effectively treat cancer while reducing heart damage will be a future research direction. Our study also has some limitations; we studied the protective effect of CRA only on male mice; the protective effect of CRA on Dox-induced heart damage may be different between genders. Moreover, CRA may also promote autophagosome formation independently of TFEB by activating AMPKα2-mTORC 1 signaling pathway. However, inhibition of TFEB was sufficient to eliminate the protective effect of CRA, suggesting that TFEB played a key role in this process. Taken together, CRA has promising future clinical applications; we hope this study provides a new theoretical basis for the treatment of Dox-induced cardiotoxicity and the clinical applications of Dox.
Availability of data and material
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Code availability
Not applicable.
Abbreviations
- CRA:
-
Corosolic acid
- TFEB:
-
Transcription factor EB
- ROS:
-
Reactive oxygen species
- Lamp2A:
-
Lysosomal-associated protein 2a
- CMA:
-
Chaperone-mediated autophagy
- CK-MB:
-
Creatine kinase Isoenzyme
- LDH:
-
Lactate dehydrogenase
- Noxs:
-
NADPH oxidases
- Bcl2:
-
B cell lymphoma 2
- Bax:
-
Bcl-2-associated X protein
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- DHE:
-
Dihydroethidium
- FBS:
-
Fetal bovine serum
- AMPKα2:
-
AMP-activated protein kinase α2
- OCR:
-
Oxidative consumption rate
References
Alshabanah OA, Hafez MM, Al-Harbi MM, Hassan ZK, Al RS, Asiri YA, et al. Doxorubicin toxicity can be ameliorated during antioxidant L-carnitine supplementation. Oxidative Med Cell Longev. 2010;6:428–33.
Bartlett JJ, Trivedi PC, Yeung P, Kienesberger PC, Pulinilkunnil T. Doxorubicin impairs cardiomyocyte viability by suppressing transcription factor EB expression and disrupting autophagy. Biochem J. 2016;21:3769–89. https://doi.org/10.1042/BCJ20160385.
Brady OA, Martina JA, Puertollano R. Emerging roles for TFEB in the immune response and inflammation. Autophagy. 2018;2:181–9. https://doi.org/10.1080/15548627.2017.1313943.
Braunersreuther V, Montecucco F, Asrih M, Pelli G, Galan K, Frias M, et al. Role of NADPH oxidase isoforms NOX1, NOX2 and NOX4 in myocardial ischemia/reperfusion injury. J Mol Cell Cardiol. 2013:99–107. https://doi.org/10.1016/j.yjmcc.2013.09.007.
Cai X, Zhang H, Tong D, Tan Z, Han D, Ji F, et al. Corosolic acid triggers mitochondria and caspase-dependent apoptotic cell death in osteosarcoma MG-63 cells. Phytother Res. 2011;9:1354–61. https://doi.org/10.1002/ptr.3422.
Cheng QL, Li HL, Li YC, Liu ZW, Guo XH, Cheng YJ. CRA(Crosolic Acid) isolated from Actinidia valvata Dunn. Radix induces apoptosis of human gastric cancer cell line BGC823 in vitro via down-regulation of the NF-kappaB pathway. Food Chem Toxicol. 2017:475–85. https://doi.org/10.1016/j.fct.2017.05.021.
Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;5274:501–3. https://doi.org/10.1126/science.273.5274.501.
Damiani RM, Moura DJ, Viau CM, Caceres RA, Henriques J, Saffi J. Pathways of cardiac toxicity: Comparison between chemotherapeutic drugs doxorubicin and mitoxantrone. Arch Toxicol. 2016;9:2063–76. https://doi.org/10.1007/s00204-016-1759-y.
De Beer EL, Bottone AE, Voest EE. Doxorubicin and mechanical performance of cardiac trabeculae after acute and chronic treatment: A review. Eur J Pharmacol. 2001;1:1–11. https://doi.org/10.1016/s0014-2999(01)00765-8.
Fujiwara Y, Komohara Y, Ikeda T, Takeya M. Corosolic acid inhibits glioblastoma cell proliferation by suppressing the activation of signal transducer and activator of transcription-3 and nuclear factor-kappa B in tumor cells and tumor-associated macrophages. Cancer Sci. 2011;1:206–11. https://doi.org/10.1111/j.1349-7006.2010.01772.x.
Fukushima M, Matsuyama F, Ueda N, Egawa K, Takemoto J, Kajimoto Y, et al. Effect of corosolic acid on postchallenge plasma glucose levels. Diabetes Res Clin Pract. 2006;2:174–7. https://doi.org/10.1016/j.diabres.2006.01.010.
Gallagher LE, Williamson LE, Chan EY. Advances in autophagy regulatory mechanisms. Cells. 2016;5:24. https://doi.org/10.3390/cells5020024.
Ghosh J, Das J, Manna P, Sil PC. The protective role of arjunolic acid against doxorubicin induced intracellular ROS dependent JNK-p38 and p53-mediated cardiac apoptosis. Biomaterials. 2011;21:4857–66. https://doi.org/10.1016/j.biomaterials.2011.03.048.
Govender J, Loos B, Marais E, Engelbrecht AM. Mitochondrial catastrophe during doxorubicin-induced cardiotoxicity: a review of the protective role of melatonin. J Pineal Res. 2014;4:367–80. https://doi.org/10.1111/jpi.12176.
Inpanathan S, Botelho RJ. The lysosome signaling platform: adapting with the times. Front Cell Dev Biol. 2019;113. https://doi.org/10.3389/fcell.2019.00113.
Jin L, Piao ZH, Sun S, Liu B, Kim GR, Seok YM, et al. Gallic acid reduces blood pressure and attenuates oxidative stress and cardiac hypertrophy in spontaneously hypertensive rats. Sci Rep. 2017;1:15607. https://doi.org/10.1038/s41598-017-15925-1.
Kim KH, Lee MS. Autophagy--a key player in cellular and body metabolism. Nat Rev Endocrinol. 2014;6:322–37. https://doi.org/10.1038/nrendo.2014.35.
Koleini N, Kardami E. Autophagy and mitophagy in the context of doxorubicin-induced cardiotoxicity. Oncotarget. 2017;28:46663–80. https://doi.org/10.18632/oncotarget.16944.
Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;3:181–9. https://doi.org/10.1038/nri1312.
Li Y, Chen Y. AMPK and autophagy. Adv Exp Med Biol. 2019:85–108. https://doi.org/10.1007/978-981-15-0602-4_4.
Li DL, Wang ZV, Ding G, Tan W, Luo X, Criollo A, et al. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation. 2016;17:1668–87. https://doi.org/10.1161/CIRCULATIONAHA.115.017443.
Liu H, Liu S, Qiu X, Yang X, Bao L, Pu F, et al. Donor MSCs release apoptotic bodies to improve myocardial infarction via autophagy regulation in recipient cells. Autophagy. 2020:1–16. https://doi.org/10.1080/15548627.2020.1717128.
Ma X, Liu H, Murphy JT, Foyil SR, Godar RJ, Abuirqeba H, et al. Regulation of the transcription factor EB-PGC1alpha axis by beclin-1 controls mitochondrial quality and cardiomyocyte death under stress. Mol Cell Biol. 2015;6:956–76. https://doi.org/10.1128/MCB.01091-14.
Ma X, Mani K, Liu H, Kovacs A, Murphy JT, Foroughi L, et al. Transcription factor EB activation rescues advanced alphaB-Crystallin Mutation-Induced cardiomyopathy by normalizing desmin localization. J Am Heart Assoc. 2019;4:e010866. https://doi.org/10.1161/JAHA.118.010866.
Markby GR, Sakamoto K. Transcription Factor EB (TFEB): New metabolic coordinators mediating adaptive responses to exercise in skeletal muscle? Am J Physiol Endocrinol Metab. 2020. https://doi.org/10.1152/ajpendo.00339.2020.
Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;6:903–14. https://doi.org/10.4161/auto.19653.
Settembre C, Ballabio A. TFEB regulates autophagy: an integrated coordination of cellular degradation and recycling processes. Autophagy. 2011;11:1379–81. https://doi.org/10.4161/auto.7.11.17166.
Shen HM, Mizushima N. At the end of the autophagic road: an emerging understanding of lysosomal functions in autophagy. Trends Biochem Sci. 2014;2:61–71. https://doi.org/10.1016/j.tibs.2013.12.001.
Singal PK, Deally CM, Weinberg LE. Subcellular effects of adriamycin in the heart: a concise review. J Mol Cell Cardiol. 1987;8:817–28.
Slade L, Biswas D, Ihionu F, El HY, Kienesberger PC, Pulinilkunnil T. A lysosome independent role for TFEB in activating DNA repair and inhibiting apoptosis in breast cancer cells. Biochem J. 2020;1:137–60. https://doi.org/10.1042/BCJ20190596.
Stohs SJ, Miller H, Kaats GR. A review of the efficacy and safety of banaba (Lagerstroemia speciosa L.) and corosolic acid. Phytother. Res. 2012;3:317–24. https://doi.org/10.1002/ptr.3664.
Sung B, Kang YJ, Kim DH, Hwang SY, Lee Y, Kim M, et al. Corosolic acid induces apoptotic cell death in HCT116 human colon cancer cells through a caspase-dependent pathway. Int J Mol Med. 2014;4:943–9. https://doi.org/10.3892/ijmm.2014.1639.
Terman A, Brunk UT. Autophagy in cardiac myocyte homeostasis, aging, and pathology. Cardiovasc Res. 2005;3:355–65. https://doi.org/10.1016/j.cardiores.2005.08.014.
Wang X, Li C, Wang Q, Li W, Guo D, Zhang X, et al. Tanshinone IIA restores dynamic balance of autophagosome/autolysosome in doxorubicin-induced cardiotoxicity via targeting Beclin1/LAMP1. Cancers (Basel). 2019a;7. https://doi.org/10.3390/cancers11070910.
Wang Y, Yang Z, Zheng G, Yu L, Yin Y, Mu N, et al. Metformin promotes autophagy in ischemia/reperfusion myocardium via cytoplasmic AMPKalpha1 and nuclear AMPKalpha2 pathways. Life Sci. 2019b:64–71. https://doi.org/10.1016/j.lfs.2019.04.002.
Wang ZP, Shen D, Che Y, Jin YG, Wang SS, Wu QQ, et al. Corosolic acid ameliorates cardiac hypertrophy via regulating autophagy. Biosci Rep. 2019c;12. https://doi.org/10.1042/BSR20191860.
Wang X, Wang Q, Li W, Zhang Q, Jiang Y, Guo D, et al. TFEB-NF-kappaB inflammatory signaling axis: A novel therapeutic pathway of dihydrotanshinone I in doxorubicin-induced cardiotoxicity. J Exp Clin Cancer Res. 2020a;1:93. https://doi.org/10.1186/s13046-020-01595-x.
Wang ZP, Che Y, Zhou H, Meng YY, Wu HM, Jin YG, et al. Corosolic acid attenuates cardiac fibrosis following myocardial infarction in mice. Int J Mol Med. 2020b;5:1425–35. https://doi.org/10.3892/ijmm.2020.4531.
Woo SM, Seo SU, Min KJ, Im SS, Nam JO, Chang JS, et al. Corosolic acid induces non-apoptotic cell death through generation of lipid reactive oxygen species production in human renal carcinoma caki cells. Int J Mol Sci. 2018:5. https://doi.org/10.3390/ijms19051309.
Wosniak JJ, Santos CX, Kowaltowski AJ, Laurindo FR. Cross-talk between mitochondria and NADPH oxidase: effects of mild mitochondrial dysfunction on angiotensin II-mediated increase in Nox isoform expression and activity in vascular smooth muscle cells. Antioxid Redox Signal. 2009;6:1265–78. https://doi.org/10.1089/ars.2009.2392.
Zachari M, Ganley IG. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017;6:585–96. https://doi.org/10.1042/EBC20170021.
Zhang L, Jaswal JS, Ussher JR, Sankaralingam S, Wagg C, Zaugg M, et al. Cardiac insulin-resistance and decreased mitochondrial energy production precede the development of systolic heart failure after pressure-overload hypertrophy. Circ Heart Fail. 2013;5:1039–48. https://doi.org/10.1161/CIRCHEARTFAILURE.112.000228.
Zhang J, Cui L, Han X, Zhang Y, Zhang X, Chu X, et al. Protective effects of tannic acid on acute doxorubicin-induced cardiotoxicity: involvement of suppression in oxidative stress, inflammation, and apoptosis. Biomed Pharmacother. 2017:1253–60. https://doi.org/10.1016/j.biopha.2017.07.051.
Zhang J, Sun Z, Lin N, Lu W, Huang X, Weng J, et al. Fucoidan from Fucus vesiculosus attenuates doxorubicin-induced acute cardiotoxicity by regulating JAK2/STAT3-mediated apoptosis and autophagy. Biomed Pharmacother. 2020a;110534. https://doi.org/10.1016/j.biopha.2020.110534.
Zhang YJ, Zhang M, Zhao X, Shi K, Ye M, Tian J, et al. NAD(+) administration decreases microvascular damage following cardiac ischemia/reperfusion by restoring autophagic flux. Basic Res Cardiol. 2020b;5:57. https://doi.org/10.1007/s00395-020-0817-z.
Zhao XX, Cho H, Lee S, Woo JS, Song MY, Cheng XW, et al. BAY60-2770 attenuates doxorubicin-induced cardiotoxicity by decreased oxidative stress and enhanced autophagy. Chem Biol Interact. 2020;109190. https://doi.org/10.1016/j.cbi.2020.109190.
Zheng D, Zhang Y, Zheng M, Cao T, Wang G, Zhang L, et al. Nicotinamide riboside promotes autolysosome clearance in preventing doxorubicin-induced cardiotoxicity. Clin Sci (Lond). 2019;13:1505–21. https://doi.org/10.1042/CS20181022.
Funding
This work was supported by grants from the Key Project of the National Natural Science Foundation (No. 81530012), the National Key R&D Program of China (2018YFC1311300), the Fundamental Research Funds for the Central Universities (2042018kf1032), the Development Center for Medical Science and Technology National Health and Family Planning Commission of the People’s Republic of China (2016ZX-008-01), and the Science and Technology Planning Projects of Wuhan (2018061005132295).
Author information
Authors and Affiliations
Contributions
Yan Che and Zhaopeng Wang designed this research and drafted the manuscript; Yuan Yuan revised this research critically for important intellectual content; Heng Zhou, Haiming Wu and Shasha Wang performed the experiments and analyzed the data; Qizhu Tang approved the version to be published. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
All animal experiments were approved by the Animal Care and Use Committee of Renmin Hospital of Wuhan University and adhered to the Guidelines for the Care and Use of Laboratory Animals (NIH publication, revised 2011).
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(DOCX 12 kb)
Rights and permissions
About this article

Cite this article
Che, Y., Wang, Z., Yuan, Y. et al. By restoring autophagic flux and improving mitochondrial function, corosolic acid protects against Dox-induced cardiotoxicity. Cell Biol Toxicol 38, 451–467 (2022). https://doi.org/10.1007/s10565-021-09619-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10565-021-09619-8