
Abstract
Polystyrene supported palladium catalysts were synthesized and their chemical and morphological nature were studied using NMR, XRD, TEM, EDX, and XPS analyses. Using the supported catalyst, the first palladium catalyzed acceptorless dehydrogenative coupling of secondary alcohols in water is reported. This method is green, sustainable, phosphine free, and carried out under aerobic condition. Reusability of the catalyst was shown for both alkylation and quinoline reactions till 7 cycles with marginal decrease in yield. Metal leaching was found to be the cause of decrease in yield in both instances.
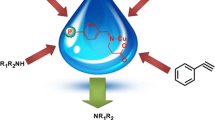
Graphical Abstract
Polystyrene anchored palladium catalysts have been synthesized and used in the acceptorless dehydrogenative coupling of secondary alcohols in aqueous condition. Stability and recyclability of the catalyst was also studied up to 7th cycle in water.

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Palladium-based catalysts have become a ubiquitous part of synthetic chemistry because of their widespread application in the synthesis of important natural and medicinal substrates [1,2,3,4,5,6,7,8]. They are successfully employed to promote a wide variety of reactions such as arylation [9,10,11,12,13,14,15,16], alkylation [17,18,19,20,21,22,23,24,25,26,27], hydrogenation [17, 28,29,30,31,32,33,34,35], oxidation [5, 36,37,38], cyclization [39,40,41,42,43,44,45,46], cross-coupling [47,48,49,50,51,52], isomerization [53,54,55,56,57,58,59], and so on. The exceptional catalytic activity of palladium-based catalysts/precatalysts motivates researchers to find new and exciting discoveries every day. Palladium catalysts have also made their impact in the hydrogen borrowing catalysis [60,61,62,63,64,65,66,67,68,69,70,71].
The hydrogen borrowing catalysis has become a green and powerful methodology to construct carbon–carbon and carbon-heteroatom bonds that uses alcohols as alkylating agents [60,61,62,63,64,65,66,67,68,69,70,71]. As this technique has the advantage to use readily available, easy-to-handle and sustainable alcohols as alkylating agents over traditional methods that uses mutagenic alkyl halides. Although, the hydrogen borrowing methodology provides high atom economy and green by-product (water), use of organic solvents limits its sustainability. In the context of developing greener and sustainable synthetic approach; reactions which uses water as the reaction medium, has gained tremendous interest [72,73,74,75,76,77,78]. Water is a cheap, abundant, environmentally friendly, non-toxic and non-flammable unlike organic solvents [79, 80]. In addition, water has a large temperature window, high heat capacity and inherently low oxygen solubility which favors the aerobic reaction with sensitive transition metal catalysts. Furthermore, the high polarity, low viscosity and immiscibility of water makes the reaction workup and product purification easy [81, 82]. Additionally, hydrophobic effect accelerates the organic reactions in water which results in unusual reactivity and selectivity [83,84,85,86].
Immobilization of catalyst makes the separation simpler and offers reusability of the expensive metal catalyst [87]. Among the several supports, polymer anchored palladium catalysts [88,89,90,91,92], especially polystyrene [89] based palladium catalysts have been studied to a greater extent. Polystyrene (PS) anchored palladium catalysts are borderline catalyst which offers the advantages of reusability and simpler purification of the heterogeneous system while retaining selectivity of the homogeneous system. However, in many instances upon longer stirring with organic solvents, polystyrene supported palladium have shown leaching of metal. This problem can be avoided with the use of water as a solvent.
There have been numerous reports for the synthesis of α-alkylated ketones starting from ketones and primary alcohols [93,94,95]. Recently, more abundant secondary alcohols are being used for the β-alkylation of secondary alcohols leading to α-alkylated ketones. However, most of these reports are based on ruthenium- or iridium-based catalysts [96,97,98,99,100,101,102,103,104,105] and are heavily relied on the use of phosphine based ligands and therefore the reaction is carried out under anaerobic and anhydrous condition. Moreover, the application of noble metal catalyst in solvent free or in aqueous condition is also very limited. Realizing the aforementioned needs from a sustainability perspective to apply in hydrogen borrowing catalysis, herein, we report the synthesis and catalytic activity of new polystyrene supported pyrazole-based palladium catalysts/precatalysts with the experience that we gained to utilize pyrazole-based palladacycles for C–C [68, 69] and C–N [67, 69] bond forming reactions via hydrogen borrowing methodology. These catalysts were used in the synthesis of quinoline and α-alkylated ketones from secondary alcohols using double dehydrogenative approach in water. The catalytic recyclability was also studied.
2 Results and Discussion
Our studies to use homogeneous palladacycles as catalysts/precatalysts reveal that -CF3 functionalized triaryl pyrazoles act as better catalysts/precatalysts. Keeping this in mind we designed and synthesized two styryl-substituted monomers M1 and M2 following the literature reported procedure [106]. Reaction of 1,3-diketone with phenylhydrazine (or) (2- (trifluoromethyl)phenylhydrazine in methanol and acetic acid produced the desired triaryl pyrazoles in good yield. The triaryl pyrazoles were brominated using N-bromosuccinimide. The bromopyrazoles thus formed were reacted with 4-vinylphenyl boronic acid under Suzuki reaction condition to afford the styryl pyrazole monomers M1 and M2. The monomers were then subjected to free radical polymerisation using azobisisobutyronitrile (AIBN) as an initiator in 1,2-dichloroethane (DCE) at 80 °C to produce the desired polymers P1 and P2 (Scheme 1a). The monomers M1 & M2 and the polymers P1 & P2 were characterized by 1H & 13C NMR spectroscopy. The 1H NMR of P1 & P2 showed complete disappearance of the AMX multiplet observed in M1 & M2 which indicates the formation of the polymers (Fig. S12 and S15). Gel permeation chromatography (GPC) (with respect to polystyrene standards) gave a molecular weight (Mn) of 23,500 with a PDI = 2.3 for P1 and 62,300 with a PDI of 1.8 for P2. (see SI, Fig. S79 and S80). Insoluble resin IP1 was prepared by reacting monomer M2 with divinylbenzene in 1:2 ratio under free radical polymerisation condition using AIBN as an initiator in DCE at 80 °C (Scheme 1b). All three polymers P1, P2 and IP1 were subjected to palladation in acetic acid at 100 °C for 1 h using palladium acetate resulting in Pd@P1, Pd@P2 and Pd@IP1 respectively. Although, we expected that Pd@P1 and Pd@P2 acts as soluble polymers, to our surprise they turned out to be insoluble in common organic solvents. Infrared spectroscopic studies reveal the presence of carbonyl functional groups at 1411 and 1564 cm−1 for Pd@P1 and at 1411 and 1568 cm−1 for Pd@P2 which are absent in case of P1 and P2 respectively (Fig. S81 and S82). Similar peaks at 1411 and 1589 cm−1 for Pd@IP1 implies the presence of carbonyl group (Fig. 1). Elemental analyses of Pd@P1, Pd@P2 and Pd@IP1 indicated a palladium loading of 1.12, 1.60 and 0.972 mmol/g respectively. These materials were further characterized using 13C cross-polarization magic-angle spinning (CPMAS) (Fig. S17-20). To further support the presence of palladium, the fresh catalyst Pd@IP1 was analyzed using powder XRD. The XRD pattern showed four peaks at 2θ = 40.2°, 46.7°, 68.3°, and 82.3° which can be assigned to (111), (200), (220), and (311) planes of the cubic crystal system (PDF No. 01–087-0639) for the metallic palladium [107,108,109,110]. The broad peaks at 2θ = 10 o and 20 o can be attributed to the C and N atom of the polymeric pyrazole unit (Fig. 1). Similar observations were made for the catalyst Pd@P1 (PDF No. 01–087-0645) and Pd@P2 (PDF No. 03–065-2867) in powder XRD (Fig. S81 and S82). Since Pd@IP1 showed better catalytic activity among the supported catalysts (see-discussion in next section), it was further characterized by transmission electron microscopy (TEM), energy dispersive X-ray analysis (EDX), and X-ray photoelectron spectroscopy (XPS). The TEM image of the fresh catalyst showed well dispersed nanoparticle with an average particle size in the range of 2–3 nm which indicates that during the metalation process the polymer stabilizes the formation of Pd-nanoparticles (Fig. 2). Further insight into the structural integrity of the nanoparticle were studied using EDX elemental mapping analysis. As seen in the EDX images, Pd atoms are well dispersed over a cloudy substrate of C atoms which is the polymeric pyrazoles (Fig. 2). The chemical state of the Pd atom were investigated using X-ray photoelectron spectroscopy. The Pd 3d spectrum (Fig. 3) shows two different chemical states of the Pd atom. In the fresh catalyst, the lower binding energies (335 eV and 340.2 eV) can be attributed to metallic palladium (Pd(0)) [111,112,113,114]. The higher components of the binding energy (336.5 and 341.5) can be assigned to Pd(II). Further the molar ratio of Pd(0) and Pd(II) present in both fresh & reused catalyst has been found to be 1.081 and 1.005 respectively.

(a) Synthesis of soluble polymers P1 and P2 and insoluble precatalysts Pd@P1 and Pd@P2, (b) Synthesis of insoluble resin IP1 and pre-catalyst Pd@IP1

IR spectra for IP1 and Pd@IP1 (left). Powder XRD pattern of Pd@IP1 (right; PDF No. 01–087-0639)

TEM images of (a) fresh Pd@IP1; (c) after 7th run of alkylation (Pd@IP1); and (e) after 7th run of quinoline reaction (Pd@IP1). EDX elemental mapping of (b) fresh Pd@IP1; (d) after 7th run of alkylation (Pd@IP1); and (f) after 7th run of quinoline reaction (Pd@IP1

XPS spectra for the Pd 3d of the (left) fresh Pd@IP1 and (right) Pd@IP1 after 7th run of alkylation
Having synthesized and characterized the catalysts, their catalytic efficiency for C–C bond formation was studied. The reaction of 1-phenylethanol and benzyl alcohol was chosen as a model reaction. Initial screening of the three PS-supported catalysts (Table 1, entry 1–3) revealed that the activity follows Pd@IP1 > Pd@P2 > Pd@P1, which can be attributed to the electronic as well as steric factors as we observed in the homogeneous system [67,68,69]. Among the different bases screened, KOtBu resulted in 59% yield at 100 °C (Table 1, entry 4–7). Further elevation in temperature to 120 °C caused an increase in yield to 76% (Table 1, entry 8). Finally, by using 2 mL of water as the solvent resulted 92% of the α-alkylated ketone (Table 1, entry 9). Decrease of metal loading or base loading resulted in substantial decrease in the product yield (Table 1, entry 10 and 11). The reaction does not proceed without the catalyst (Table 1, entry 12). With the optimised conditions in hand, the scope and limitations of this protocol were screened. Electron donating as well as electron withdrawing groups were well tolerated and resulted in good yields. 4-Methylbenzyl alcohol, 2-methylbenzyl alcohol, 4-methoxybenzyl alcohol and 4-fluorobenzyl alcohol afforded the α-alkylated products in good to excellent yields (81 to 91%) (Table 2, 2b–2e). Varying the secondary alcohol also had little impact on the reactivity of this protocol. 1-(2-Methylphenyl)ethanol and 1-(4-methylphenyl)ethanol afforded the alkylated product in 81 and 79% yield respectively (Table 2, 2f, 2h). 4-Fluoro-α-methylbenzyl alcohol produced the ketone product in 87% yield (Table 2, 2g). Increase of aliphatic chain in the secondary alcohol did not impact the reactivity as 1-phenylpropan-1-ol afforded the desired ketone product in good yield (Table 2, 2i). Cyclic system such as 1,2,3,4-tetrahydronaphthalen-1-ol gave the corresponding α-alkylated product in 75% yield (Table 2, 2k). This protocol has good tolerance towards heteroaryl substituted secondary alcohol such as 1-(thiophen-2-yl)ethan-1-ol which resulted in 71% yield of the ketone product (Table 2, 2j).
With this success, we focused our attention for the synthesis of quinoline using the optimized conditions and catalyst mentioned above. 2-Aminobenzyl alcohol is made to react with 1.5 equivalent of 1-phenylethanol in aqueous condition (1 mL H2O) which afforded the desired quinoline product in 93% yield. Substitutions such as –CH3 and –OCH3 on the aryl group of secondary alcohol afforded excellent yield of the quinoline product (Table 3, 3b (87%) and 3c (83%). In general, halo substituted compounds at elevated temperature undergoes dehalogenation and thereby making them incompatible in most of the protocols involving palladium catalysis [67,68,69,70,71]. It is worth mentioning that use of aqueous medium helped in this protocol tolerate fluoro and chloro substituted secondary alcohols underthe experimental conditions and produced the quinoline compounds in 72–89% (Table 3, 3d–3g).
Heteroaryl substrate such as 1-thiophenylethanol afforded the quinoline product in 77% isolated yield (Table 3, 3i). 2-Phenyl-1,8-naphthyridine was also prepared using this protocol in 78% yield (Table 3, 3j). It should be noted that 2-methylquinoline was produced in 63% yield (Table 3, 3k) using an excess amount of isopropyl alcohol (0.5 mL). To our delight 3-aminopropan-1-ol was converted to 2-phenylpyridine in 65% yield (Table 3, 3l). 1-Phenylpropan-1-ol which has one more carbon and cyclic secondary alcohol viz., 1,2,3,4-tetrahydronaphthalen-1-ol generated a good yield (Table 3, 81% (3m) and 74%(3n)) of the quinoline products.
The heterogeneous nature of the catalyst was tested by hot filtration. Two reactions were performed separately, in the first reaction, alkylation of alcohol was tested under the optimized conditions after 5 and 24 h. While, in the second case after 5 h the reaction mixture was filtered at hot condition, and the filtrate was further heated for 24 h after adding the required base. The first reaction gave a 93% of the product yield after 24 h, whereas in the second case 43% of product was observed after 5 h which remained same till 24 h (Fig. 4). This result suggest that the reaction does not proceed through the dissolved palladium and thus implies the heterogeneous nature of the catalyst.

Recycling experiment (Top). Hot filtration test (Bottom)
Recycling experiments were performed for both alkylation and quinoline synthesis. After the first run, the catalyst was filtered using glass filter and was washed with water, methanol and acetone three times prior to the next run. The catalyst showed good activity for both the reactions till the fourth run (Fig. 4; yield ranges from 92–89% in case of α-alkylation and 91–88% in case of quinoline synthesis). After the fourth run, a decrease in the yields were observed in both the cases which eventually dropped down to 77 and 81% in case of alkylation reaction and quinoline synthesis respectively.
To understand the reason behind the decrease in the yield, we analysed the spent catalyst Pd@IP1 using different techniques. The TEM reveal a marginal increase in the average particle size after the seventh run of alkylation (4.5–5 nm) and quinoline (3–4 nm) reactions. (Fig. 2). A similar binding energy for Pd@IP1 was observed after 7th run of alkylation using X-ray photoelectron spectroscopy. Elemental analysis reveal that there was considerable palladium loss observed in both the reactions after the seventh cycle (0.972 mmol/g fresh catalyst; 0.834 mmol/g after 7th cycle-alkylation reaction; 0.85 mmol/g after 7th cycle-quinoline reaction). These observations suggest that palladium leaching is responsible for the slight decrease in activity. Presence of oxygen in the EDX elemental analysis further supports the presence of carbonyl group in the catalyst (See Fig. S74, S76, and S78).
3 Conclusion
In conclusion, we have designed and synthesized polystyrene supported palladium catalysts. Among the three catalysts studied, Pd@IP1 showed better activity. The catalysts were characterized using 13C CPMAS NMR, TEM, XPS, XRD and ICP techniques. Effective use of Pd@IP1 in dehydrogenative cross coupling of secondary alcohols which involves synthesis of quinoline and α-alkylated ketones from secondary alcohols using water as a sustainable medium. This protocol tolerates different substrates including halogen substituted aryl compounds. Interestingly, the recycling experiments reveal that the catalyst is stable under the experimental conditions with slight decrease in yield which is due to the metal leaching. Further studies to use this catalytic system for different organic transformation are currently in progress.
4 Experimental Section
4.1 General Information
Reagents and starting materials were purchased from Alfa-Aesar, Sigma-Aldrich and Spectrochem chemical companies and used as received unless otherwise noted. Chlorinated solvents were distilled from CaH2. THF was distilled from Na/benzophenone prior to use. 1,3,5-Triphenyl-1H-pyrazole and 3,5-diphenyl-1-(2-(trifluoromethyl)phenyl)-1H-pyrazole were prepared according to literature procedure15. All 400 or 700 MHz 1H and 100 or 176 MHz 13C NMR, and 377 MHz 19F spectra were recorded on a spectrometer operating at 400 or 700 MHz referenced internally to solvent signals. 19F NMR spectra were externally referenced to α,α,α- trifluorotoluene in CDCl3 (δ = -63.73 ppm). High-resolution mass spectra (HRMS) were recorded on a Bruker microTOF-QII mass spectrometer. Gel permeation chromatography (GPC) analyses were performed on a Shimadzu-LC20AD system referenced to poly(styrene) standards. THF was used as the mobile phase with a flow rate of 1.0 mL min−1. Morphological study and elemental mapping analyses of the samples were performed using a transmission electron microscope (equipped with HRTEM, JEOL 2100F, operated at 200 kV). Particle size was measured using ImageJ software. The elemental composition of the synthesized catalysts were verified by using an inductively coupled plasma-optical emission spectrophotometer (iCAP 7000 ICP-OES). The powder X-ray diffraction data were collected on a Bruker D8 Advance X-ray powder diffractometer with Cu Kα radiation (λ = 1.5418 Å) as the X-ray source. XPS measurements were carried out using Thermo Kalpha + spectrometer using micro focused and monochromated AlKα radiation with energy 1488.6 eV.
Synthetic procedure for tetraarylpyrazole M1: Monomer M1 was prepared by following the literature reported method [1,2,3,4,5,6,7,8]. The quantities involved are as follows. Bromopyrazole 3 (1.00 g, 2.67 mmol), Pd(PPh3)4 (0.09 g,0.08 mmol) Na2CO3 ( 0.56 g, 5.34 mmol) and 4-vinylbenzeneboronic acid (0.47 g, 3.20 mmol) were taken in a 100 mL two neck round bottom flask. The whole set up was evacuated and nitrogen was purged into it. A degassed dimethoxyethane and water in 20:8 was added to it and was refluxed for 24 h. Aqueous layer was separated and the organic layer was collected using dichloromethane (3 × 20 mL). The organic phase was dried with Na2SO4 and concentrated under vacuum. After purification by column chromatography, the compound was isolated as white solid (0.76 g, 72%). 1H NMR (400 MHz, CDCl3) δ 7.55–7.47 (m, 2H, Ar–H), 7.32–7.26 (m, 7H, Ar–H), 7.25–7.21 (m, 4H, Ar–H), 7.21–7.16 (m, 2H, Ar–H), 7.04 (t, J = 8.0 Hz, 4H, Ar–H), 6.65 (dd, J = 17.6, 10.9 Hz, 1H, Ph-CH = CH2), 5.69 (d, J = 17.6 Hz, 1H, Ph-CH = CH2), 5.19 (d, J = 10.9 Hz, 1H, Ph-CH = CH2) ppm. 13C NMR (101 MHz, CDCl3) δ 150.37(CPz), 141.54(CPz), 140.01(CPz), 136.75(Ph-CH = CH2), 135.86(CHAr), 133.19 (CAr), 132.76 (CAr), 130.90(CHAr), 130.59(CHAr), 130.17(CAr), 128.91(CHAr), 128.62(CHAr), 128.47(CHAr), 128.37(CHAr), 127.84 (CAr), 127.41 (CAr), 126.24(CHAr), 125.49(CHAr), 120.45(CHAr), 113.67 (Ph-CH = CH2) ppm.
Synthetic procedure for tetra aryl pyrazole M2: Monomer M2 was prepared using the procedure used for the preparation of M1. The quantities involved are as follows: Bromopyrazole 4 (1.5 g, 3.38 mmol), Pd(PPh3)4 (0.12 g, 0.10 mmol), Na2CO3 (0.72 g, 6.76 mmol) and 4-vinylbenzeneboronic acid (0.60 g, 4.05 mmol). After purification by column chromatography, the compound as isolated as white solid (1.02 g, 65%). 1H NMR (700 MHz, CDCl3) δ 7.78–7.72 (m, 1H, Ar–H), 7.59–7.53 (m, 2H, Ar–H), 7.51 (t, J = 6.9 Hz, 2H, Ar–H), 7.42–7.37 (m, 1H, Ar–H), 7.32–7.26 (m, 5H, Ar–H), 7.20–7.04 (m, 7H, Ar–H), 6.69 (dd, J = 17.6, 10.9 Hz, 1H, Ph-CH = CH2), 5.73 (d, J = 17.6 Hz, 1H, Ph-CH = CH2), 5.23 (d, J = 10.9 Hz, 1H, Ph-CH = CH2) ppm. 19F decoupled 13C NMR (176 MHz, CDCl3) δ 150.30(CPz), 143.53(CPz), 137.82(CPz), 136.71(Ph-CH = CH2), 135.94(CHAr), 133.10(CAr), 132.65(CAr), 132.29(CHAr), 131.04(CHAr), 130.91(CHAr), 130.4(CAr), 129.46(CHAr), 129.31(CHAr), 128.59(CHAr), 128.32(CHAr), 128.27(CHAr), 127.85(CHAr), 126.26(CHAr), 124.47(CAr), 121.74(CAr), 119.81(CHAr), 113.70(Ph-CH = CH2) ppm. 19F NMR (376 MHz, CDCl3) δ -59.61 ppm.
Synthetic procedure for soluble polymeric tetra aryl pyrazole P11: A schlenk tube was charged with the monomer M1 (0.500 g, 1.25 mmol) and free radical initiator azobisisobutyronitrile (0.004 g, 0.02 mmol). The entire system was purged with nitrogen followed by the addition of 1 mL of DCE. The entire system was degassed using the freeze–pump–thaw cycle for three times. Then the reaction was stirred for 24 h at 80 °C. Then the reaction mixture slowly added to the distilled hexane in order to precipitate the polymeric material. The precipitate was then dissolved in dichloromethane and reprecipitated from the hexane. This process was reiterated three times. The resulting solid was dried under high vacuum to obtain the white solid. Yield = 0.400 g, (80%). 1H NMR (400 MHz, CDCl3) 7.6–5.9 (aromatic H) and 2.39–0.51 (polymeric backbone) ppm. 13C NMR (101 MHz, CDCl3) δ 150.16, 141.18, 139.90, 133.37, 130.32, 128.81, 128.24, 127.70, 127.24, 125.25, 120.37, 39–41 (polymeric backbone) ppm.
Synthetic procedure for soluble polymeric tetra aryl pyrazole P2: Polymer P2 was prepared following the procedure used for the preparation of P1. The quantities involved are as follows: Monomer M2 (0.5 g, 1.07 mmol) and azobisisobutyronitrile (0.004 g, 0.02 mmol). Yield = 0.38 g (76%). 1H NMR (400 MHz, CDCl3) 7.8–5.9 (Aromatic H) and 2.12–0.57 (b, polymeric backbone) ppm. 13C NMR (176 MHz, CDCl3) δ 150.04, 143.22, 137.63, 133.12, 132.17, 130.80, 130.14, 129.36, 129.05, 128.02, 127.57, 125.36, 123.80, 122.25, 120.70, 119.79 and 39–41 (polymeric backbone) ppm. 19F NMR (376 MHz, CDCl3) δ -59.48 ppm.
Synthetic procedure for insoluble resin IP1: A 100 mL two neck round bottom flask was charged with the monomer M2 (0.5 g, 1.07 mmol) and azobisisobutyronitrile (0.004 g, 0.02 mmol). Divinyl benzene (0.28 g, 2.14 mmol) was added to it. The set up was purged with nitrogen and then 1 mL DCE was added to it. The whole set up was degassed using freeze–pump–thaw cycle for three times. The reaction was stirred at 80 °C for 48 h which resulted in the formation insoluble yellow compound. The insoluble compound was transferred to a frit connected to a conical flask and was washed three times with dichloromethane, methanol, and acetone. The yellowish white compound obtained was grounded to a fine powder using mortar and pestle and was dried under high vacuum. Yield = 0.47 g, (94%). 13C CPMAS NMR (101 MHz) δ 153.47–109.49 and 62.77–23.49 (polymeric backbone) ppm.
Synthetic procedure for catalyst Pd@P1: 0.5 g of polymer P1 was suspended in 10 mL acetic acid at 100 °C. Palladium acetate (0.28 g) was added to it and was stirred for 1 h. Then insoluble precipitates were filtered from the reaction mixture under hot condition. The residue was again washed with dichloromethane, methanol, and acetone three times each. Then the brownish black solid obtained was grounded using mortar and pestle and was dried under high vacuum. Palladium content of the catalyst was estimated from ICP-AES analysis using HCl:HNO3:H2O2 (37%) in 3:1:1 ratio as the digesting solution under microwave digestion. Palladium loading = 1.12 mmolg−1. 13C CPMAS NMR (101 MHz) δ 152.35–108.76 and 60.43–20.35 (polymeric backbone) ppm.
Catalyst Pd@P2 was prepared following the similar procedure as used for Pd@P1. Palladium loading = 1.6 mmolg−1. 13C CPMAS NMR (101 MHz) δ 152.60–106.33 and 51.88–20.13 (polymeric backbone) ppm.
Catalyst Pd@IP1 was prepared following the similar procedure as used for Pd@P1. Palladium loading = 0.972 mmolg−1. 13C CPMAS NMR (101 MHz) δ 161.11–106.33 and 57.58–10.42 (polymeric backbone) ppm.
General procedure for alkylation reaction: Pd@IP1 (1 mol% of Pd), KOtBu (0.5 mmol), primary alcohol (0.5 mmol) and secondary alcohol (0.75 mmol) were added to a seal tube. Deionised water (2 mL) was added to it and the tube was sealed. The reaction was stirred at 120 °C. After 24 h, the reaction mixture was filtered using a filter paper. Aqueous layer was separated from the reaction mixture and the organic layer was collected using dichloromethane (3 × 10 mL). The organic layer was dried on Na2SO4 and concentrated under vacuum. Product was purified by column chromatography.
General procedure for quinoline synthesis: Pd@IP1 (1 mol% of Pd), KOtBu (0.5 mmol), 2-aminobenzyl alcohol (0.5 mmol) and secondary alcohol (0.75 mmol) were added to a seal tube. Deionised water (1 mL) was added to it and the tube was sealed. The reaction was stirred at 120 °C. After 24 h, the reaction mixture was filtered using a filter paper. Aqueous layer was separated from the reaction mixture and the organic layer was collected using dichloromethane (3 × 10 mL). The organic layer was dried on Na2SO4 and concentrated under vacuum. Product was purified by column chromatography.
General procedure for recycling experiment: Pd@IP1 (1 mol% Pd), KOtBu (10 mmol), primary alcohol/aminobenzyl alcohol (10 mmol) and secondary alcohol (15 mmol) were added to a seal tube. Deionised water (15 mL) was added to it. Then the tube was sealed and the reaction was stirred at 120 °C. After 24 h, the reaction mixture was filtered using frit apparatus. Then the catalyst was washed with water to remove excess base followed by washing with dichloromethane, methanol and acetone three times each. The catalyst was the dried under high vacuum for 3 h and reused for the next cycle. This process was reiterated 7 times.
References
Selander N, Szabó KJ (2011) Catalysis by palladium pincer complexes. Chem Rev 111(3):2048–2076
Wu X-F, Neumann H, Beller M (2013) Synthesis of heterocycles via palladium-catalyzed carbonylations. Chem Rev 113(1):1–35
Hazari N, Melvin PR, Beromi MM (2017) Well-defined nickel and palladium pre-catalysts for cross-coupling. Nat Rev Chem 1(3):1–16
He J, Wasa M, Chan KS, Shao Q, Yu J-Q (2017) Palladium-catalyzed transformations of alkyl C-H bonds. Chem Rev 117(13):8754–8786
Wang D, Weinstein AB, White PB, Stahl SS (2017) Ligand-promoted palladium-catalyzed aerobic oxidation reactions. Chem Rev 118(5):2636–2679
Devendar P, Qu R-Y, Kang W-M, He B, Yang G-F (2018) Palladium-catalyzed cross-coupling reactions: a powerful tool for the synthesis of agrochemicals. J Agric Food Chem 66(34):8914–8934
Nicolaou K, Bulger PG, Sarlah D (2005) Palladium-catalyzed cross-coupling reactions in total synthesis. Angew Chem Int Ed 44(29):4442–4489
Phan NT, Van Der Sluys M, Jones CW (2006) On the nature of the active species in palladium catalyzed Mizoroki-Heck and Suzuki-Miyaura couplings–homogeneous or heterogeneous catalysis, a critical review. Adv Synth Catal 348(6):609–679
Culkin DA, Hartwig JF (2003) Palladium-catalyzed α-arylation of carbonyl compounds and nitriles. Acc Chem Res 36(4):234–245
Daugulis O, Do H-Q, Shabashov D (2009) Palladium-and copper-catalyzed arylation of carbon− hydrogen bonds. Acc Chem Res 42(8):1074–1086
Brusoe AT, Hartwig JF (2015) Palladium-catalyzed arylation of fluoroalkylamines. J Am Chem Soc 137(26):8460–8468
Sivanandan ST, Shaji A, Ibnusaud I, Seechurn CCJ (2015) Colacot TJ (2015) Palladium-Catalyzed α-Arylation Reactions in Total Synthesis. Eur J Org Chem 1:38–49
Ruiz-Castillo P, Buchwald SL (2016) Applications of palladium-catalyzed C-N cross-coupling reactions. Chem Rev 116(19):12564–12649
Gou B-B, Liu H-F, Chen J, Zhou L (2019) Palladium-catalyzed site-selective C (sp3)–H arylation of phenylacetaldehydes. Org Lett 21(17):7084–7088
Hagui W, Doucet H, Soulé J-F (2019) Application of palladium-catalyzed C (sp2)–H bond arylation to the synthesis of polycyclic (hetero) aromatics. Chem 5(8):2006–2078
Gildner PG, DeAngelis A, Colacot TJ (2016) Palladium-Catalyzed N-Arylation of Cyclopropylamines. Org Lett 18(6):1442–1445
Hong AY (2013) Stoltz BM (2013) The construction of all-carbon quaternary stereocenters by use of Pd-catalyzed asymmetric allylic alkylation reactions in total synthesis. Eur J Org Chem 14:2745–2759
Pritchett BP, Stoltz BM (2018) Enantioselective palladium-catalyzed allylic alkylation reactions in the synthesis of Aspidosperma and structurally related monoterpene indole alkaloids. Nat Prod Rep 35(6):559–574
James J, Jackson M, Guiry PJ (2019) Palladium-catalyzed decarboxylative asymmetric allylic alkylation: development, mechanistic understanding and recent advances. Adv Synth Catal 361(13):3016–3049
Noreen S, Zahoor AF, Ahmad S, Shahzadi I, Irfan A, Faiz S (2019) Novel chiral ligands for palladium-catalyzed asymmetric allylic alkylation/asymmetric Tsuji-Trost reaction: a review. Curr Org Chem 23(11):1168–1213
Premi C, Dixit A, Jain N (2015) Palladium-Catalyzed Regioselective Decarboxylative Alkylation of Arenes and Heteroarenes with Aliphatic Carboxylic Acids. Org Lett 17(11):2598–2601
Yadav S, Ramasastry SSV (2021) Palladium-catalysed annulative allylic alkylation for the synthesis of benzannulated heteroarenes. Chem Commun 57(1):77–80
Trost BM, Brennan MK (2007) Palladium-catalyzed regio-and enantioselective allylic alkylation of bis allylic carbonates derived from Morita−Baylis−Hillman adducts. Org Lett 9(20):3961–3964
Fu L, Chen Q, Wang Z, Nishihara Y (2020) Palladium-catalyzed decarbonylative alkylation of acyl fluorides. Org Lett 22(6):2350–2353
Shen Y, Dai Z-Y, Zhang C, Wang P-S (2021) Palladium-catalyzed allylic alkylation via photocatalytic nucleophile generation. ACS Catal 11(12):6757–6762
Craig RA, Loskot SA, Mohr JT, Behenna DC, Harned AM, Stoltz BM (2015) Palladium-catalyzed enantioselective decarboxylative allylic alkylation of cyclopentanones. Org Lett 17(21):5160–5163
Luo Y-C, Yang C, Qiu S-Q, Liang Q-J, Xu Y-H, Loh T-P (2019) Palladium (II)-Catalyzed Stereospecific Alkenyl C-H Bond Alkylation of Allylamines with Alkyl Iodides. ACS Catal 9(5):4271–4276
Zhu J, Wood J, Deplanche K, Mikheenko I, Macaskie LE (2016) Selective hydrogenation using palladium bioinorganic catalyst. ApplCatal B 199:108–122
Mao Z, Gu H, Lin X (2021) Recent advances of Pd/C-catalyzed reactions. Catalysts 11(9):1078
Liu Y, He S, Quan Z, Cai H, Zhao Y, Wang B (2019) Mild palladium-catalysed highly efficient hydrogenation of C [triple bond, length as m-dash] N, C-NO 2, and C [double bond, length as m-dash] O bonds using H 2 of 1 atm in H 2 O. Green Chem 21(4):830–838
Advani JH, Noor-ul HK, Bajaj HC, Biradar AV (2019) Stabilization of palladium nanoparticles on chitosan derived N-doped carbon for hydrogenation of various functional groups. Appl Surf Sci 487:1307–1315
Vilches-Herrera M, Werkmeister S, Junge K, Börner A, Beller M (2014) Selective catalytic transfer hydrogenation of nitriles to primary amines using Pd/C. Catal Sci Technol 4(3):629–632
Lévay K, Madarász J, Hegedűs L (2022) Tuning the chemoselectivity of the Pd-catalysed hydrogenation of pyridinecarbonitriles: an efficient and simple method for preparing pyridyl-or piperidylmethylamines. Catal Sci Technol 12(8):2634–2648
Guo Y, Li J, Zhao F, Lan G, Li L, Liu Y, Si Y, Jiang Y, Yang B, Yang R (2016) Palladium-modified functionalized cyclodextrin as an efficient and recyclable catalyst for reduction of nitroarenes. RSC Adv 6(10):7950–7954
Hegedűs L, Máthé T (2005) Selective heterogeneous catalytic hydrogenation of nitriles to primary amines in liquid phase: Part I. Hydrogenation of benzonitrile over palladium. Appl Catal A Gen 296(2):209–215
Gligorich KM, Sigman MS (2009) Recent advancements and challenges of palladium II-catalyzed oxidation reactions with molecular oxygen as the sole oxidant. Chem Commun 26:3854–3867
Hu M, Wu W, Jiang H (2019) Palladium-catalyzed oxidation reactions of alkenes with green oxidants. Chemsuschem 12(13):2911–2935
Wu W, Jiang H (2012) Palladium-catalyzed oxidation of unsaturated hydrocarbons using molecular oxygen. Acc Chem Res 45(10):1736–1748
Hess W, Burton JW (2010) Palladium-Catalysed Cyclisation of N-Alkynyl Aminomalonates. Chem Eur J 16(41):12303–12306
Ye J, Ma S (2014) Palladium-catalyzed cyclization reactions of allenes in the presence of unsaturated carbon–carbon bonds. Acc Chem Res 47(4):989–1000
Li J, Yang S, Wu W, Jiang H (2019) Palladium-Catalyzed Cascade Cyclization/Alkynylation Reactions. Chem Asian J 14(23):4114–4128
Wang J, Li D, Li J, Zhu Q (2021) Advances in palladium-catalysed imidoylative cyclization of functionalized isocyanides for the construction of N-heterocycles. Org Biomol Chem 19(31):6730–6745
Zhu C, Zhao Y, Wang D, Sun W-Y, Shi Z (2016) Palladium-catalyzed direct arylation and cyclization of o-iodobiaryls to a library of tetraphenylenes. Sci Rep 6(1):33131
Zou S, Gao B, Huang Y, Zhang T, Huang H (2019) Palladium-catalyzed hydrocarbonylative cyclization of 1, 5-dienes. Org Lett 21(16):6333–6336
Yan F, Liang H, Song J, Cui J, Liu Q, Liu S, Wang P, Dong Y, Liu H (2017) Palladium-catalyzed cyclization-Heck reaction of allenamides: an approach to 3-Methylene-5-phenyl-1, 2, 3, 4-tetrahydropyridine derivatives. Org Lett 19(1):86–89
Liu YZ, Wang Z, Huang Z, Zheng X, Yang WL, Deng WP (2020) Palladium-catalyzed asymmetric [4+3] cyclization of trimethylenemethane: regio-, diastereo-, and enantioselective construction of benzofuro [3, 2-b] azepine skeletons. Angew Chem Int Ed 59(3):1238–1242
Js CC, Kitching M, Colacot T, Snieckus V (2012) Palladium-catalyzed cross-coupling: a historical contextual perspective to the 2010 Nobel Prize. Angew Chem Int Ed 51(21):5062–5085
Rullah K, Mohd Aluwi MFF, Yamin BM, Juan JC, Wai LK (2019) Palladium-catalysed cross-coupling reactions for the synthesis of chalcones. Asian J Org Chem 8(8):1174–1193
Helbert H, Visser P, Hermens JG, Buter J, Feringa BL (2020) Palladium-catalysed cross-coupling of lithium acetylides. Nat Catal 3(8):664–671
Vila Descals C, Giannerini M, Hornillos V, Fañanás-Mastral M, Feringa BL (2014) Palladium-catalysed direct cross-coupling of secondary alkyllithium reagents. Chem Sci 5(4):1361–1367
Sore HF, Galloway WR, Spring DR (2012) Palladium-catalysed cross-coupling of organosilicon reagents. Chem Soc Rev 41(5):1845–1866
Türtscher PL, Davis HJ, Phipps RJ (2018) Palladium-catalysed cross-coupling of benzylammonium salts with boronic acids under mild conditions. Synthesis 50(04):793–802
Wen J-H, Li Q, Nie S-Z, Ye J-J, Xu Q, Zhao C-Q (2018) Palladium-catalyzed isomerization-coupling reactions of allyl chloride with amines to generate functionalized phosphorus derivatives. Catalysts 8(5):194
Ren W, Sun F, Chu J, Shi Y (2020) A Pd-catalyzed site-controlled isomerization of terminal olefins. Org Lett 22(5):1868–1873
Hong-Chao Chen YW, Yang Yu, Wang P (2022) Pd-Catalyzed Isomerization of Alkenes. Chinese J Org Chem 42(3):742–757. https://doi.org/10.6023/cjoc202109045
Kocen AL, Brookhart M, Daugulis O (2017) Palladium-catalysed alkene chain-running isomerization Chem Commun 53(72):10010–10013
Larionov E, Lin L, Guenee L, Mazet C (2014) Scope and mechanism in palladium-catalyzed isomerizations of highly substituted allylic, homoallylic, and alkenyl alcohols. J Am Chem Soc 136(48):16882–16894
Biswal P, Samser S, Meher SK, Chandrasekhar V, Venkatasubbaiah K (2022) Palladium-catalyzed synthesis of α-methyl ketones from allylic alcohols and methanol. Adv Synth Catal 364(2):413–419
Lin L, Romano C, Mazet C (2016) Palladium-catalyzed long-range deconjugative isomerization of highly substituted α, β-unsaturated carbonyl compounds. J Am Chem Soc 138(32):10344–10350
Corma A, Navas J, Ródenas T, Sabater MJ (2013) One-Pot Palladium-catalyzed borrowing hydrogen synthesis of thioethers. Chem Eur J 19(51):17464–17471
Hikawa H, Imamura H, Kikkawa S, Azumaya I (2018) A borrowing hydrogen methodology: palladium-catalyzed dehydrative N-benzylation of 2-aminopyridines in water. Green Chem 20(13):3044–3049
Hikawa H, Koike T, Izumi K, Kikkawa S, Azumaya I (2016) Borrowing hydrogen methodology for N-benzylation using a π-benzylpalladium system in water. Adv Synth Catal 358(5):784–791
Xie Y, Liu S, Liu Y, Wen Y, Deng G-J (2012) Palladium-catalyzed one-pot diarylamine formation from nitroarenes and cyclohexanones. Org Lett 14(7):1692–1695
Dang TT, Ramalingam B, Shan SP, Seayad AM (2013) An efficient palladium-catalyzed N-alkylation of amines using primary and secondary alcohols. ACS Catal 3(11):2536–2540
Shiraishi Y, Fujiwara K, Sugano Y, Ichikawa S, Hirai T (2013) N-monoalkylation of amines with alcohols by tandem photocatalytic and catalytic reactions on TiO2 loaded with Pd nanoparticles. ACS Catal 3(3):312–320
Yu X, Jiang L, Li Q, Xie Y, Xu Q (2012) Palladium-catalyzed N-alkylation of amides and amines with alcohols employing the aerobic relay race methodology. Chin J Chem 30(10):2322–2332
Mamidala R, Mukundam V, Dhanunjayarao K, Venkatasubbaiah K (2017) Cyclometalated palladium pre-catalyst for N-alkylation of amines using alcohols and regioselective alkylation of sulfanilamide using aryl alcohols. Tetrahedron 73(16):2225–2233
Mamidala R, Samser S, Sharma N, Lourderaj U, Venkatasubbaiah K (2017) Isolation and characterization of regioisomers of pyrazole-based palladacycles and their use in α-alkylation of ketones using alcohols. Organometallics 36(17):3343–3351
Mamidala R, Biswal P, Subramani MS, Samser S, Venkatasubbaiah K (2019) Palladacycle-phosphine catalyzed methylation of amines and ketones using methanol. J Org Chem 84(16):10472–10480
Samser S, Mohapatra O, Biswal P, Venkatasubbaiah K (2021) Palladium-mediated tandem isomerization-methylenation of allyl alcohols: one-pot synthesis of 1, 5-diketones. J Org Chem 86(19):13744–13753
Muzart J (2015) Pd-catalyzed hydrogen-transfer reactions from alcohols to C=C, C=O, and C=N Bonds. Eur J Org Chem 2015(26):5693–5707
Ansari TN, Gallou F, Handa S (2020) Cross‐couplings in water: a better way to assemble new bonds organometallic chemistry in industry: A practical approach. p. 203–238
Dixneuf P, Cadierno V (2013) Metal-catalyzed reactions in water. John Wiley & Sons
Lu S-M, Wang Z, Li J, Xiao J, Li C (2016) Base-free hydrogenation of CO 2 to formic acid in water with an iridium complex bearing a N, N′-diimine ligand. Green Chem 18(16):4553–4558
Fujita K-i, Tamura R, Tanaka Y, Yoshida M, Onoda M, Yamaguchi R (2017) Dehydrogenative oxidation of alcohols in aqueous media catalyzed by a water-soluble dicationic iridium complex bearing a functional N-heterocyclic carbene ligand without using base. ACS Catal 7(10):7226–7230
Vivancos A, Beller M, Albrecht M (2018) NHC-based iridium catalysts for hydrogenation and dehydrogenation of N-heteroarenes in water under mild conditions. ACS Catal 8(1):17–21
Huang M, Li Y, Liu J, Lan X-B, Liu Y, Zhao C, Ke Z (2019) A bifunctional strategy for N-heterocyclic carbene-stabilized iridium complex-catalyzed N-alkylation of amines with alcohols in aqueous media. Green Chem 21(2):219–224
Verma A, Hazra S, Dolui P, Elias AJ (2021) Ruthenium-catalyzed synthesis of α-alkylated ketones and quinolines in an aqueous medium via a hydrogen-borrowing atrategy using ketones and alcohols. Asian J Org Chem 10(3):626–633
Lindstrom UM (2008) Organic reactions in water: principles, strategies and applications. John Wiley & Sons
Prat D, Hayler J, Wells A (2014) A survey of solvent selection guides. Green Chem 16(10):4546–4551
Cornils B, Herrmann WA (2004) Aqueous-phase organometallic catalysis: concepts and applications. Aqueous-phase organometallic catalysis: concepts and applications.
Sharma S, Buchbinder NW, Braje WM, Handa S (2020) Fast amide couplings in water: Extraction, column chromatography, and crystallization not required. Org Lett 22(15):5737–5740
Breslow R (2006) The hydrophobic effect in reaction mechanism studies and in catalysis by artificial enzymes. J Phys Org Chem 19(12):813–822
Butler RN, Coyne AG (2016) Organic synthesis reactions on-water at the organic–liquid water interface. Org Biomol Chem 14(42):9945–9960
Lipshutz BH, Ghorai S, Cortes-Clerget M (2018) The hydrophobic effect applied to organic synthesis: recent synthetic chemistry “in water.” Chem Eur J 24(26):6672–6695
Kitanosono T, Masuda K, Xu P, Kobayashi S (2018) Catalytic organic reactions in water toward sustainable society. Chem Rev 118(2):679–746
Benaglia M (2009) Recoverable and recyclable catalysts. John Wiley & Sons
Nasrollahzadeh M, Motahharifar N, Ghorbannezhad F, Bidgoli NSS, Baran T, Varma RS (2020) Recent advances in polymer supported palladium complexes as (nano) catalysts for Sonogashira coupling reaction. Mol Cat 480:110645
Leadbeater NE, Marco M (2002) Preparation of polymer-supported ligands and metal complexes for use in catalysis. Chem Rev 102(10):3217–3274
McNamara CA, Dixon MJ, Bradley M (2002) Recoverable catalysts and reagents using recyclable polystyrene-based supports. Chem Rev 102(10):3275–3300
Drabina P, Svoboda J, Sedlák M (2017) Recent advances in C-C and C–N bond forming reactions catalysed by polystyrene-supported copper complexes. Molecules 22(6):865
Bai L, Wang J-X (2005) Environment friendly Suzuki aryl-aryl cross-coupling reaction. Curr Org Chem 9(6):535–553
Reed-Berendt BG, Latham DE, Dambatta MB, Morrill LC (2021) Borrowing hydrogen for organic synthesis. ACS Cent Sci 7(4):570–585
Irrgang T, Kempe R (2018) 3d-Metal catalyzed N-and C-alkylation reactions via borrowing hydrogen or hydrogen autotransfer. Chem Rev 119(4):2524–2549
Corma A, Navas J, Sabater MJ (2018) Advances in one-pot synthesis through borrowing hydrogen catalysis. Chem Rev 118(4):1410–1459
Wang R, Ma J, Li F (2015) Synthesis of a-alkylated ketones via tandem acceptorless dehydrogenation/a-alkylation from secondary and primary alcohols catalyzed by metal-ligand bifunctional iridium complex [Cp* Ir (2, 2′-bpyO)(H2O)]. J Org Chem 80(21):10769–10776
Musa S, Ackermann L, Gelman D (2013) Dehydrogenative cross-coupling of primary and secondary alcohols. Adv Synth Catal 355(14–15):3077–3080
Sahoo AR, Lalitha G, Murugesh V, Bruneau C, Sharma GV, Suresh S, Achard M (2017) Ruthenium phosphine–pyridone catalyzed cross-coupling of alcohols to form α-alkylated ketones. J Org Chem 82(19):10727–10731
Chang W, Gong X, Wang S, Xiao L-P, Song G (2017) Acceptorless dehydrogenation and dehydrogenative coupling of alcohols catalysed by protic NHC ruthenium complexes. Org Biomol Chem 15(16):3466–3471
Jumde VR, Gonsalvi L, Guerriero A, Peruzzini M (2015) Taddei M (2015) A Ruthenium-Based Catalytic System for a Mild Borrowing-Hydrogen Process. Eur J Org Chem 8:1829–1833
Genç S, Günnaz S, Çetinkaya B, Sl G, Gülcemal D (2018) Iridium (I)-catalyzed alkylation reactions to form α-alkylated ketones. J Org Chem 83(5):2875–2881
Akhtar WM, Cheong CB, Frost JR, Christensen KE, Stevenson NG, Donohoe TJ (2017) Hydrogen borrowing catalysis with secondary alcohols: a new route for the generation of β-branched carbonyl compounds. J Am Chem Soc 139(7):2577–2580
Bhattacharyya D, Sarmah BK, Nandi S, Srivastava HK, Das A (2021) Selective catalytic synthesis of α-alkylated ketones and β-disubstituted ketones via acceptorless dehydrogenative cross-coupling of alcohols. Org Lett 23(3):869–875
Thiyagarajan S, Vijaya Sankar R, Gunanathan C (2020) Ruthenium-catalyzed α-alkylation of ketones using secondary alcohols to β-disubstituted ketones. Org Lett 22(20):7879–7884
Chakraborty P, Garg N, Manoury E, Poli R, Sundararaju B (2020) C-alkylation of various carbonucleophiles with secondary alcohols under CoIII-catalysis. ACS Catal 10(14):8023–8031
Mukundam V, Kumar A, Dhanunjayarao K, Ravi A, Peruncheralathan S, Venkatasubbaiah K (2015) Tetraaryl pyrazole polymers: versatile synthesis, aggregation induced emission enhancement and detection of explosives. Polym Chem 6(44):7764–7770
Eghbali P, Nişancı B, Metin Ö (2018) Graphene hydrogel supported palladium nanoparticles as an efficient and reusable heterogeneous catalysts in the transfer hydrogenation of nitroarenes using ammonia borane as a hydrogen source. Pure Appl Chem 90(2):327–335
Mondal J, Gomes R, Modak A, Bhaumik A (2013) Pd-anchored functionalized mesoporous materials as robust and recyclable heterogeneous catalysts for a series of CC bond forming reactions. Recyclable Catalysis 1:10–33
Demir MM, Gulgun MA, Menceloglu YZ, Erman B, Abramchuk SS, Makhaeva EE, Khokhlov AR, Matveeva VG, Sulman MG (2004) Palladium nanoparticles by electrospinning from poly (acrylonitrile-co-acrylic acid)−PdCl2 solutions. Relations between preparation conditions, particle size, and catalytic activity. Macromolecules 37(5):1787–1792
Ovezova M, Eroğlu Z, Metin Ö, Çetinkaya B, Gülcemal S (2021) Unveiling the catalytic nature of palladium-N-heterocyclic carbene catalysts in the α-alkylation of ketones with primary alcohols. Dalton Trans 50(31):10896–10908
Lisowski W, Keim EG (2010) Vacuum annealing phenomena in ultrathin TiD y/Pd bi-layer films evaporated on Si (100) as studied by TEM and XPS. Anal Bioanal Chem 396(8):2797–2804
Farooq MU, Novosad V, Rozhkova EA, Wali H, Ali A, Fateh AA, Neogi PB, Neogi A, Wang Z (2018) Gold nanoparticles-enabled efficient dual delivery of anticancer therapeutics to HeLa cells. Sci Rep 8(1):1–12
Yu W, Hou H, Xin Z, Niu S, Xie Y, Ji X, Shao L (2017) Nanosizing Pd on 3D porous carbon frameworks as effective catalysts for selective phenylacetylene hydrogenation. RSC Adv 7(25):15309–15314
Li F, Han M, Dai P, Xu W, He J, Tao X, Wu Y, Tong X, Xia X, Guo W (2021) Distinct mechanisms for TMPRSS2 expression explain organ-specific inhibition of SARS-CoV-2 infection by enzalutamide. Nat Commun 12(1):1–14
Acknowledgements
KV thank Department of Atomic Energy (DAE) for financial support. S.S. thanks DST for an INSPIRE fellowship. P.B and SKM thank CSIR for a research fellowship. We thank Marimuthu Rajendiran and Dr. Jiban Krushna Das for XPS measurement and Abhishek Padhy and Ranjit Mishra for TEM and EDX measurement respectively.
Funding
Department of Atomic Energy, Government of India
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shaikh, S., Biswal, P., Meher, S.K. et al. Polystyrene Supported Pyrazole-based Palladium Catalysts/Precatalysts for Acceptorless Dehydrogenative Coupling of Alcohols in Water. Catal Lett 154, 737–748 (2024). https://doi.org/10.1007/s10562-023-04316-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-023-04316-z