
Abstract
Ternary Cu2SnS3 (CTS) and Cu2SnSe3 (CTSe) quantum dots were grown in-situ on reduced graphene oxide (rGO) nanosheets by a facile one-pot colloidal synthesis. Possessing abundant active sites and having the ability to harvest solar energy and to facilitate charge separation, the CTS(e)/rGO 0D/2D nanocomposites showed remarkably enhanced photocatalytic activities for degradation of rhodamine B and methyl orange aqueous solutions upon visible light irradiation. The nanocomposites of rGO supported monodispersed quantum dots were proved to be a suitable platform for photocatalytic applications due to the well-maintained 0D/2D nanostructure.
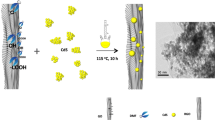
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Persistent organic pollutants (POPs) in water cause severe environmental problems. Biomagnification of organic contaminants in plants and small animals poses a great health risk to their consumers—predators on the top of the food chain, and ultimately humans [1, 2]. It is therefore desirable to develop an advanced technology for POPs removal from the water supply chain. So far, photocatalytic degradation has been proved as an effective, economic and environmental-friendly approach to organic pollutants removal [3]. Photocatalytic nanostructured materials synthesized by simple innovative and economical methods (one-pot colloidal synthesis, hydrothermal and solvothermal synthesis), display impressive characteristics and efficiency in pollutant removal [4]. However, typical photocatalysts such as TiO2 and ZnO face two major issues: they require use of the high energy ultraviolet (UV) light, and the photo-generated charge carriers recombine fast [5].
The urgent need for combating POPs has led to the development of various visible light-response photocatalysts [6,7,8,9,10]. Lately, narrow-bandgap photocatalysts, copper-based ternary and quaternary chalcogenide nanocrystals, such as Cu2SnS3 [11], CuInxGa1−xSe2 [12, 13], and Cu2ZnSnS4 [14, 15] have enjoyed a great deal of attention owing to their large absorption coefficients (> 104 cm−1), good chemical and thermal stability, and suitable band energy level and bandgap (1.0–1.5 eV), meeting the specific redox potentials of active species generated during photocatalytic reactions. Low-cost compounds Cu2SnS3 (CTS) and Cu2SnSe3 (CTSe) feature excellent optoelectronic properties [16, 17] and are intensively investigated for photovoltaic cells [18], thermoelectric generators [19], electrochemical energy storage [20], photocatalytic pollutant degradation [11, 21] and water splitting [22]. Inspired by graphene common utilization in nanocomposites as an electron acceptor that hinders carrier recombination and enhances photocatalytic performance, reduced graphene oxide (rGO), a well-known two-dimensional (2D) material with a large specific area, high conductivity and carrier mobility, was implemented in our nanocomposites to overcome the fast charge recombination [23].
In the present work, we take advantage of properties of both zero-dimensional (0D) copper-based ternary and quaternary chalcogenide quantum dots (QDs) and 2D graphene nanosheets, the combination of which yields nanocomposites of interesting characteristics. Up to date, the research was conducted on CTS microspheres/rGO composites that were prepared by hydrothermal and solvothermal methods, and on CTSe matrix/graphene composite that was prepared by ball-milling method [24,25,26]. Traditionally, QDs comprising three or more elements were prepared first, and then deposited on rGO through physical adsorption [27]. In this study, we report a facile one-pot synthesis of CTS(e)/rGO 0D/2D nanocomposites by growing CTS(e) QDs directly on rGO nanosheets using long-chain oleylamine (OLA) capping ligand, which helps to control uniform distribution of QD sizes. The photocatalytic activities of the 0D/2D nanocomposites are subjected to further investigation.
2 Experimental
2.1 Synthesis of CTS(e) QDs and CTS(e)/rGO
All chemicals used in this work were purchased from Aladdin and used directly without further purification. CTS(e) QDs synthesis, CTS precursors: Sulfur powder (96.2 mg, 3 mmol) dissolved in 3 mL OLA, Cu(OAc)2·H2O (399.3 mg, 2 mmol) and SnCl2⋅2H2O (225.7 mg, 1 mmol) were added to 27 mL OLA. The CTSe precursors were prepared by dissolving selenium powder (236.9 mg, 3 mmol) in a solution of 1.5 mL OLA and 1.5 mL 1-dodecanethiol [28]. The precursor solutions were degassed at 100 °C for 1 h, purged with nitrogen for 30 min, and consecutively heated at 240 °C for additional 5 min. The black products were dissolved in hexane and precipitated with ethanol followed by centrifugation at 8000 rpm for 5 min. This step was repeated three times. Finally, the purified products were dried at 60 °C under vacuum. The CTS(e)/rGO nanocomposites were prepared by adding 10 mg of GO into the QDs precursor solutions followed by the same procedure as described for QDs. To remove OLA ligands and reduce GO, the samples were annealed at 300 °C in H2/N2 (5:95) for 1 h.
2.2 Characterization
The crystal phase was analyzed by a powder X-ray diffraction spectroscopy (XRD, SmartLab, Rigaku, Japan) with Cu Ka radiation (40 kV, 30 mA). The microstructure was characterized by a transmission electron microscopy (TEM, JEM-2100F, JEOL., Japan). The chemical state was identified by an X-ray photoelectron spectroscopy (XPS, AXIS Ultrabld, Kratos, UK) using monochromatic Al Kα radiation (150 W, 15 kV, 1486.6 eV). The UV–Vis–NIR absorption spectrum was measured by a UV/Vis/NIR spectrometer (JASCO, V570, Japan). The OLA ligands removal was confirmed by the Fourier transform infrared spectroscopy (FTIR, Nicolet 6700, Thermo Scientific, USA). The total organic carbon (TOC) was measured by a TOC analyzer (liquiTOC II, Elementar, Germany).
2.3 Photocatalytic Activity Measurement
Samples’ photocatalytic activity driven by visible light were evaluated by photodegradation of organic dyes rhodamine B (Rh B) and methyl orange (MO) in aqueous solutions (10 mg/L, simulating for POPs), using UV–Vis spectroscopy and TOC analyzer as investigation tools. A 300 W Xe lamp equipped with a 400 nm cutoff filter was used as the visible light source. Typically, 50 mg photocatalyst powder was added into 100 mL dye solution in a 150 mL beaker with water-cooling at ambient temperature. Before irradiation, the mixture was stirred to ensure adsorption–desorption equilibrium was established. The whole mixture was exposed to a constant photon flux (100 mW/cm2) for 2 h (for Rh B) and 3 h (for MO) with a stirring speed 500 rpm. Every 20 min (for Rh B) and 30 min (for MO), 2 mL mixture was taken out and removed the solid sediment (catalyst) by centrifugation for a UV–Vis analysis. The UV–Vis spectrum was referenced by pure water. The maximum absorption peaks of the dyes were monitored at 554 nm (for Rh B) and 463 nm (for MO), and changes in optical density were plotted as a function of time.
To identify the active species in the photocatalytic reaction, oxygen was removed from the system by bubbling the solutions with pure Argon (Ar), and 0.1 mmol ammonium oxalate (AO), tert-butanol (TBA) and benzoquinone (BQ) were applied to quench hole (h+), hydroxyl radical (⋅OH) and superoxide radical (⋅O2−) in consecutive order.
3 Results and Discussion
The strategy for one-pot synthesis of copper-tin-chalcogenide QDs on rGO is illustrated by Fig. 1a. Figure S1 shows the effective removal of the OLA ligands. Cu2+ and Sn2+ ions can be attracted by oxygen-containing functional groups on GO, which provides physical support and nucleation sites necessary for QDs growth, making it possible for the QDs to attach firmly on GO surface. Moreover, the particle size on rGO is small due to steric hindrance of the long-chain OLA capping ligands. As a result, the QDs are distributed uniformly on rGO. As shown in Fig. 1b, all diffraction peaks in XRD patterns of the CTS(e) and CTS(e)/rGO samples can be clearly assigned to the monoclinic structure of Cu2SnS3 (JCPDS No. 89-4714) and Cu2SnSe3 (JCPDS No. 89-2879). The diffraction peaks at 28.3°, 33.4°, 46.7°, 55.9°, 69.1° and 76.2° are consistent with (111), (200), (220), (311), (400) and (331) facets of CTS, respectively. The diffraction peaks at 27.1°, 45.0°, 53.4°, 65.6°and 72.3° can be assigned to (111), (220), (311), (400) and (331) facets of CTSe, respectively. The broad diffraction peaks in all samples indicate the small size of the particles. Besides, no obvious diffraction peak of rGO is observed in the nanocomposites, which may be attributed to the relatively low diffraction intensity of rGO [24]. The average crystallite size of CTS, CTSe, CTS/rGO and CTSe/rGO determined from XRD pattern by Scherrer equation is 10.2 nm, 7.4 nm, 9.4 nm and 6.6 nm, in consecutive order (further details are included in Electronic Supplementary Material).

a Scheme of one-pot synthesis of copper-tin-chalcogenide QDs on rGO. b XRD patterns of CTS, CTSe, CTS/rGO and CTSe/rGO
TEM images of CTS and CTSe QDs in Fig. 2a, b, show an average particle size of 9.8 nm and 7.1 nm, respectively (Fig. S2a, b), matching well the XRD results. The selected area electron diffraction (SAED) patterns suggest that the QDs are highly crystalline. The optical absorption of QDs covers the entire visible region, and the bandgaps of CTS and CTSe QDs are estimated to be 1.52 eV and 1.31 eV (Fig. S3). As depicted in Fig. 2c, d, the average particle size of CTS and CTSe QDs on rGO is 9.6 nm and 6.0 nm (Fig. S2c, d), and almost all QDs are strongly adhered to the rGO nanosheets without aggregation. The hexagonal diffraction points of SAED patterns are attributed to rGO. Figure 2e, f display high-resolution TEM (HRTEM) images of the nanocomposites, indicating that the QDs have primarily ordered crystalline structure with 0.31 nm and 0.33 nm interplanar distance, which is in agreement with (111) facet of CTS and CTSe. The clear lattice fringes with 0.35 nm interplanar distance can be assigned to the (110) facet of rGO.

TEM images of a CTS, b CTSe, c CTS/rGO, d CTSe/rGO, insets: the corresponding SAED patterns. HRTEM images of e CTS/rGO and f CTSe/rGO
Figure 3 shows XPS spectra of CTS/rGO and CTSe/rGO nanocomposites. In the Cu 2p region, the two peaks separated by 19.8 eV lacking satellites were assigned to Cu 2p3/2 and Cu 2p1/2 of Cu+ oxidation state. Two such peaks found in Sn 3d region, were assigned to Sn4+ oxidation state. S 2p core splits into S 2p3/2 and S 2p1/2 with a peak separation of 1.1 eV, consistent with S2− [12]. Additionally, there is also a weak S2− signal appearing in CTSe/rGO, which indicates a small S2− doping, likely caused by 1-dodecanethiol solvent (Fig. S4). Se 3d peak at 54.5 eV is assigned to Se2− [14]. C 1s spectra consist of four peaks at binding energy of 284.8 eV, 285.2 eV, 286.4 eV and 287.8 eV, corresponding to C–C or C=C bonds, C–OH group, C–O–C and C=O bonds of rGO, respectively, suggesting the presence of residual oxygen-containing groups in the rGO [24]. It can be inferred that the Cu2+ was successfully reduced to Cu+ while Sn2+was oxidized to Sn4+, which are expected chemical states in CTS(e).

XPS spectra of CTS/rGO and CTSe/rGO: a Cu 2p, b Sn 3d, c S 2p and Se 3d, and d C 1s
The nanocomposites have stronger dye adsorption ability than that of the QDs, which is attributed to the aggregation of the QDs (Fig. S5). As shown in Fig. 4a, b, no photodegradation is observed without photocatalysts. In the presence of a photocatalyst, the following percentages of the degraded dyes are obtained: For both dyes, the percentage of dye degradation caused by individual catalysts are given in this order: titanium dioxide P25 (Degussa), CTS, CTSe, CTS/rGO and CTSe/rGO. Rh B − 23.1%, 65.1%, 49.3%, 96.3% and 88.7% and MO − 10.3%, 57.3%, 45.1%, 94.3% and 84.5%. The high numbers indicate significantly enhanced photocatalytic activities of the 0D/2D nanocomposites. A linear relation of photodegradation data can be fitted for all samples (Fig. S6), suggesting a pseudo-first-order kinetics for the photocatalytic degradation process: − ln(C/C0) = kt, where C refers to the dye concentration at time t, C0 refers to the initial concentration of the dye, and k represents the apparent degradation rate constant. The calculated k values are summarized in Table S1. The k values for CTS/rGO catalyzed photodegradation are approximately 3.3 times (for Rh B) and 3.5 times (for MO) higher than that of CTS QDs, and the CTSe/rGO catalyzed photodegradation gives 3.2 times higher rates for both dyes compared to their degradation catalyzed by CTSe QDs only. Compared with other similar copper-based ternary and quaternary materials, the 0D/2D nanocomposites synthesized by the facile one-pot method have the smallest particle size and the fastest photodegradation rate for Rh B (Table S2). The decomposition of Rh B and MO was followed by TOC measurement, which reveals that the organic carbon was turned into inorganic carbon products over time (Fig. S7). The enhanced photocatalytic activity could be attributed to the monodispersion of QDs on rGO. The rGO with large specific surface area enables better QDs adhesion, which leads to their uniform dispersion and prevents QDs aggregation, and stronger adsorption of O2 molecules, which provides more active sites for photocatalytic reaction. Additionally, rGO with high conductivity and electron mobility facilitates a fast electron transport from the CTS(e) e−–h+ pairs while prolonging the exciton lifetime.

The degradation rate of a Rh B and b MO as a function of irradiation time. c Test of photocatalytic performance of CTS/rGO over 10 cycles. d Effects of various quenchers on degradation efficiency. e Schematic illustration of the photocatalytic degradation process
Figure 4c summarizes the photocatalytic degradation performance of CTS/rGO over 10 cycles. The catalysts maintain high photocatalytic activity, their crystal structure and morphology (Fig. S8) after 10 cycles, indicating high stability in the experimental conditions. It is well-known that the photodegradation process relies on several active species, such as ⋅OH, ⋅O2−, and h+. Because both the conduction band (CB) energy level ECB and the valence band (VB) energy level EVB of CTS(e) are more negative than the redox potentials of Eθ (O2/⋅O2−) and Eθ (H2O/⋅OH) [22], theoretically the ⋅O2− can be generated in the photocatalytic process rather than ⋅OH. Figure 4d shows a great decrease of the dye degradation extend in the absence of O2, and in the presence of ⋅O2− and h+ scavengers. Contrary, the presence of ⋅OH scavenger has very little effect on the degradation rate. It can be concluded, that both the ⋅O2− and h+ are actively participating in the photodegradation process. Figure 4e depicts the proposed photocatalytic degradation process. Upon photoexcitation of e− in CTS(e) from VB to CB, the excited e− is transferred to rGO, where it is captured by the adsorbed O2 molecules and ⋅O2−, a highly oxidative species, is formed. Simultaneously, h+ in the VB can directly function as an oxidant. This way the improved visible light-driven photocatalytic activity is achieved.
4 Conclusion
CTS(e)/rGO 0D/2D nanocomposites were successfully prepared by a facile one-pot synthesis. The nanocomposites were used as visible light response photocatalyst for Rh B and MO dye degradation, simulating for POPs. The enhanced photocatalytic performance of the nanocomposites can be attributed to a synergistic effect between 0D QDs and 2D nanosheets, namely the effective visible light absorption by the CTS(e) QDs, abundant active sites provided by the well-dispersed QDs on rGO, and effective charge separation between the QDs and rGO interface. This work is expected to provide an innovative and facile route to the development of 0D/2D nanocomposites for solar energy conversion applications.
References
Bolong N, Ismail AF, Salim MR, Matsuura T (2009) Desalination 239:229–246
World Health Organization (2010) Persistent organic pollutants: impact on child health. WHO, Geneva
Li H, Zhou Y, Tu W, Ye J, Zou Z (2015) Adv Func Mater 25:998–1013
Kershaw SV, Susha AS, Rogach AL (2013) Chem Soc Rev 42:3033–3087
Wang Y, Wang Q, Zhan X, Wang F, Safdar M, He J (2013) Nanoscale 5:8326–8339
Zinatloo-Ajabshir S, Salehi Z, Salavati-Niasari M (2016) RSC Adv 6:107785–107792
Zinatloo-Ajabshir S, Salehi Z, Salavati-Niasari M (2018) Ceram Int 44:3873–3883
Yuan YP, Ruan LW, Barber J, Joachim Loo SC, Xue C (2014) Energy Environ Sci 7:3934–3951
Zinatloo-Ajabshir S, Salehi Z, Salavati-Niasari M (2018) J Clean Prod 192:678–687
Zinatloo-Ajabshir S, Mortazavi-Derazkola S, Salavati-Niasari M (2018) Ultrason Sonochem 42:171–182
Guo Y, Yin X, Yang Y, Que W (2016) RSC Adv 6:104041–104048
Yang Y, Que W, Zhang X, Xing Y, Yin X, Du Y (2016) J Hazard Mater 317:430–439
Zhang Z, Xiao A, Yan K, Liu Y, Yan Z, Chen J (2017) Catal Lett 147:1631–1639
Yang Y, Que W, Zhang X, Yin X, Xing Y, Que M, Zhao H, Du Y (2017) Appl Catal B 200:402–411
Guan H, Shen H, Raza A (2017) Catal Lett 147:1844–1850
Sun W, Ye Y, You Y, Xu J (2018) J Mater Chem A 6:8221–8226
Jeong J, Chung H, Ju YC, Moon J, Roh J, Yoon S, Do YR, Kim W (2010) Mater Lett 19:2043–2045
Liu F, Zhu J, Li Y, Wei J, Lv M, Xu Y, Zhou L, Hu L, Dai S (2015) J Power Sources 292:7–14
Ibáñez M, Cadavid D, Anselmi-Tamburini U, Zamani R, Gorsse S, Li W, López AM, Morante JR, Arbiol J, Cabot A (2013) J Mater Chem A 4:1421–1426
Tao HC, Zhu SC, Yang XL, Zhang LL, Ni SB (2016) J Electroanal Chem 760:127–134
Beshkar F, Zinatloo-Ajabshir S, Bagheri S, Salavati-Niasari M (2017) PLoS ONE 12:e0158549
Wang W, Ding T, Chen G, Zhang L, Yu Y, Yang Q (2015) Nanoscale 37:15106–15110
Moon IK, Lee J, Ruoff RS, Lee H (2010) Nat Commun 73:1–6
Vadivel S, Maruthamani D, Paul B, Dhar SS, Habibi-Yangjeh A, Balachandran S, Saravanakumar B, Selvakumar A, Selvam K (2016) RSC Adv 78:74177–74185
Yao S, Xu L, Gao Q, Wang X, Kong N, Li W, Wang J, Li G, Pu X (2017) J Alloys Compd 704:469–477
Zhao D, Wang X, Wu D (2017) Crystals 7:71–83
Xu T, Hu J, Yang Y, Que W, Yin X, Wu H, Chen L (2018) J Alloys Compd 734:196–203
Liu Y, Yao D, Shen L, Zhang H, Zhang X, Yang B (2012) J Am Chem Soc 134:7207–7210
Acknowledgements
This work was supported by the National Natural Science Foundation of China under Grant No. 61774122 and the 111 Project of China (B14040). The SEM and TEM work was conducted at International Center for Dielectric Research, Xi’an Jiaotong University, China. We also thank MSci. Natalie Uhlikova from Emory University for her help with language editing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Han, Y., Yang, Y., Zhao, J. et al. Facile One-Pot Synthesis of Ternary Copper-Tin-Chalcogenide Quantum Dots on Reduced Graphene Oxide for Enhanced Photocatalytic Activity. Catal Lett 148, 3112–3118 (2018). https://doi.org/10.1007/s10562-018-2525-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-018-2525-y