
Abstract
The novel, non-carbene complex [RuCl2(PPh3)2(azocane)] (1) of trigonal bipyramid geometry was synthesized, characterized (CHN elementary analysis, FTIR, EPR, and 31P NMR), and applied as catalyst precursor for ring opening metathesis polymerization (ROMP) and atom transfer radical polymerization (ATRP). ROMP of norbornene (NBE) resulted in 70% yield at 50 °C in 60 min, with [NBE]/[1] = 5000, and 15% yield from ROMP of norbornadiene was obtained under the same conditions. ATRP of methyl methacrylate (MMA) resulted in ca. 20% conversion at 85 °C in 9 h, with [MMA]/[1] = 1000, and increased to ca. 50% in the presence of Al(OiPr)3, with narrower PDI values. With [MMA]/[1] = 200, ca. 50% yield was obtained at 85 °C in 6 h either in the absence or presence of Al(OiPr)3. ATRP of styrene resulted in 9 and 16% conversion at 110 °C in 7 h in the absence or presence of Al(OiPr)3, respectively, with [St]/[1] = 1000. Results are discussed considering the five-coordinated molecular geometry of the starting complex and the arrangements required in ROMP and ATRP to initiate the reactions.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
There is large interest in the development of dual complexes for Ring Opening Metathesis Polymerization (ROMP) and Atom Transfer Radical Polymerization (ATRP) to produce well-defined copolymers through the combination of both controlled polymerization techniques [1]. The difficulty in developing complexes capable of catalysing both of these polymerizations resides in the fact that ROMP and ATRP occur via different mechanisms. A metal-carbene complex catalyses ROMP via a metalocyclobutane intermediate without a redox pathway, [2] whereas ATRP is driven by a metal-complex redox couple capable of activating/deactivating a propagating radical moiety, involving halogen atom exchange [3]. Scheme 1 illustrates the mechanisms of these reactions.

Illustration of the ATRP and ROMP reactions; the termination steps were omitted for simplification and clarity
An example of ROMP catalyst precursor is the ruthenium benzylidene complex of the [RuCl2(= CHPh)(PCy3)L] type, with L = PCy3 or an N-heterocyclic carbene [4–7]. The Ru = CHPh moiety interacts with a cyclic olefin to produce metalocyclobutane, followed by a rearrangement of the bonds to result in a new olefin and in a new Ru-carbene species. This type of complex is also active for ATRP, where the reduced-metal complex reacts with a halide-dormant species to generate an oxidized-metal-halide species and a radical species that initiates the reaction with the main monomer [5]. The oxidized-metal-halide complex must be able to transfer the halogen atom to the propagating radical polymer to control the growing chain, recovering the reduced-metal complex. Therefore, ROMP needs a carbene-metal complex, whereas ATRP does not, and non-carbene complexes that are active for ATRP of methyl methacrylate (MMA) are also active for ROMP of norbornene (NBE) and norbornadiene (NBD), such as the [RuCl2(PPh3)3] complex [8–10].
We have proposed the development of non-carbene complexes of type [RuCl2(PR3)x(amine)y] combining amines and phosphines as ancillary ligands to promote ROMP and improve the characteristics of the polymers produced [11–16]. The electronic combination of a π-acceptor phosphine with a σ-donor amine is a convenient modular approach to tune the reactivity of such pre-catalyst when varying the amine nature, in addition to promoting appropriate steric hindrance. These features have successfully avoided the formation of dimeric species in solution, which decreases the catalytic activity, as it occurs with [RuCl2(PPh3)3] [10]. The metal-carbene moiety is generated in situ from reaction with a diazo compound. This usually requires an induction period, but in many cases reaction occurs after a few minutes at room temperature. These complexes do not need special conditions of storage and manipulation and they are not very sensitive to air, moisture, light, and heat, which makes them suitable for use at a preparative scale. It is worth emphasising that this type of compound uses cheap, simple ligands.
Few studies have discussed bulky ligands and basic amines as additives to improve ATRP with [RuCl2(PPh3)3] [17, 18]. In contrast, we have studied the [RuCl2(PR3)x(amine)y] complex where the amine is already coordinated. This is quite interesting for ATRP because the complex is coordinatively and electronically unsaturated (five-coordinated, 16-electron complex) and is composed of a combination of strongly bound ligands (amine) and bulky ligands (PPh3 or PCy3, for example). The σ-donor amine increases electron density on the metal centre, decreasing the reduction potential, and a free vacancy permits coordination of the halogen in the metal to start ATRP.
This study reports the synthesis and application of a novel, non-carbene, amine-phosphine, Ru-based complex, namely [RuCl2(PPh3)2(azocane)] (1), as a versatile catalyst precursor for ROMP of norbornene (NBE) and norbornadiene (NBD) and for ATRP of MMA and styrene (St), as illustrated in Scheme 2. The investigation leads to the development of alternative, non-carbene complexes, considering that a carbene complex is not needed to initiate the ATRP reaction and an in-situ carbene compound can be produced in the reaction medium to initiate the ROMP reaction. Therefore, a dual complex can be planned and the azocane ligand becomes a candidate to act as ancillary ligand to increase the electron density at the ruthenium, where it is an σ-donor amine, and promote steric hindrance, providing an effective reduction process to avoid an uncontrollable growth of the polymer chain. Different conditions of reaction time, monomer concentration, solvent, and the use of Al(OiPr)3 as additive were investigated. Al(OiPr)3 is known as an additive that accelerates ATRP mediated by various metal complexes, but it is particularly effective for Ru-compounds [17, 18]. Ethyl 2-bromoisobutyrate (EBiB) or 2-phenylethyl bromide (PEB) were used as initiators for ATRP of MMA or for St, respectively, whereas ethyl diazoacetate (EDA) was used as starting source of carbene for ROMP.

Illustration of [RuCl2(PPh3)2(azocane)] (1) complex as catalyst precursor for ATRP of vinyl monomers and ROMP of cyclic olefin
2 Experimental
2.1 General Procedure
All reactions and manipulations were performed under a nitrogen atmosphere by using standard Schlenk techniques. Toluene was dried overnight over calcium chloride, filtered and distilled from sodium/benzophenone ketyl and degassed by three vacuum–nitrogen cycles under nitrogen before use. Methyl methacrylate (MMA) and styrene (St) were washed with 5% NaOH solution, dried over anhydrous Mg2SO4, vacuum distilled from CaH2 and stored under nitrogen at −18 °C before use. 1,2-dichloroethane (DCE), CHCl3, RuCl3×H2O, anisole, n-Bu4NPF6, norbornene (NBE), norbornadiene (NBD), ethyl diazoacetate (EDA), azocane, 1-phenylethylbromide (PEB), ethyl 2-bromoisobutyrate (EBiB) and aluminium isopropoxide (Al(OiPr)3) were used as acquired from Aldrich. The [RuCl2(PPh3)3] complex was prepared following the literature and its purity was checked by satisfactory elemental analysis and spectroscopic analyses (31P{1H} and 1H NMR, FTIR and EPR) [19, 20]. Room temperature (RT) is 23 ± 1 °C.
2.2 Synthesis of [RuCl2(PPh3)2(azocane)] (1)
Azocane (0.34 mmol; 0.028 g) was added to a solution of [RuCl2(PPh3)3] (0.26 mmol; 0.25 g) in acetone (10 mL). The resulting dark green solution was stirred for 2 h at RT. A green precipitate was then filtered and washed with methanol and ethyl ether, and then dried in a vacuum. Yield: 73%. Elemental analysis for RuCl2P2NC42H44 experimental: C, 62.6; H, 5.76; N, 1.75%; calculated: C, 63.8; H, 5.56; N, 1.73%. FTIR in CsI: ν(Ru–Cl) = 320 and 299 cm− 1; ν(N–H) = 3050 cm− 1. EPR: g = 2.45, 2.24 and 1.84. 1H NMR (CDCl3, 400 MHz): δ(ppm) = 1.35–1.21 (m, 8 H, CH2—azocane), 0.92 − 0.83 (m, 6 H, CH2—azocane), 1.78 (s, 1 H, NH—azocane), 7.73–7.61 (m, 12 H, meta-PPh3); 7.57–7.37 (m, 18 H, ortho and para—PPh3). 31P{1H} NMR (CDCl3, 162 MHz, δ): 45.1 (s).
2.3 Equipment
Elemental analysis was performed in a Perkin-Elmer CHN 2400 in the Institute of Chemistry of the University of São Paulo. Electron Paramagnetic Resonance (EPR) measurements from solid sample were carried out at 77 K using a Bruker EPR 300 C apparatus (X-band) equipped with a TE102 cavity and HP 52,152 A frequency counter. FTIR spectra in CsI pellets were recorded using a Bomem FTIR MB 102. The 1H and 31P{1H}NMR spectra were obtained in CDCl3 at 25.0 ± 0.1 °C using a Bruker AVANCE-III spectrometer operating at 400.13 and 161.98 MHz, respectively. The obtained chemical shifts were reported in ppm relative to TMS or 85% H3PO4.
Conversion in ATRP was determined from the concentration of residual monomer measured by gas chromatography (GC) using a Shimadzu GC-2010 gas chromatograph equipped with flame ionization detector and a 30 m SPB-1 Supelco fused silica capillary column (0.53 mm I.D., 0.5 µm film thickness). Anisole was added in polymerization and used as an internal standard. Analysis conditions: injector and detector temperature, 250 °C; temperature program, 40 °C (4 min), 20 °C min−1 until 200 °C, 200 °C (2 min).
The molecular weights and the molecular weight distribution of the polymers were determined by gel permeation chromatography using a Shimadzu Prominence LC system equipped with a LC-20AD pump, a DGU-20A5 degasser, a CBM-20A communication module, a CTO-20A oven at 40 °C and a RID-10A detector equipped with two Shimadzu column (GPC-805: 30 cm, Ø = 8.0 mm). The retention time was calibrated with standard monodispersed polystyrene using HPLC-grade THF as an eluent at 40 °C with a flow rate of 1.0 mL min−1.
Electrochemical measurements were performed using an Autolab PGSTAT204 potentiostat with a stationary Pt disk (ϕ= 3 mm) and a Pt wire as working and auxiliary electrodes, respectively. The reference electrode was Ag/AgCl. The measurements were at 25 °C ± 0.1 in DCE with 0.1 mol.L−1 of n-Bu4NPF6.
2.4 ROMP Reactions
In a typical ROMP experiment, norbornene (NBE) or norbornadiene (NBD) was dissolved in 2 mL of degassed CHCl3 and 1 µmol of 1 was added to the reaction solution, followed by addition of 5 µL of ethyl diazoacetate (EDA). The molar ratio [monomer]/[Ru] was 5000. The reaction mixture was stirred for different periods of times (5, 10, 15, 30, 45 or 60 min) at 25 or 50 °C in a silicon oil bath. At RT, 5 mL of methanol was added to the reaction flask and the polymer was filtered, washed with methanol and dried in a vacuum oven at 40 °C up to constant weight. The reported yields are average values from catalytic runs performed at least three times with 10% error at most.
2.5 ATRP Reactions
In a typical ATRP experiment, 12.3 µmol of 1 was placed in a Schlenk flask containing a magnetic stirrer bar and capped by a rubber septum. Air was expelled by three vacuum–nitrogen cycles before addition of MMA or St, toluene (1 mL) and EBiB or PEB (22.4 µmol). All liquids were handled with dried syringes under nitrogen. The flask was capped under N2 atmosphere using Schlenk techniques, and then, the reaction mixture was immediately immersed in an oil bath at desired temperature. The polymerizations were conducted as a function of time at 85 °C for MMA and 110 °C for St. Samples were removed from the tube after certain time intervals using syringes. Conversion was determined from the concentration of residual monomer measured by gas chromatography. Calculated molecular weight (Mn;calcd) was determined by ([Monomer]/[initiator]) × (MWmonomer) × conversion.
3 Results and Discussion
3.1 Synthesis and Characterization of [RuCl2(PPh3)2(azocane)] (1)
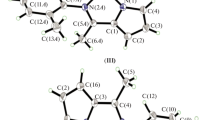
Complex 1 was synthesised by a procedure similar to that of the parent [RuCl2(PPh3)2(amine)] complexes, from one-pot synthesis between azocane and [RuCl2(PPh3)3] [10–16]. The proposed formulation of a five-coordinated complex agrees with the CHN elemental analysis. A trigonal, bipyramidal geometry (TBP) can be supported by the solid-state EPR spectrum, with signals associated with a triplet d6 electronic distribution, in agreement with the 31P-NMR spectrum in CDCl3, with one signal at 45.1 ppm of two PPh3 ligands localized in the axial axis of the TBP geometry (Fig. 1). In this case, the azocane molecule and the Cl− ions are in the TBP equatorial plane.

Illustration of possible TBP geometry arrangement for 1 in CDCl3
3.2 ROMP Reactions
Plot of yield for ROMP of NBE as a function of time at 25 °C can be described by an induction period followed by a saturation curve profile with a maximum yield of 20% (Fig. 2). At 50 °C, 70% yield was obtained in 60 min with a non-linear behaviour. The Mn values increased with reaction time and were in the order of magnitude of 104 g mol−1 either at 25 or at 50 °C (Fig. 3). The PDI values were ca. 2.0 independent of reaction time and temperature. Considering the improvement in the yields at 50 °C and the increase in the Mn values with time, an induction period must be accelerated at higher temperature to initiate ROMP. As previously discussed in the case of parent [RuCl2(PPh3)(amine)] complexes with a square pyramidal arrangement, the TBP geometry is not favourable to coordinate EDA and monomer [21]. Thus the induction period probably also involves a change in the geometry favoured at 50 °C, with yield improvement owing to chain-growing polymer occurrence, but the reaction is limited by a stationary state at 25 °C.

Dependence of yield on reaction time and temperature for ROMP of NBE with 1 (1.1 µmol in CHCl3); [NBE]/[1] = 5000 and 5 µL of EDA

Tendency of the Mn of polyNBE obtained from ROMP with 1 as a function of reaction time and temperature; [NBE]/[1] = 5000 with 1.1 µmol of 1 and 5 µL of EDA in CHCl3
For ROMP of NBD with [NBD]/[1] molar ratio of 5000 at 50 °C, the polyNBD yield presented a linear relationship as a function of time up to 60 min (Fig. 4). The resulting polymers were insoluble in CHCl3, probably because of the occurrence of cross-linked chain, owing to polyNBD type network. Yields lower than those obtained with NBE under the same conditions could be associated with a double coordination of NBD to the metal coordination sphere, which represents an additional step in the reaction, retarding ROMP initiation [13].

Dependence of yield on reaction time for ROMP of NBD with 1; [NBD]/[1] = 5000 with 1.1 µmol of complex and 5 µL of EDA in CHCl3 at 50 °C
3.3 ATRP Reactions
Figure 5 shows the conversion percentage of MMA as a function of time in different media, when the [MMA]/[1] molar ratio was 1000/1 at 85 °C, with [EBiB]/[1] = 2/1. The reactions performed in toluene or in bulk resulted in similar consumptions of monomer, with 17–19% of conversion in 9 h. The Mn values were in the order of magnitude of 104 g mol−1 and roughly doubled when the reactions were performed in bulk compared with those in the presence of solvent (Table 1; entries 1–6). The Mn values from reactions in toluene were similar to the calculated values (entries 1–3). The conversions increased in the presence of Al(OiPr)3 in toluene when increasing the reaction time, reaching 52% in 9 h (Fig. 5). The molecular weights were double those in toluene without the additive and the PDI values were lower regardless of reaction time (entries 7–9). The Al(OiPr)3 additive improved conversion, but the Mn values were different from the calculated ones. Considering the literature, this means that the radical concentration did not remain constant throughout the polymerization reaction [22, 23]. However, improvement has occurred in conversion with the additive, which could be associated with bromine abstraction from the initiator or from the propagating chain, but the RuIII–Br moiety was not sufficiently efficient to drive the bromine back to the radical chain.

Studies with [MMA]/[1] molar ratio = 200 in toluene showed higher MMA conversion compared with that of studies with [MMA]/[1] = 1000 either in the absence or in the presence of Al(OiPr)3 (Fig. 6; top). Conversion in the absence of additive follows a linear increase as a function of time up to 5 h, followed by a saturation behaviour, whereas conversion in the presence of additive follows a linear increase as a function of time up to 9 h. Linear correlations of ln([M]0/[M]) vs time are obtained up to 5 h (Fig. 6; bottom) with similar pseudo first-order rate constants (k obs). The Mn values agreed with the calculated ones, indicating a constant radical concentration throughout the polymerization process.

In the presence of Al additive with [MMA]/[1] = 200 (Fig. 7), a linear growth in the polyMMA chain with MMA conversion is observed up to 9 h, with significant decrease in the PDI values. The polydispersity of all polymers obtained in the presence of Al additive remained low; e.g., between 1.4 and 1.3 (Table 2). This suggests the occurrence of more controlled molecular weight with low [MMA]/[1] molar ratio in the presence of Al(OiPr)3. Higher Mn values were obtained (105 g mol−1) compared with those in the absence of Al additive (104 gmol− 1). Experimental Mn vs calculated Mn plot from data in the absence of additive a linear plot, with slope of the straight line equals 0.9574, which could indicate high initiator efficiency (Figure S1).24 With Al additive, the experimental Mn values also show a linear relationship with the calculated Mn values and narrower PDI values, but with slope equals 0.3005.

ATRP of St with [St]/[1] molar ratio = 1000 in toluene at 110 °C showed increased consumption of St as a function of time either in the absence or in the presence of additive (Fig. 8), with higher conversion when additive was present. In both cases, the Mn values increased with reaction time and narrower PDI values were observed in the presence of additive (Table 3). With [St]/[1] = 200, a linear increase in the St conversion is observed, with 30% in 7 h (Fig. 8). The Mn values also increased and the PDI decreased as a function of time (PDI = 1.5 in 7 h; Table 3). In general, the radical concentration did not remain constant throughout the polymerization reaction considering the difference between the theoretical and experimental molecular weights. In the presence of additive, styrene polymerization appeared to be better controlled, as evidenced by narrower polydispersity and more similar theoretical and experimental molecular weight values.

3.4 Characterization of 1 by Cyclic Voltammetry
The voltammogram of 1 shows an irreversible redox process with anodic peak at E pa =1.14 V vs Ag/AgCl (Fig. 9; solid line). The literature suggests that the absence of cathodic peak can affect the equilibrium between oxidized and reduced metal species necessary for a living ATRP [24–29]. Obviously, the consequence of an irreversible redox process is a decrease in the RuII concentration in the medium. This fact would increase the concentration of propagating free radical species in the reaction medium due to the absence of RuIII species shifting back to RuII. This circumstance could explain the moderate catalytic activity of 1 for ATRP with broad PDI values, where the growing chain control is missed because of the low stability of the oxidized RuIII species. However, the voltammogram was not recorded under ATRP conditions. Measurements under similar ATRP conditions have failed, i.e., only an irreversible process was observed.

Cyclic voltammogram of 1 in DCE at 25 °C in the absence (solid line) and in the presence (dashed line) of Al(OiPr)3; 10 mmol L−1 of 1, [Al]/[1] = 4, [nBu4NPF6] = 0.10 mol L−1, scan rate = 100 mV s−1
A cathodic process at E pc = 0.79 V vs Ag/AgCl appears in the voltammogram when Al(OiPr)3 is present in the medium (Fig. 9; dashed line), which could indicate partial recovery of RuII species from the generated RuIII. Reversibility of 1 can improve the control over the concentration of propagating radical species. In fact, a more controlled process was reached where the Mn values were closer to their calculated values and PDI values were narrower when the Al(OiPr)3 was present as additive in the reaction mixture. An interaction between Al(OiPr)3 and the complex and the metal in high oxidation state has been proposed as an improvement factor to retrieve the reduced species, decreasing the concentration of the radical growing chain [30]. In part, the 17-electron oxidized species presents lower electron density to support the original coordination sphere. An (OiPr)3Al-Br adduct moiety may drive the reaction to restore equilibrium [31].
4 Conclusions
The amine azocane replaced one PPh3 molecule in the [RuCl2(PPh3)3] complex to combine electronic and steric features in the activity for ROMP of NBE and NBD and for ATRP of MMA and St. From studies conducted under different reaction conditions, moderate to high yields of polymers were obtained from both types of reactions.
The control of the catalytic activity of 1 for ATRP of MMA and St is still under investigation. It was not clear whether there was a relationship of the conversions with the presence or absence of Al(OiPr)3 as additive, as discussed in the literature. Some Mn results correlate well with the calculated values and, in other cases, narrow PDI values were obtained. In addition, the electrochemical behaviour of 1 did not correlate to control in ATRP. Some increase in the stability of RuIII species in the presence of Al(OiPr)3 was observed, which could result in better control of polymerization. In contrast, good catalytic activity of 1 for the ROMP of NBE connects well to the family of complexes of the [RuCl2(PPh3)(amine)] type, where the type of geometry in solution and a greater amine\(\mathop{\rightarrow}\limits^{\sigma}\)Ru\(\mathop{\rightarrow}\limits^{\pi}\)monomer electronic synergism explain the results.
References
Matson J-B, Grubb R-H (2008) Macromolecules 41:5626–5631
Nargarkar A-A, Kilbinger A-F-M (2015) Nat Chem 7:718–723
Matyjaszewski K, Coca S, Gaynor S-G, Wei M, Woodworth B-E (1998) Macromolecules 31:5967–5969
Grubbs R-H (2004) Tetrahedron 60:7117–7140
Lee J, Grandner J-M, Engle K-M, Houk K-N, Grubbs R-H (2016) J Am Chem Soc 138:7171–7177
Airaud C, Ibarboure E, Gaillard C, Héroguez V (2009) Macromol Symp 281: 31–38
Nguyen M-N, Mougnier S-J, Ibarboure E, Heroguez V (2011) J Polym Sci Part A 49: 1471–1482
Simal F, Delfosse S, Demonceau A, Noels A-F, Denk K, Kohl F-J, Weskamp T, Herrmann W A (2002) Chem—A Eur J 8: 3047–3052
Kato M, Kamigaito M, Sawamoto M, Higashimura T (1995) Macromolecules 28:1721–1723
Matos J-M-E, Lima Neto B-S (2004) J Mol Catal A 222:81–85
Matos J-M-E, Lima Neto B-S (2006) J Mol Catal A 259:286–291
Sá J-L-S, Lima Neto B-S (2009) J Mol Catal A 304:187–190
Sá J-L-S, Vieira L-H, Nascimento E-S-P, Lima Neto B-S (2010) Appl Catal A 374:194–200
Carvalho V-P Jr, Ferraz C-P, Lima Neto B-S (2010) J Mol Catal A 333:46–53
Chaves H-K, Ferraz C-P, Carvalho V-P Jr, Lima Neto B-S (2014) J Mol Catal A 385:46–53
Fonseca L-R, Nascimento E-S-P, Sá J-L-S, Lima Neto B-S (2015) New J Chem 39:4063–4069
Hamasaki S, Kamigaito M, Sawamoto M (2002) Macromolecules 35:2934–2940
Melis K, Verpoort F (2003) J Mol Catal A 201:33–41
Armit P-W, Boyd A-S-F, Stephenson T-A (1975) J Chem Soc Dalton Trans 1663–1672 doi: 10.1039/DT9750001663
Hoffman P-R, Caulton K-G (1975) J Am Chem Soc 97:4221–4228
Fernandes R-J, Silva T-B, Lima Neto B-S, Haiduke R-L (2015) J Mol Catal A 410:58–65
Kamigaito M, Ando T, Sawamoto M (2001) Chem Rev 101:3689–3746
Kamigaito M, Watanabe Y, Ando T, Sawamoto M (2002) J Am Chem Soc 124:9994–9995
Matyjaszewski K, Xia J (2001) Chem Rev 101:2921–2990
Siegwart D-J, Oh J-K, Matyjaszewski K (2012) Prog Polym Sci 37: 18–37
Wever D-A-Z, Raffa P, Picchioni F, Broekhuis A-A (2012) Macromolecules 45:4040–4045
Matyjaszewski K, Tsarevsky N-V (2014) J Am Chem Soc 136:6513–6533
Wu W, Wang W, Li J (2015) Prog Polym Sci 46: 55–85
Chmielarz P, Park S, Simakova A, Matyjaszewski K (2015) Polymer 60:302–307
Ando T, Kamigaito M, Sawamoto M (2000) Macromolecules 33:6732–6737
Aguilar Lugo C, le Lagadec R, Ryabov A-D, Valverde G-C, Morales S-L, Alexandrova L (2009) J Polym Sci Part A 47: 3814–3828
Acknowledgements
The authors are indebted to the financial support from CAPES, CNPq, and FAPESP (Process numbers: 2013/10002-0, 2011/12571-7 and 06/57577-4).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Silva, R.A.N., Borim, P., Fonseca, L.R. et al. Non-carbene Complex [RuCl2(PPh3)2(azocane)] as Active Catalyst Precursor for ROMP and ATRP. Catal Lett 147, 1144–1152 (2017). https://doi.org/10.1007/s10562-017-2003-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-017-2003-y