
Abstract
In prostate to bone metastases, the “vicious cycle” paradigm has been traditionally used to illustrate how metastases manipulate the bone forming osteoblasts and resorbing osteoclasts in order to yield factors that facilitate growth and establishment. However, recent advances have illustrated that the cycle is far more complex than this simple interpretation. In this review, we will discuss the role of exosomes and hematopoietic/mesenchymal stem/stromal cells (MSC) that facilitate the establishment and activation of prostate metastases and how cells including myeloid-derived suppressor cells, macrophages, T cells, and nerve cells contribute to the momentum of the vicious cycle. The increased complexity of the tumor–bone microenvironment requires a system level approach. The evolution of computational models to interrogate the tumor–bone microenvironment is also discussed, and the application of this integrated approach should allow for the development of effective therapies to treat and cure prostate to bone metastases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Prostate to bone metastases stimulate enhanced osteoblast and osteoclast activity that in turn promotes the release of growth factors from the bone matrix creating a “vicious cycle” of cancer growth. This feedback loop results in mixed lesions that are comprised of areas of extensive osteolysis (bone degradation) and osteogenesis (bone formation) with the latter being a hallmark of the disease [1]. While prostate to bone metastases remain incurable, therapeutic developments have begun to improve patient quality of life and begun push overall survival rates higher. Denosumab (Xgeva®), a monoclonal antibody to receptor activator of nuclear factor κB (RANKL), has recently been approved for the treatment of prostate cancer patients. Denosumab has been shown in clinical trials to significantly increase the time to SREs in comparison to bisphosphonates but does not appear to improve overall survival [2]. Another breakthrough has come in the form of radium-233 chloride (Alpharadin/Xofigo®) [3]. This alpha emitting radiopharmaceutical is similar to calcium in structure and targets the bone tissue. The short wavelength of the radiation ensures less damage to surrounding tissues when compared to beta or gamma emitting sources of radiation. In clinical trials of men with castrate-resistant prostate cancer (CRPC) bone metastases, alpharadin was found to significantly improve overall survival by 3–4 months and is the first therapy to achieve this milestone [4]. Despite these advances, there is room for improvement. New studies have begun to define the precise cellular and molecular players that drive the establishment and evolution of prostate to bone metastases, thus presenting novel targets for therapeutic intervention (Table 1). These advances and how to integrate them on a global level are the focus of the current review.
2 Preparing the “soil”
In 1889, Stephen Paget classically described the “seed and soil” hypothesis as the propensity of circulating tumor cells (CTCs) to home to specific organs, irrespective of the nearest anatomical location to the primary tumor [5]. CTCs preferentially metastasize and “seed” organs with the proper “soil.” While empirical evidence has identified genetic programs that facilitate the homing and initial establishment of prostate and breast cancer cells to bone such as interleukin-11 (IL-11), connective tissue growth factor, chemokine receptor 4 (CXCR4), matrix metalloproteinase-1 (MMP-1), and vascular cellular adhesion molecule-1 (VCAM-1), studies have also shown that primary tumor-derived signals can prepare the distant soil in pre-ordained areas for metastases [6–9]. This has been particularly true for lung metastasis in which studies using a B16 melanoma model identified that primary tumor-secreted factors induced the mobilization of immune-derived cells to niches in the lung where they promote angiogenesis and extracellular matrix remodeling [10,11]. These niches then become the sites of future metastases. Understanding the factors that control the formation of the pre-metastatic niche in bone could yield insights into identifying therapeutic targets that will prevent metastasis and therefore have a significant impact on cancer-related deaths. Recently, studies have implicated tumor-derived microvesicular liposomes known as exosomes in facilitating the initial communication between the primary tumor and the site of metastasis [12,13].
2.1 Exosomes and oncosomes
Exosomes were first identified in the mid-1980s as recycled portions of intravesicular membranes released by reticulocytes following endocytosis of the membrane receptor, transferrin [14,15]. Exosomes are utilized for the packaging and recycling of membrane and cytosolic proteins [16]. Produced by a variety of cells, exosomes vary in size from 30 to 100 nm in diameter and can contain numerous proteins that control, for example, membrane fusion and transport (annexins, small GTPase Rab proteins, and Lamp proteins) [17]. Despite the variability in exosome contents and membrane proteins, components can be transferred both homo- and heterotypically between multiple cell types [18].
Significant roles for exosomes in the establishment of the pre-metastatic niche and metastasis have been recently described [13]. Systemic delivery of fluorescent exosomes from metastatic B16F10 melanoma cells has been shown to localize to lungs and other tissues including bone. Subsequent experiments identified that the growth and metastasis of subcutaneous B16F10 metastatic melanoma tumors was significantly increased in C57BL/6J mice that received exosomes from B16F10 cells, compared to mice that received exosomes from congenic non-metastatic B16F1 cells [13]. Furthermore, the developing metastases co-localized at the sites of exosome deposition with the underlying mechanism being implicated as exosome-derived MET, receptor for hepatocyte growth factor, transfer to bone marrow-derived cells. These data illustrate a role for exosomes in the development of metastatic lesions. Supporting this observation, knockdown of Rab27a, a gene necessary for exosome release, significantly reduced the number of circulating exosomes and the number of metastases, demonstrating that metastatic burden was dependent on the number of circulating exosomes [13,19]. Clinically, the detection of TYRP2 and MET in exosomes derived from melanoma patient plasma is already proving informative as a prognostic indicator of disease progression [13]. MET is involved in normal prostate development and promotes progression of malignant prostate cells. Androgen ablation therapy increases MET expression by bone marrow stromal, osteoblastic, and prostate tumor cells promoting the transition of localized prostate cancer to a more metastatic phenotype [20]. Thus, exosome-mediated transfer of MET to bone marrow stromal cells may play a large role in prostate tumor progression in bone.
In prostate cancer, the number of oncosomes (vesicles larger than exosomes that range in size from 1 to 10 μm) in patient plasma was found to correlate with Gleason scores of 7 and higher while exosomes isolated from LNCaP, DU145, and PC3 prostate cancer cell lines have been found to enhance osteoblast differentiation and to induce the differentiation of bone marrow-derived mesenchymal stem cells into myofibroblasts that promote tumor growth [21–23]. Collectively, these data suggest a role for oncosomes in prostate cancer malignancy and potentially in the establishment of pre-metastatic niches in the bone microenvironment.
Advances in our understanding of how exosomes can contribute to tumor progression have provided new angles for prognostic evaluation and potentially for therapeutic intervention [13]. Cabozantinib (XL184), a vascular endothelial growth factor (VEGF) and MET inhibitor, has had very promising results in phase II clinical trials of men with advanced prostate cancer. Indications suggest significant decreases in tumor burden and increases in overall survival [24]. MET transfer from exosomes to bone marrow-derived cells is an important step in generating the pre-metastatic niche so it is possible cabozantinib may also interfere with the process. Approval of cabozantinib will add to the clinician’s ability to extend overall survival of patients with bone metastases and also supports the approach of developing targeted therapies to cells and factors that control the vicious cycle (Fig. 1). Preventing exosome release by targeting proteins involved in the process such as lysosome-associated membrane proteins (Lamps) could have a significant impact on cancer progression. Exosomes are non-immunogenic and therefore could be the perfect means through which to deliver therapies. In this regard, cationic liposomes that release their contents via exosome release were loaded with siRNA to Plekho1, a factor responsible for tagging osteogenic genes such as Runx2 for ubiquitination, and were found to increase osteoblast-mediated bone formation and demonstrate the potential feasibility of the approach in the clinical setting [25].

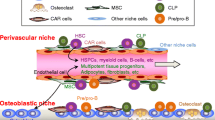
Role of exosomes, HPCs, and MSCs in the establishment and outgrowth of prostate to bone metastases. 1 Prior to the arrival of the prostate metastases in bone, exosomes released from the primary tumor can facilitate the establishment of the pre-metastatic niche via mechanisms such as transfer of MET. 2 Activated metastases establish and grow in the bone microenvironment via the expression of VCAM-2 and the recruitment of osteoclast pre-cursors. 3 Tumor-derived CXCL16 and CCL2 can promote MSC recruitment and differentiation into CAFs or osteoblasts. 4 Enhanced osteoblast activity and expression of RANKL can in turn drive osteoclastogenesis and bone resorption. 5 Cancer recruitment of HPCs can also lead to osteoclast mediated bone resorption resulting in a release of growth factors that further stimulate the growth of the metastases. Inhibitors of the various molecules that control cell–cell interactions are highlighted in red
3 New roles for stem cells in the establishment of prostate to bone metastases
Extravasation and colonization in bone is tightly regulated by adhesion molecules such as VCAM-1, which has been shown to be critical in the initial interaction between CTCs and stromal cells [26,27]. Additionally, VCAM-1-mediated osteoclast activity releases disseminated tumor cells (DTCs) from dormancy, converting indolent metastasis to actively growing lesions demonstrating an important role for VCAM-1 in both the initial entrance and activation of DTCs in bone [6]. Once in the bone marrow, DTCs must survive in the new microenvironment and communicate with the host cells in order to generate active bone metastases. A number of new exciting studies illustrate how metastatic prostate cancer cells can interact with hematopoietic stem cell niches and MSCs to facilitate establishment in the bone microenvironment (Fig. 2).

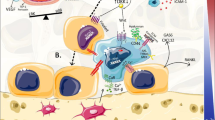
Immune regulation of bone metastasis. 1 Dendritic cells in the bone microenvironment can contribute to T cell polarization resulting in an immunoevasion of the prostate metastases. Dendritic cells can also influence osteoclast behavior via a number of molecules. 2 T cells can act directly or indirectly through the regulation of osteoclast function on the growth of prostate to bone metastases. 3 MDSCs can impact the function of several cell types in the tumor–bone microenvironment and can also directly contribute to prostate cancer growth and osteoclastogenesis. 4 Macrophages are commonly found in the bone microenvironment and can be recruited by cancer-derived factors such as CCL2. They can promote cancer cell invasion and angiogenesis while in parallel suppress cytotoxic T cell activity. Macrophages have also been identified to play roles in regulating bone resorption. Therapies targeting the cellular and molecular mediators are highlighted in red
3.1 Hematopoietic stem cells
Hematopoietic stem cells (HSCs) reside in specialized niches within the bone marrow and are the source of myeloid and lymphoid cell lineages [28]. HSCs can mobilize from the marrow to distant regions in response to injury or primary cancer and subsequently return to bone marrow stem cell niches where they remain quiescent until needed again [29]. Niches in the bone marrow can be found proximal to the sinusoidal vasculature or the endosteal surface at the bone marrow interface. Lining the endosteal surface is osteoblasts to which HSCs adhere via interactions with VCAM-1, selectins (P- and E-selectin), angiopoietin (via interactions with Tie2), or Kit-ligand [30,31]. Osteoblasts are critical for the maintenance of HSC quiescence in the endosteal niches but can activate HSC expansion via the expression of chemokine ligand 12 (CXCL12) and its interaction with CXCR4 on the HSC cell surface. Osteoclasts also play a role in mobilizing HSCs from the endosteal niche via the expression of MMP-9 that in turn processes Kit-ligand, a mechanism that facilitates the release of the HSCs from the niche and increases CXCL12 and VEGF expression in the bone marrow compartment. Newly arriving prostate to bone metastases can utilize the factors involved in the maintenance of the HSC niche. For example, SUMO-specific protease 1, a member of the deSUMOylation protease family, has been recently identified as a regulator of MMP-9 expression by prostate tumors and promotes prostate cancer metastasis to bone [32]. Additionally, CXCR4 expression by the prostate cancer cells is involved in the homing of prostate metastases to the bone, in particular to areas that are rich in CXCL12 such as the endosteal HSC niche [33].
Recent evidence has identified how metastatic prostate cancer cells directly compete with HSCs to engraft within the bone marrow compartment [34]. Disseminated cells shed from implanted subcutaneous prostate cancers (PC3 or C42B) were found to inhibit HSCs engraftment after bone marrow transplant [35]. Results show that the HSCs and disseminated prostate metastases co-localized to osteoblasts expressing CXCL12 and annexin 2, an adhesion molecule utilized by HSCs to adhere to osteoblasts in the endothelial stem cell niche. Increasing osteoblast number via parathyroid hormone treatments and thereby the number of niches also increased the number of metastatic cells that engrafted within the bone marrow. Likewise, ganciclovir-mediated ablation of the osteoblasts decreased the number of endothelial stem cell niches and thus the number of engrafted metastatic cells in osteoblast specific (Col2.3Δ-TK) transgenic mice, demonstrating the critical role of osteoblasts and the HSC stem cell niche in prostate cancer engraftment within the bone marrow. Interestingly, prostate metastases occupying the endosteal niche in tumor-bearing mice can also be mobilized back into the circulation by administrating CXCL12 blocking antibodies or treatment with granulocyte colony-stimulating factor [35].
In addition to utilizing the same pathways as HSCs to home and graft into bone niches, prostate cancer cells can also manipulate HSC differentiation to facilitate the induction of the vicious cycle. Using osteolytic and osteogenic cell lines, PC3 and C42B, respectively, studies have shown that in the presence of RANKL, HSCs derived from mice bearing subcutaneous PC3 tumors can promote the differentiation of naïve HSCs into osteoclasts compared to HSCs derived from C42B-bearing mice with PC3-derived IL-6 being implicated as the underlying mechanism involved [36]. IL-6 is important for prostate tumor growth in bone as well as for progression of primary prostate tumors via cross-activation of insulin-like growth factor (IGF) signaling, one of the most abundant factors released from the bone matrix upon osteolysis [37,38]. These data demonstrate the effects that prostate cancer cells can elicit on the HSC compartment to initiate the lytic compartment of the vicious cycle. Intriguingly, the authors also show in converse experiments that HSCs isolated from C42B tumor-bearing animals exhibited increased BMP2 and BMP6 expression and promoted the differentiation of naïve bone marrow-derived MSCs into osteoblasts more efficiently than HSCs derived from PC3 tumor-bearing animals, thus implicating potential roles for MSCs in prostate cancer-induced osteogenesis.
These advances offer a means to therapeutically prevent bone metastasis formation by targeting underlying molecular mechanisms such as VCAM-1 and CXCL12. In recent years, there have been an increasing number of clinical trials for CXCL12/CXCR4 antagonists as anticancer agents. Nox-A12, a CXCL12 antagonist, is currently in clinical development as an anticancer agent and has recently been introduced into phase III clinical trials of myeloma patients, a cancer known to promote increased osteolysis in bone. Additionally, AMD3100 was recently FDA-approved for the use of stem cell mobilization in non-Hodgkin’s lymphoma and multiple myeloma along with other CXCL12 inhibitors including BKT140, POL6326, and TG-0054 [39,40]. However, MSX-122 is the first reported CXCL12 inhibitor in phase 1 clinical trials for solid tumor cancers. Activation of peroxisome proliferator-activated receptor-α with Wy14643 has been shown to inhibit VCAM-1, abrogating HSC interactions with the endosteal niche, and promote HSC mobilization [41]. Inhibition of VCAM-1 in prostate cancer patients with early stage disease may prevent both DTC extravasation into bone and localization to HSC niches and should be further investigated. Therapeutic targeting of the CXCL12/CXCR4 or VCAM-1 pathway in combination with cytokine targeted therapies (e.g., Tocilizumab, monoclonal antibody to IL-6) may be beneficial for preventing DTC localization to the HSC niche and prostate tumor progression in bone. It is currently unknown whether these CXCL12-targeted therapies will mobilize tumor cells that may metastasize to other areas or “re-seed” the primary tumor as has been noted [35]. This, however, is a profound observation that has implications for not only preventing bone metastases but also in considering how applied therapies may affect disseminated prostate cancer cells.
3.2 Mesenchymal stromal cells
Approximately 0.001 to 0.01 % of the marrow compartment is comprised of MSCs [42]. Given the correct cues, MSCs can differentiate into fibroblasts, chondrocytes, myocytes, adipocytes, or osteoblasts and are involved in the maintenance and regeneration of skeletal tissues, including connective tissues, cartilage, muscle, fat tissue, and bone [43]. MSCs are multipotent, have limitless proliferative abilities, maintain the HSC niche, and can be mobilized from the marrow to sites of tissue injury for repair. Virchow has described cancer as a wound that does not heal, and therefore, it is unsurprising that MSCs have been identified as contributing to the progression of primary cancers. In breast cancer, chemokine ligand 5 (CCL5) plays a key role in the recruitment of MSCs to the primary tumor microenvironment [44]. Heightened MSC infiltration results in enhanced expression of lysyl oxidase (LOX) and angiogenesis that in turn promotes the invasion and metastasis of the cancer cells [44,45]. In primary prostate cancer, CXCL16 facilitates MSC recruitment via the receptor CXCR6 to the tumor microenvironment and their subsequent differentiation into cancer-associated fibroblasts (CAFs). The MSC-derived CAFs were found to be critical for promoting tumor growth, epithelial to mesenchymal transition (EMT), and metastasis to bone [46]. CAFs exhibit CD44-mediated increased TWIST expression, a downstream target of transforming growth factor β (TGFβ) that promotes EMT, differentiation of MSCs into CAFS, and osteoclastogenesis [47–49]. CD44 and TWIST may play a large role in recruiting MSCs and in stimulating the differentiation of tumor-promoting CAFs in bone.
It is important to note that while these studies have highlighted pro-tumorigenic roles for MSCs in cancer progression, other reports have documented protective roles. For example, MSCs have been found to home to, and potently inhibit, cancer proliferation and angiogenesis by decreasing Akt signaling and causing endothelial cell death via reactive oxygen species, respectively [50]. In contrast, VEGF is produced by MSCs and promotes angiogenesis, MSC migration, and osteoblast differentiation, demonstrating the multiplicity of MSC function [51–53]. Expression of Wnt antagonist Dickkopf-1 (Dkk-1), by MSCs, can suppress breast cancer growth but conversely enhances prostate cancer metastasis [54,55]. Clearly, more investigation is required to define how MSCs contribute to disease progression in different contexts. Complicating this picture is the fact that the “stemness” of MSCs has been difficult to define despite the availability of many markers. Recent studies have also identified that the source of tissue from which stem cells are derived (adipose vs. bone marrow for example) should be a critical consideration for testing the roles of MSCs in cancer progression [56].
Since bone is a major reservoir for MSCs and the MSC niche is thought to be proximal to sinusoidal vasculature, prostate cancer metastases arriving in the bone marrow have a unique opportunity to interact with MSCs. MSC-derived factors such as IL-6 and VEGF directly promote secretion of tumor-derived factors that induce precursor osteoblast proliferation and differentiation (such as endothelin 1 and TGFβ). However, despite the ability of MSCs to differentiate into osteoblasts and prostate cancers to express potent mediators of osteoblast differentiation, no studies to date have examined the interplay between prostate to bone metastases and MSCs in the generation of osteogenic lesions. For example, Wnt7B, a mediator of MSC differentiation, was recently found to be highly expressed by CRPC cells and in osteoblastic xenografts and is important for their proliferation, implicating a dual role for Wnt signaling in promoting MSC-mediated bone formation as well as tumor growth [57]. The differentiation of MSCs into other lineages such as adipocytes for example could also impact prostate cancer progression in bone [58]. Bone is a rich source of adipocytes, and adipokines such as leptin and adiponectin can promote osteoblast differentiation, proliferation, and mineralization while inhibiting osteoclastogenesis [59]. Understanding the roles of MSCs in the prostate cancer-bone vicious cycle will be challenging but has the exciting potential to yield new insights into how prostate metastases establish and grow in bone.
Given the multiple roles MSCs and MSC-derived factors in primary and bone metastatic prostate cancer, a clear opportunity for therapeutic intervention exists (Fig. 2). For example, VEGF is produced by MSCs in large quantities and, in addition to promoting angiogenesis, directly promotes MSC migration and osteoblast differentiation. Thus, targeting of VEGF may inhibit tumor growth as well as the tumor-promoting effects of MSCs in the tumor–bone microenvironment [51–53]. Preclinical studies investigating VEGF-targeted therapies, such as bevasuzimab, have successfully inhibited prostate cancer growth in bone. However, MET expression can be increased by VEGF inhibition; thus, dual targeting of both pathways with carbozantenib, for example, could overcome these effects [60–62]. The use of MSCs for targeted gene therapy is also a potentially powerful way to specifically deliver therapies to prostate to bone metastases, but key to this approach is ensuring that the MSCs do not exacerbate prostate cancer progression in bone. Therapies such as autologous stem cell transplant could be used to isolate individual patient MSCs and induce or manipulate them to express anti-cancer factors. For example, injection of bone marrow-derived MSCs exogenously expressing osteoprotegerin into PC3 prostate cancer lesions significantly inhibited osteoclastogenesis [63]. Further characterization will be required to understand the optimal approach for the therapeutic targeting or use of MSCs in the treatment of prostate to bone metastases.
4 Integrating immune cell functions into the vicious cycle
Compelling evidence has emerged in recent years indicating that immune cell polarization in response to factors in the tumor microenvironment is a powerful regulator of cancer progression and metastasis. The bone marrow is a reservoir for immune cells such as macrophages, myeloid-derived suppressor cells (MDSCs), dendritic cells (DCs), and various T cell subsets that can directly impact the steps of the vicious cycle (Fig. 3).

Megakaryocytes/platelets and the sympathetic nervous system contribute to tumor progression in bone. 1 Megakaryocyte-derived platelets produce tumor-promoting growth factors and cytokines promoting extravasation and colonization of the bone compartment by prostate to bone metastases. 2 Resident megakaryocytes in the bone marrow stimulate osteoblast activity and inhibit osteoclast differentiation by secreting osteoprotegerin. 3 Nerve cells have recently been shown to play major roles in the cancer–bone microenvironment by regulating osteoblast differentiation, osteoclast activity, and tumor growth. Events for potential therapeutic intervention and respective available therapies are highlighted in red
4.1 MDSC and macrophages
In the bone marrow of healthy individuals, naïve immature myeloid cells (iMCs) do not have immune suppressive capabilities [64]. iMCs are poised to rapidly differentiate into mature macrophages, granulocytes, and neutrophils. However, in response to tumor-derived factors (e.g., VEGF, SDF-1, and IL-3), differentiation is blocked, giving rise to MDSCs that have immune suppressive capabilities (widely defined as CD11b+GR1+ in mice), and can be identified in the bone marrow, spleen, blood, and solid tumors [65–67]. MDSCs are defined as a heterogeneous population of activated, immature myeloid cells with the ability to suppress T cells and promote tumor growth [68]. This heterogeneous population of cells can be divided into two major subgroups in mice, granulocytic (CD11b+Ly6G+Ly6Clow) and monocytic MDSCs (CD11b+Ly6G-Ly6Chigh) that differ not only by their phenotype and morphology but also in their mechanism of suppression [68]. In addition to immune suppression, MDSCs can directly promote tumor growth by releasing pro-angiogenic factors and incorporating into developing vasculature [69]. MDSCs can also directly influence the vicious cycle due to their ability to differentiate into bone-resorbing osteoclasts in vivo [70–72]. Although iMC and MDSC can differentiate into osteoclasts in vitro, MDSCs isolated from the bone marrow of tumor-bearing mice were found to be significantly primed for osteoclastogenesis compared to MDSCs derived from non-skeletal tissue [70]. Additionally, independent of their ability to differentiate into osteoclasts, MDSCs produce TGFβ that in turn can further promote cancer cell-derived parathyroid hormone-related protein expression thereby accelerating the vicious cycle. Myeloid-derived TGFβ has been shown to be essential for tumor metastasis, with mice deficient in myeloid-specific TGFβ, showing a significant reduction in metastasis due to interferon (IFN)-γ activation of CD8+ T cells [73]. MDSCs can play major roles in facilitating cancer progression in several sites including the metastatic bone microenvironment. Given the importance of MDSCs in cancer progression, they provide an interesting therapeutic target in the treatment of bone metastasis. Such therapeutic strategies include inhibiting MDSC expansion (e.g., gemcitabine), stimulating their differentiation into mature antigen presenting cells (e.g., transretinoic acid) and inhibiting their function (e.g., Cox-2 inhibitors) [74–77]. These therapies, and others, are currently under investigation as adjuvants for immunotherapies in a variety of cancer types, with a significant enhancement of immune interventions by reversing MDSC-induced immune suppression [78].
Tumor-associated macrophages (TAMs) play pivotal roles in cancer progression and metastasis with the density of TAM infiltration correlating with a poor prognosis for several cancer types, including prostate cancer [79,80]. Naïve macrophages polarize in response to microenvironment signals into an “anti”- or “pro”-tumor phenotypes that have a broad spectrum of functions but are classically defined as M1 and M2 macrophages, respectively. However, in cancer, the majority of TAMS are M2-orientated and largely contribute to tumor immunoevasion via the secretion of anti-inflammatory cytokines such as IL-10 [81]. Independent of their immune regulatory effects, TAMs can promote angiogenesis by expressing proteinases including MMP-9 that in turn regulates the bio-availability of growth factors such as VEGF [82]. Osteal macrophages (osteomacs) also contribute to bone healing [83]. In the context of prostate to bone metastases, the expression of CCL2, also known as monocyte chemoattractant protein-1, by prostate cancer cells promotes the recruitment of TAMs and osteoclast precursors to the prostate bone microenvironment and the growth of the metastases implicating important roles for macrophages in the prostate cancer vicious cycle of bone metastasis [84]. In addition, bone marrow macrophages (BMMs) have been shown to highly express osteogenic-genes CCL2, COX-2, and cathepsin K. The overexpression of cathepsin K by BMMs in bone tumors has been shown to promote tumor progression in a prostate cancer model by upregulating CCL2 and COX-2 pathways in the bone microenvironment [85]. Such potential influences include the stimulation of αvβ3 integrin expression on prostate cancer cells increasing tumor cell migration and invasion by CCL2 [86]. Targeting tumor and macrophage-derived CCL2 significantly increases survival of prostate cancer-bearing mice [87,88]. Preclinical studies are investigating the reprogramming of macrophage polarization from an “M2” to a “M1” phenotype by targeting NFκB and COX-2 as a potential therapeutic strategy [89–91]. Inhibition of NFκB in TAMs resulted in the expression of pro-inflammatory cytokines such as IL-12 that were cytotoxic to tumor cells [89]. Several animal studies have indicated that nitrogen-containing bisphosphonates also reduce pro-angiogenic MMP-9, as well as skewing tumor-associated macrophages to a M1 phenotype, by a yet to be understood mechanism [92,93].
4.2 Dendritic cells
DCs are derived from the myeloid lineage and act as messengers between the adaptive and innate arms of the immune system, specifically by presenting antigens primarily to adaptive T cells. DCs are divided into two main groups: myeloid (mDC) and plasmacytoid (pDC) with the latter resembling plasma cells and expressing large amounts of interferon alpha. Recently, pDCs have been implicated in regulating osteolysis. Primary cancers including prostate are often heavily infiltrated by pDCs [94]. Activation of pDCs led to elevated levels of systemic circulating cytokines such as IL-15, CCL5, and CCL2 that in turn stimulate osteoclast activity in bone [84,95]. These data imply roles for pDCs in the primary tumor microenvironment regulating systemic bone turnover and suggest that pDCs may play a role in the generation of pre-metastatic niches in the bone microenvironment as depletion of the pDCs in vivo also decreases the number of bone metastases [96]. Tumor-derived CXCL12 is important for the recruitment of pDCs to the tumor site, providing a potential therapeutic strategy by inhibiting the CXCR4 receptor (e.g., small molecule inhibitor AMD3100 [97]). Autologous dendritic cell transplant therapies with “educated” DCs (Provenge®) has proven an effective way to stimulate T cell-mediated recognition and attack of the prostate cancer cells, an approach that has been successful in extending the overall survival of prostate cancer patients [94]. It will be interesting to determine if the re-education of the dendritic cells also impacts bone remodeling and prostate to bone metastases.
4.3 T cells
T cell subclasses (e.g., CD4+, CD8+ T cells) originate from HSCs in the bone marrow and are activated in the thymus prior to migrating to their target tissue [98]. T cell control of normal bone homeostasis and osteolytic inflammatory bone diseases such as rheumatoid arthritis and cancer-induced bone disease has been described [99]. In normal bone remodeling, CD4+/CD8+ T cell-derived cytokines such as IL-17 and TNF-α can act in an autocrine manner to stimulate RANKL expression. Thus, T cells negate the requirement for the interaction of osteoclast precursors with osteoblasts and directly stimulate osteoclastogenesis [100]. However, IFN-γ expression by T cells can prevent osteoclastogenesis and therefore is a potential mechanism through which T cells can dampen osteoclastogenesis and bone resorption [101,102]. In the tumor–bone microenvironment, T cells can either promote or protect against cancer progression. Phospholipase C gamma-2 (PLCγ-2−/−) null mice have defects in osteoclast function and an impairment in T cell activation. Surprisingly, the inoculation of tumors into these animals stimulated tumor growth in bone, despite less osteolysis. Rescue experiments in which the delivery of activated CD8+ T cells into PLCγ-2−/− resulted in reduced tumor burden and suggested a protective role for this T cell subset [103]. Patients whose primary tumors have high levels of IFN type I were also found to be protected from bone metastasis. The secretion of IFN type I by tumor cells results in the expansion of CD8+ T cells and NK cells, which is associated with a decrease in metastasis to the bone. The importance of NK cells and CD8+ T cells was confirmed as mice lacking the IFN receptor, or depletion in these cells, had increased bone metastasis [104]. Regulatory T cell (Treg CD4+/CD25+) infiltration of tumors can suppress immune-mediated cytotoxicity and result in an immune-privileged tumor microenvironment. Tregs are recruited to the bone marrow of prostate cancer patients with bone metastasis via CXCR4/CXCL12 signaling, where dendritic cells induce their expansion within the marrow. Tregs inhibit osteoclast differentiation in vitro, and adoptive transfer of Tregs in a mouse model of prostate cancer also increased bone mineral density [105]. The authors suggest that this inhibitory effect of Tregs on osteoclasts may be responsible for the progression of osteoblastic prostate cancer. This interaction between T cells and osteoclasts is not solely one-sided, with osteoclasts capable of influencing T cell function. For example, osteoclasts secrete chemokines (in particular CCL4, CXCL5, CXCL1) that can recruit CD8+ T cells. Upon recruitment, osteoclasts activate the CD8+ T cells via antigen presentation, to express cytokines including IL-2 and IFN-γ. These activated T cells express FOXP3 that in turn inhibits the priming of naive CD8+ T cell activation by dendritic cells. This interaction provides evidence of a feedback loop between osteoclasts and T cells [101]. It has been suggested that patients with bone metastasis may benefit from IFN-γ-based therapies, with mice deficient in IFN-γ receptor, or NK and CD8+ T cells more susceptible to bone metastasis [106]. In addition, treatment with standard of care nitrogen-containing bisphosphonates induces the expansion of a particular subset of γδ T cells in some patients receiving the drugs intravenously. γδ T cells are involved in tumor surveillance and upon target recognition can express pro-inflammatory cytokines that promotes cancer cell elimination. Interestingly, γδ T cells themselves have the potential to inhibit osteoclast formation, via the secretion of IFN-γ [107]. Prostate cancer patients receiving zoledronate treatment that exhibited expansion in γδ T cells were found to have a reduction in disease progression, indicating a potential therapeutic approach [108]. In a similar manner, dendritic cell autologous therapies such as Provenge® can promote the activation of cytotoxic T cells and may prove important in preventing prostate metastases and/or cancer progression in bone.
5 Blood-derived contributors to metastasis to bone
Immune cells such as B cells and mast cells have been described as playing critical roles in metastasis, which is not surprising given the importance of other immune cells in the process [109,110]. Interestingly, cells in the bone marrow microenvironment previously thought to be bystanders such as megakaryocytes have been identified as playing active roles in the vicious cycle of prostate to bone metastases.
5.1 Megakaryocytes
Mature megakaryocytes are primarily located adjacent to blood vessels within the bone, where they migrate or protrude through the endothelium and deliver platelets directly into the circulating blood. Emerging evidence suggests that megakaryocytes can also regulate bone homeostasis. Mice with abnormal megakaryocytopoiesis have high bone content due to an increased number of osteoblasts, but a reduced number of osteoclasts. Megakaryocytes have also been shown to inhibit osteoclast differentiation via the release of soluble factors such as osteoprotegerin (OPG), an endogenous inhibitor of RANKL [111,112]. Additionally, megakaryocytes influence bone formation by stimulating the proliferation and differentiation of osteoblasts via a cell-contact-dependent mechanism suggesting that the number and activity of megakaryocytes within the bone marrow could potentially be important in the prostate to bone metastasis vicious cycle [113]. This potential has recently been explored. In an in vivo prostate cancer model, the expansion of megakaryocytes (following treatment with thrombopoietin) within the bone marrow significantly reduced tumor burden [114]. Further, direct co-culture of megakaryocytes with prostate cancer cells reduced the prostate cancer growth by decreasing proliferation and inducing apoptosis. These data suggest that high numbers of megakaryocytes could play protective roles in prostate cancer to bone metastasis, but it is possible that megakaryocytes could also promote cancer metastasis given the roles of platelets in the process. Several therapies are currently under clinical investigation for megakaryocyte expansion, predominately for treatment of thrombopenia associated with diseases such as acute myeloid leukemia and chronic hepatitis C. These therapies include thrombopoietin mimetics such as Romiplostim that also acts to increase megakaryocyte colony forming cells and Elthombopag that increases megakaryocyte proliferation and maturation [115].
5.2 Platelets
Platelets are small anuclear cells derived from megakaryocytes that function primarily to induce coagulation at sites of tissue injury. Injury-induced platelet activation results in aggregation at the injury site and adhesion to the endothelium (via integrin and selectin interactions) as well as a subsequent release of growth-promoting and wound-healing factors including VEGF, CXCL12, PDGF, IGF, and TGFβ from platelet granules. However, platelet aggregation and interaction with tumor cells enhances invasion and tumor metastasis via the formation of tumor emboli thereby, allowing the evasion of immune surveillance in circulation, adhesion of the emboli to the endothelium, and facilitating extravasation at distant tissue sites [116,117]. Tumor-induced platelet cell activation can promote the release of platelet-derived growth factors that further aid in tumor progression at secondary tissue sites. Understanding the mechanisms that control platelet–cancer cell interaction could offer unique opportunities to prevent prostate to bone metastasis. The platelet membrane glycoprotein complex GPIIb/IIIa (now known as integrin αIIβ3) is the most abundant integrin solely expressed by platelets and megakaryocytes. αIIβ3 facilitates platelet binding to fibronectin and von Willebrand factor on B16 melanoma cells with αIIβ3 blocking antibodies decreasing platelet binding to melanoma cells by ∼60 % and significantly reducing the number of pulmonary metastases [118]. αIIβ3 expression has been implicated as an important facilitator of bone metastasis. Mice that are null for β3 integrin and thus unable to form heteromeric complexes with aIIb and/or av integrins exhibited fewer metastases after intracardiac injection of B16 melanoma cells in immunocompetent mice [119]. Additionally, β3 null platelets significantly abrogated platelet aggregation with circulating tumor cells and resulted in reduced cell “seeding” within bone. There was also a significant reduction in tumor-induced osteolysis, a result of a dual role of β3 integrins in both platelet aggregation and osteoclastogenesis. Expression of E-selectin and integrins by platelet-tumor emboli is critical for prostate cancer extravasation. CCL2, a chemokine that promotes tumor progression in bone via osteoclast recruitment, has recently been shown to induce avβ3 expression, thereby promoting prostate cancer migration [84]. These and other findings highlight the importance of cell adhesion molecules in platelet interaction with both tumor cells and stromal cells in the bone [86,120,121].
Understanding the factors that control platelet activation can lead to therapeutic intervention opportunities. Targeting β3 integrins may prove an effective means at not only preventing metastasis by reducing the ability of cancer cells to form emboli by inhibiting platelet activation and osteoclast-mediated bone resorption. Originally approved for prevention of thrombosis associated with cardiovascular disease, Abciximab (ReoPro®) and XV454 are potent antagonists of platelet GPIIb/IIIa receptors (including avβ3) and have recently been shown to prevent angiogenesis, tumor growth, and metastasis in melanoma and lung cancer models [122]. Therefore, targeting β3 integrins may be an effective means to prevent prostate cancer metastasis and bone lesions. Platelets derived from prostate cancer patients with metastases were found to be more reactive than those with only primary disease and platelet depletion in an orthotopic LNCaP-C42 model of metastatic prostate carcinoma-prevented bone metastasis and tumor-induced osteogenesis [123]. Additionally, platelet ablation reduced serum levels of osteocalcin and alkaline phosphatase in tumor-bearing mice suggesting decreases in bone-matrix turnover. Analysis of platelet granules derived from tumor-bearing mice revealed a significant increase in VEGF and TGFβ, factors both shown to modulate osteoblast maturation and bone formation, as well as several MMPs, including MMP-1, MMP-3, and MMP-13, also known to be important in bone matrix remodeling. Collectively, these studies implicate that cancer induced release of platelet-stored factors contributes to bone remodeling and may be critical for the colonization and progression of prostate cancer to bone metastases. By proxy, the data also suggest indirect roles for megakaryocytes in promoting bone metastasis.
6 Innervation of the vicious cycle
Cancer-associated bone pain is common in patients with prostate to bone metastases since the bone tissue is considerably innervated by both sensory and sympathetic nerve fibers [124,125]. Studies have highlighted that heightened osteoclast activity can lead to acidosis and the resultant protons can stimulate nociceptors on the surface of sensory neurons [126]. Recently, new data have also revealed the importance of neuronal factors and the sympathetic nervous system (SNS) in controlling bone homeostasis and cancer progression in the bone microenvironment.
6.1 Sympathetic nervous system
The SNS has been shown to be primarily involved in early stages of tumor development, with the parasympathetic nervous system important for metastasis to the bone. The density of sympathetic and parasympathetic nerve fibers within and surrounding the human prostate samples is associated with the aggressiveness of the tumor and a poor clinical outcome [127]. Recent studies have highlighted the roles of the SNS and the PNS in driving prostate cancer progression and metastasis [127]. However, once tumor cells metastasize to the bone, the SNS may also play a role in cancer progression. The SNS maintains homeostasis of many essential functions, as well as responding to stress signals as a part of the “fight or flight” response. The role of the sympathetic nervous system in bone homeostasis was first observed due to the effect of adipocyte secreted hormone leptin on decreasing bone mass [128]. Leptin stimulates the sympathetic nerves to release the neurotransmitter noradrenaline that in turn acts on beta-androgenic receptors (βAR) on the osteoblast surface leading to reduced activity. βARs are expressed on a variety of cells involved in the vicious cycle including: tumor cells, osteoclasts, osteoblasts, and macrophages. Activation of the sympathetic nervous system in a stress-induced breast cancer model resulted in a significant increase in the number of osteolytic lesions. This was primarily due to the βAR activation on osteoblasts leading to elevated levels of RANKL [129]. These findings are in agreement with studies illustrating the direct effects of βAR stimulation on osteoclastogenesis via reactive oxidase species [130]. Activation of βAR can also impact prostate cancer cells by inhibiting apoptosis and increasing migration in vitro. Induction of stress in a xenograft model of prostate cancer enhanced cancer growth and could be inhibited via the application of a βAR inhibitor [131]. Targeting the sympathetic nervous system, in particular βAR receptors, with agents such as beta-blockers is likely to significantly contribute to our ability to treat prostate to bone metastases. In several recent retrospective studies, the use of beta-blockers was shown to increase survival in a number of solid tumors, including in prostate cancer patients receiving androgen deprivation therapy [132].
7 Interrogating the expanded vicious cycle
While our understanding of the cellular and molecular factors that control the vicious cycle has become clearer, the degree of complexity as to how the multiple cell types interact with each other over time represents a major challenge to determining the impact of specific growth factors/cytokines or targeted therapies in the progression of prostate to bone metastases. Recent advances in computational modeling, however, and their integration with biological models of the disease may provide a way forward for researchers to reduce background noise and focus in on major processes being utilized by cancer cells to establish and grow in the bone microenvironment.
Computational modeling of cancer has become a practical tool to understanding fundamental principles and specific features in cancer [133]. These models have been proven useful in the development of new hypotheses, in the tackling of complex interactions and testing putative therapies [134,135]. With the increasing power of computer technology, it is now feasible and practical to directly address the interactions between the individual components of the prostate cancer bone microenvironment over clinically relevant periods [136]. In this regard, hybrid cellular automata (HCA) are beginning to be implemented as a means to model key aspects of cancer dynamics. HCA are a class of spatially and temporally discrete dynamic systems [137]. They can incorporate many features of self-organizing, complex systems and are able to exhibit emergent behavior. Cancer models that rely on cellular automata consider the simulation of a biological process that utilizes known biological parameters to inform rules of interaction [138]. Actions in the model are carried out by these predefined rules whose conditions may or may not be satisfied by a given situation but ultimately yield a deterministic outcome that can be predictive and tested biologically. For example, HCAs have been successfully used to model complex cancer phenomena such as angiogenesis and how the stroma impacts the somatic evolution of prostate cancer cells [139,140]. Computational modeling is also being utilized to understand the cellular dynamics of the bone microenvironment, and partial differential equations have been generated that describe the spatiotemporal interactions between osteoblasts, osteoclasts, and osteocytes [141–143]. In this model, RANKL and OPG were considered to be the principal biochemical factors controlling the interactions between the bone cells during trabecular bone remodeling. The output of the model hypothesizes that the spatial localization of RANKL and OPG are critical in directing the bone remodeling unit. Importantly, the model can assess bone formation over clinical relevant periods of time (6 to 12 months) in a short simulation period, a characteristic that would be difficult and expensive to determine using in vivo models. Applying a higher-level paradigm of modeling such as HCAs will allow us to assess the biological stochasticity of multiple cells and factors of interest over time in the setting of prostate to bone metastases. A major challenge, however, is that the design of the computational model is critically dependent on the quality of the biological information used for parameterization. Therefore, utilizing quality clinical and biological empirical information will be key for the development of robust models that can be used to investigate regulators of the vicious cycle of prostate to bone metastases.
8 Conclusions
For several years, the paradigm of the vicious cycle has been limited to three major cell types, namely prostate cancer cells, osteoblasts and osteoclasts, and the factors that facilitate the interaction between these cell types. These studies have yielded important therapeutic targets such as RANKL-based inhibitors and bisphosphonates. In the last 5 years, there has been a significant expansion in our knowledge of the cellular and molecular factors that drive the vicious cycle, especially in regards to stem and immune cell contributions. It stands to reason that if clinical successes such as Zometa and Xgeva® can be born out of studies into the factors that control the vicious cycle, then further dissection of how stem cells, immune cells, and neurons control the process can yield similar successes that not only improve quality of life but significantly extend overall survival. Understanding how multiple cellular interactions occur over time, especially in the presence of putative inhibitors, represents a major challenge given the current biological tools. However, the integration of global system level approaches such as computational model with biological data offers a unique means with which to tackle these complexities and accelerate the discovery of therapies that will cure prostate to bone metastases.
References
Keller, E. T., & Brown, J. (2004). Prostate cancer bone metastases promote both osteolytic and osteoblastic activity. Journal of Cellular Biochemistry, 91(4), 718–729.
Fizazi, K., et al. (2011). Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: a randomised, double-blind study. Lancet, 377(9768), 813–822.
Choudhury, A. D., & Kantoff, P. W. (2012). New agents in metastatic prostate cancer. Journal of the National Comprehensive Cancer Network, 10(11), 1403–1409.
Anonymous. (2013). FDA approves radiopharmaceutical for metastatic prostate cancer. Cancer Discovery, 3(7), OF1.
Paget, S. (1989). The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Reviews, 8(2), 98–101.
Lu, X., et al. (2011). VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging alpha4beta1-positive osteoclast progenitors. Cancer Cell, 20(6), 701–714.
Kang, Y., et al. (2003). A multigenic program mediating breast cancer metastasis to bone. Cancer Cell, 3(6), 537–549.
Sun, Y. X., et al. (2005). Skeletal localization and neutralization of the SDF-1(CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo. Journal of Bone and Mineral Research: The Official Journal of the American Society for Bone and Mineral Research, 20(2), 318–329.
Kaplan, R. N., Psaila, B., & Lyden, D. (2006). Bone marrow cells in the ‘pre-metastatic niche’: within bone and beyond. Cancer Metastasis Reviews, 25(4), 521–529.
Kaplan, R. N., et al. (2005). VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature, 438(7069), 820–827.
Kaplan, R. N., Psaila, B., & Lyden, D. (2007). Niche-to-niche migration of bone-marrow-derived cells. Trends in Molecular Medicine, 13(2), 72–81.
Janowska-Wieczorek, A., et al. (2005). Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. International Journal of Cancer, 113(5), 752–760.
Peinado, H., et al. (2012). Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nature Medicine, 18(6), 883–891.
Pan, B. T., et al. (1985). Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. The Journal of Cell Biology, 101(3), 942–948.
Couzin, J. (2005). Cell biology: the ins and outs of exosomes. Science, 308(5730), 1862–1863.
Johnstone, R. M., et al. (1987). Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). Journal of Biological Chemistry, 262(19), 9412–9420.
Thery, C., Zitvogel, L., & Amigorena, S. (2002). Exosomes: composition, biogenesis and function. Nature Reviews Immunology, 2(8), 569–579.
Schara, K., et al. (2009). Mechanisms for the formation of membranous nanostructures in cell-to-cell communication. Cellular & Molecular Biology Letters, 14(4), 636–656.
Ostrowski, M., et al. (2010). Rab27a and Rab27b control different steps of the exosome secretion pathway. Nature Cell Biology, 12(1), 19–30. sup pp 1–13.
Knudsen, B. S., & Edlund, M. (2004). Prostate cancer and the met hepatocyte growth factor receptor. Advances in Cancer Research, 91, 31–67.
Di Vizio, D., et al. (2012). Large oncosomes in human prostate cancer tissues and in the circulation of mice with metastatic disease. American Journal of Pathology, 181(5), 1573–1584.
Webber, J., et al. (2010). Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Research, 70(23), 9621–9630.
Itoh, T., et al. (2012). Microvesicles released from hormone-refractory prostate cancer cells facilitate mouse pre-osteoblast differentiation. Journal of Molecular Histology, 43(5), 509–515.
Lee, R. J., et al. (2013). A dose-ranging study of cabozantinib in men with castration-resistant prostate cancer and bone metastases. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 19(11), 3088–3094.
Zhang, G., et al. (2012). A delivery system targeting bone formation surfaces to facilitate RNAi-based anabolic therapy. Nature Medicine, 18(2), 307–314.
Chen, Q., & Massague, J. (2012). Molecular pathways: VCAM-1 as a potential therapeutic target in metastasis. Clinical Cancer Research, 18(20), 5520–5525.
Chen, Q., Zhang, X. H., & Massague, J. (2011). Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell, 20(4), 538–549.
Spain, L. M., & Mulligan, R. C. (1992). Purification and characterization of retrovirally transduced hematopoietic stem cells. Proceedings of the National Academy of Sciences of the United States of America, 89(9), 3790–3794.
Matsuzaki, Y., et al. (2004). Unexpectedly efficient homing capacity of purified murine hematopoietic stem cells. Immunity, 20(1), 87–93.
Levesque, J. P., Helwani, F. M., & Winkler, I. G. (2010). The endosteal ‘osteoblastic’ niche and its role in hematopoietic stem cell homing and mobilization. Leukemia: Official Journal of the Leukemia Society of America, Leukemia Research Fund, UK, 24(12), 1979–1992.
Heissig, B., et al. (2002). Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell, 109(5), 625–637.
Wang, Q., et al. (2013). SUMO-specific protease 1 promotes prostate cancer progression and metastasis. Oncogene, 32(19), 2493–2498.
Taichman, R. S., et al. (2002). Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Research, 62(6), 1832–1837.
Yu, C., et al. (2012). Prostate cancer and parasitism of the bone hematopoietic stem cell niche. Critical Reviews in Eukaryotic Gene Expression, 22(2), 131–148.
Shiozawa, Y., et al. (2011). Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. Journal of Clinical Investigation, 121(4), 1298–1312.
Joseph, J., et al. (2012). Disseminated prostate cancer cells can instruct hematopoietic stem and progenitor cells to regulate bone phenotype. Molecular Cancer Research, 10(3), 282–292.
Kim, S. W., et al. (2011). Consistent interactions between tumor cell IL-6 and macrophage TNF-alpha enhance the growth of human prostate cancer cells in the bone of nude mouse. International Immunopharmacology, 11(7), 862–872.
Rojas, A., et al. (2011). IL-6 promotes prostate tumorigenesis and progression through autocrine cross-activation of IGF-IR. Oncogene, 30(20), 2345–2355.
Pusic, I., & DiPersio, J. F. (2010). Update on clinical experience with AMD3100, an SDF-1/CXCL12-CXCR4 inhibitor, in mobilization of hematopoietic stem and progenitor cells. Current Opinion in Hematology, 17(4), 319–326.
de Nigris, F., et al. (2012). CXCR4 inhibitors: tumor vasculature and therapeutic challenges. Recent Patents on Anti-Cancer Drug Discovery, 7(3), 251–264.
Peng, Y., et al. (2012). Peroxisome proliferator-activated receptor alpha plays an important role in the expression of monocyte chemoattractant protein-1 and neointimal hyperplasia after vascular injury. PPAR Research, 2012, 970525.
Li, T., & Wu, Y. (2011). Paracrine molecules of mesenchymal stem cells for hematopoietic stem cell niche. Bone Marrow Research, 2011, 353878.
Pittenger, M. F., et al. (1999). Multilineage potential of adult human mesenchymal stem cells. Science, 284(5411), 143–147.
Karnoub, A. E., et al. (2007). Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature, 449(7162), 557–563.
El-Haibi, C. P., et al. (2012). Critical role for lysyl oxidase in mesenchymal stem cell-driven breast cancer malignancy. Proc Natl Acad Sci U S A, 109(43), 17460–17465.
Jung, Y., et al. (2013). Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nature Communications, 4, 1795.
Spaeth, E. L., et al. (2013) Mesenchymal CD44 expression contributes to the acquisition of an activated fibroblast phenotype via TWIST activation in the tumor microenvironment. Cancer Research, 73, 5347–5359.
Yuen, H. F., et al. (2008). TWIST modulates prostate cancer cell-mediated bone cell activity and is upregulated by osteogenic induction. Carcinogenesis, 29(8), 1509–1518.
Lee, T. K., et al. (2006). Twist overexpression correlates with hepatocellular carcinoma metastasis through induction of epithelial–mesenchymal transition. Clinical Cancer Research, 12(18), 5369–5376.
Otsu, K., et al. (2009). Concentration-dependent inhibition of angiogenesis by mesenchymal stem cells. Blood, 113(18), 4197–4205.
Wang, M., et al. (2007). STAT3 mediates bone marrow mesenchymal stem cell VEGF production. Journal of Molecular and Cellular Cardiology, 42(6), 1009–1015.
Liu, Y., et al. (2012). Intracellular VEGF regulates the balance between osteoblast and adipocyte differentiation. Journal of Clinical Investigation, 122(9), 3101–3113.
Okuyama, H., et al. (2006). Expression of vascular endothelial growth factor receptor 1 in bone marrow-derived mesenchymal cells is dependent on hypoxia-inducible factor 1. Journal of Biological Chemistry, 281(22), 15554–15563.
Qiao, L., et al. (2008). Dkk-1 secreted by mesenchymal stem cells inhibits growth of breast cancer cells via depression of Wnt signalling. Cancer Letters, 269(1), 67–77.
Thudi, N. K., et al. (2011). Dickkopf-1 (DKK-1) stimulated prostate cancer growth and metastasis and inhibited bone formation in osteoblastic bone metastases. Prostate, 71(6), 615–625.
Bianco, P., et al. (2013). The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nature Medicine, 19(1), 35–42.
Zheng, D., et al. (2013). Role of WNT7B-induced noncanonical pathway in advanced prostate cancer. Molecular Cancer Research, 11(5), 482–493.
Hoda, M. R., et al. (2012). The adipocyte-derived hormone leptin has proliferative actions on androgen-resistant prostate cancer cells linking obesity to advanced stages of prostate cancer. Journal of Oncology, 2012, 280386.
Liu, Y., et al. (2013). Novel adipokines and bone metabolism. International Journal of Endocrinology, 2013, 895045.
Sennino, B., et al. (2013). Inhibition of c-Met reduces lymphatic metastasis in RIP-Tag2 transgenic mice. Cancer Research, 73(12), 3692–3703.
Sennino, B., et al. (2012). Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discovery, 2(3), 270–287.
Lee, R. J., & Smith, M. R. (2013). Targeting MET and vascular endothelial growth factor receptor signaling in castration-resistant prostate cancer. Cancer Journal, 19(1), 90–98.
Chanda, D., et al. (2009). Therapeutic potential of adult bone marrow-derived mesenchymal stem cells in prostate cancer bone metastasis. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 15(23), 7175–7185.
Kusmartsev, S., et al. (2004). Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. Journal of Immunology, 172(2), 989–999.
Ueha, S., Shand, F. H., & Matsushima, K. (2011). Myeloid cell population dynamics in healthy and tumor-bearing mice. International Immunopharmacology, 11(7), 783–788.
Lechner, M. G., Liebertz, D. J., & Epstein, A. L. (2010). Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. Journal of Immunology, 185(4), 2273–2284.
Melani, C., et al. (2003). Myeloid cell expansion elicited by the progression of spontaneous mammary carcinomas in c-erbB-2 transgenic BALB/c mice suppresses immune reactivity. Blood, 102(6), 2138–2145.
Yang, L., Edwards, C. M., & Mundy, G. R. (2010). Gr-1+CD11b+ myeloid-derived suppressor cells: formidable partners in tumor metastasis. Journal of Bone and Mineral Research: The Official Journal of the American Society for Bone and Mineral Research, 25(8), 1701–1706.
Yang, L., et al. (2004). Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell, 6(4), 409–421.
Sawant, A., et al. (2013). Myeloid-derived suppressor cells function as novel osteoclast progenitors enhancing bone loss in breast cancer. Cancer Research, 73(2), 672–682.
Zhuang, J., et al. (2012). Osteoclasts in multiple myeloma are derived from Gr-1+CD11b+myeloid-derived suppressor cells. PLoS One, 7(11), e48871.
Danilin, S., et al. (2012). Myeloid-derived suppressor cells expand during breast cancer progression and promote tumor-induced bone destruction. Oncoimmunology, 1(9), 1484–1494.
Pang, Y., et al. (2013). TGF-beta signaling in myeloid cells is required for tumor metastasis. Cancer Discovery, 3(8), 936–951.
Nefedova, Y., et al. (2007). Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Research, 67(22), 11021–11028.
Fujita, M., et al. (2011). COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Research, 71(7), 2664–2674.
Suzuki, E., et al. (2005). Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clinical Cancer Research, 11(18), 6713–6721.
Gabrilovich, D. I., Ostrand-Rosenberg, S., & Bronte, V. (2012). Coordinated regulation of myeloid cells by tumours. Nature Reviews Immunology, 12(4), 253–268.
Iclozan, C., et al. (2013). Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunology, Immunotherapy : CII, 62(5), 909–918.
Qian, B. Z., & Pollard, J. W. (2012). New tricks for metastasis-associated macrophages. Breast Cancer Research, 14(4), 316.
Joyce, J. A., & Pollard, J. W. (2009). Microenvironmental regulation of metastasis. Nature Reviews Cancer, 9(4), 239–252.
Glofcheskie, B. D., & Surgeoner, G. A. (1990). Muscovy ducks as an adjunct for the control of the house fly (Diptera: Muscidae). Journal of Economic Entomology, 83(3), 788–791.
Dirkx, A. E., et al. (2006). Monocyte/macrophage infiltration in tumors: modulators of angiogenesis. Journal of Leukocyte Biology, 80(6), 1183–1196.
Alexander, K. A., et al. (2011). Osteal macrophages promote in vivo intramembranous bone healing in a mouse tibial injury model. Journal of Bone and Mineral Research: The Official Journal of the American Society for Bone and Mineral Research, 26(7), 1517–1532.
Mizutani, K., et al. (2009). The chemokine CCL2 increases prostate tumor growth and bone metastasis through macrophage and osteoclast recruitment. Neoplasia, 11(11), 1235–1242.
Herroon, M. K., et al. (2013). Macrophage cathepsin K promotes prostate tumor progression in bone. Oncogene, 32(12), 1580–1593.
Lin, T. H., et al. (2013). CCL2 increases alphavbeta3 integrin expression and subsequently promotes prostate cancer migration. Biochimica et Biophysica Acta, 1830(10), 4917–4927.
Loberg, R. D., et al. (2007). Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Research, 67(19), 9417–9424.
Loberg, R. D., et al. (2007). CCL2 as an important mediator of prostate cancer growth in vivo through the regulation of macrophage infiltration. Neoplasia, 9(7), 556–562.
Hagemann, T., et al. (2008). “Re-educating” tumor-associated macrophages by targeting NF-kappaB. Journal of Experimental Medicine, 205(6), 1261–1268.
Rolny, C., et al. (2011). HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell, 19(1), 31–44.
Na, Y. R., et al. (2013). Cyclooxygenase-2 inhibition blocks M2 macrophage differentiation and suppresses metastasis in murine breast cancer model. PLoS One, 8(5), e63451.
Veltman, J. D., et al. (2010). Zoledronic acid impairs myeloid differentiation to tumour-associated macrophages in mesothelioma. British Journal of Cancer, 103(5), 629–641.
Coscia, M., et al. (2010). Zoledronic acid repolarizes tumour-associated macrophages and inhibits mammary carcinogenesis by targeting the mevalonate pathway. Journal of Cellular and Molecular Medicine, 14(12), 2803–2815.
Di Lorenzo, G., Buonerba, C., & Kantoff, P. W. (2011). Immunotherapy for the treatment of prostate cancer. Nature Reviews. Clinical Oncology, 8(9), 551–561.
Djaafar, S., et al. (2010). Inhibition of T cell-dependent and RANKL-dependent osteoclastogenic processes associated with high levels of bone mass in interleukin-15 receptor-deficient mice. Arthritis and Rheumatism, 62(11), 3300–3310.
Sawant, A., et al. (2012). Depletion of plasmacytoid dendritic cells inhibits tumor growth and prevents bone metastasis of breast cancer cells. Journal of Immunology, 189(9), 4258–4265.
Zou, W., et al. (2001). Stromal-derived factor-1 in human tumors recruits and alters the function of plasmacytoid precursor dendritic cells. Nature Medicine, 7(12), 1339–1346.
Masopust, D., & Schenkel, J. M. (2013). The integration of T cell migration, differentiation and function. Nature Reviews Immunology, 13(5), 309–320.
Fournier, P. G., Chirgwin, J. M., & Guise, T. A. (2006). New insights into the role of T cells in the vicious cycle of bone metastases. Curr Opin Rheumatology, 18(4), 396–404.
Nakashima, T., & Takayanagi, H. (2009). Osteoimmunology: crosstalk between the immune and bone systems. Journal of Clinical Immunology, 29(5), 555–567.
Kiesel, J. R., Buchwald, Z. S., & Aurora, R. (2009). Cross-presentation by osteoclasts induces FoxP3 in CD8+ T cells. Journal of Immunology, 182(9), 5477–5487.
Takayanagi, H. (2010). New immune connections in osteoclast formation. Annals of the New York Academy of Sciences, 1192, 117–123.
Zhang, K., et al. (2011). CD8+ T cells regulate bone tumor burden independent of osteoclast resorption. Cancer Research, 71(14), 4799–4808.
Bidwell, B. N., et al. (2012). Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nature Medicine, 18, 1224–1231.
Zhao, E., et al. (2012). Regulatory T cells in the bone marrow microenvironment in patients with prostate cancer. Oncoimmunology, 1(2), 152–161.
Bidwell, B. N., et al. (2012). Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat Med, 18(8), 1224–1231.
Pappalardo, A., Thompson, K. (2013). Activated gammadelta T cells inhibit osteoclast differentiation and resorptive activity in vitro. Clinical and Experimental Immunology, 174, 281–291.
Dieli, F., et al. (2003). Induction of gammadelta T-lymphocyte effector functions by bisphosphonate zoledronic acid in cancer patients in vivo. Blood, 102(6), 2310–2311.
DeNardo, D. G., Andreu, P., & Coussens, L. M. (2010). Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity. Cancer Metastasis Reviews, 29(2), 309–316.
Roy, L. D., et al. (2013). Arthritis augments breast cancer metastasis: role of mast cells and SCF/c-Kit signaling. Breast Cancer Research, 15(2), R32.
Bord, S., et al. (2004). Synthesis of osteoprotegerin and RANKL by megakaryocytes is modulated by oestrogen. British Journal of Haematology, 126(2), 244–251.
Kacena, M. A., Gundberg, C. M., & Horowitz, M. C. (2006). A reciprocal regulatory interaction between megakaryocytes, bone cells, and hematopoietic stem cells. Bone, 39(5), 978–984.
Ciovacco, W. A., et al. (2010). Immature and mature megakaryocytes enhance osteoblast proliferation and inhibit osteoclast formation. Journal of Cellular Biochemistry, 109(4), 774–781.
Li, X., et al. (2011). Inhibitory effects of megakaryocytic cells in prostate cancer skeletal metastasis. Journal of Bone and Mineral Research: The Official Journal of the American Society for Bone and Mineral Research, 26(1), 125–134.
Deutsch, V. R., & Tomer, A. (2013). Advances in megakaryocytopoiesis and thrombopoiesis: from bench to bedside. British Journal of Haematology, 161(6), 778–793.
Gay, L. J., & Felding-Habermann, B. (2011). Contribution of platelets to tumour metastasis. Nature Reviews Cancer, 11(2), 123–134.
Erpenbeck, L., & Schon, M. P. (2010). Deadly allies: the fatal interplay between platelets and metastasizing cancer cells. Blood, 115(17), 3427–3436.
Karpatkin, S., et al. (1988). Role of adhesive proteins in platelet tumor interaction in vitro and metastasis formation in vivo. The Journal of Clinical Investigation, 81(4), 1012–1019.
Bakewell, S. J., et al. (2003). Platelet and osteoclast beta3 integrins are critical for bone metastasis. Proceedings of the National Academy of Sciences of the United States of America, 100(24), 14205–14210.
Sottnik, J. L., et al. (2013). Integrin alpha2beta 1 (alpha2beta1) promotes prostate cancer skeletal metastasis. Clinical and Experimental Metastasis, 30(5), 569–578.
Gupta, A., Cao, W., & Chellaiah, M. A. (2012). Integrin alphavbeta3 and CD44 pathways in metastatic prostate cancer cells support osteoclastogenesis via a Runx2/Smad 5/receptor activator of NF-kappaB ligand signaling axis. Molecular Cancer, 11, 66.
King, T. E., et al. (2008). The role of alpha 6 integrin in prostate cancer migration and bone pain in a novel xenograft model. PLoS One, 3(10), e3535.
Kerr, B. A., et al. (2013) Platelets govern pre-metastatic tumor communication to bone. Oncogene, 32, 4319–4324.
Mantyh, P. W., et al. (2002). Molecular mechanisms of cancer pain. Nature Reviews Cancer, 2(3), 201–209.
Elefteriou, F. (2008). Regulation of bone remodeling by the central and peripheral nervous system. Archives of Biochemistry and Biophysics, 473(2), 231–236.
Yoneda, T., et al. (2011). Involvement of acidic microenvironment in the pathophysiology of cancer-associated bone pain. Bone, 48(1), 100–105.
Magnon, C., et al. (2013). Autonomic nerve development contributes to prostate cancer progression. Science, 341(6142), 1236361.
Mistry, T., et al. (2007). Obesity and prostate cancer: a role for adipokines. European Urology, 52(1), 46–53.
Campbell, J. P., et al. (2012). Stimulation of host bone marrow stromal cells by sympathetic nerves promotes breast cancer bone metastasis in mice. PLoS Biology, 10(7), e1001363.
Kondo, H., Takeuchi, S., & Togari, A. (2013). beta-Adrenergic signaling stimulates osteoclastogenesis via reactive oxygen species. American Journal of Physiology, Endocrinology and Metabolism, 304(5), E507–E515.
Hassan, S., et al. (2013). Behavioral stress accelerates prostate cancer development in mice. The Journal of Clinical Investigation, 123(2), 874–886.
Grytli, H. H., et al. (2013). Use of beta-blockers is associated with prostate cancer-specific survival in prostate cancer patients on androgen deprivation therapy. Prostate, 73(3), 250–260.
Macklin, P., & Lowengrub, J. (2007). Nonlinear simulation of the effect of microenvironment on tumor growth. Journal of Theoretical Biology, 245(4), 677–704.
Nagl, S. (2005). Cancer bioinformatics: from therapy design to treatment. Chichester: Wiley.
Wodarz, D., & Komarova, N. L. (2008). Computational biology of cancer: lecture notes and mathematical modeling. Hackensack: World Scientific.
Macal, C. M., & North, M. J. (2010). Tutorial on agent-based modelling and simulation. Journal of Simulation, 4(3), 151–162.
Wolfram, S. (2002). A new kind of science. Champaign: Wolfram Media.
Ribba, B., et al. (2004). The use of hybrid cellular automaton models for improving cancer therapy. Lecture Notes in Computer Science, 3305, 444–53.
Anderson, A. R. A., Chaplain, M. A. J., & Rejniak, K. A. (2007). Single-cell-based models in biology and medicine. Basel: Birkhäuser.
Basanta, D., et al. (2009). The role of transforming growth factor-beta-mediated tumor-stroma interactions in prostate cancer progression: an integrative approach. Cancer Research, 69(17), 7111–7120.
Pivonka, P., & Komarova, S. V. (2010). Mathematical modeling in bone biology: from intracellular signaling to tissue mechanics. Bone, 47(2), 181–189.
Ryser, M. D., Nigam, N., & Komarova, S. V. (2009). Mathematical modeling of spatio-temporal dynamics of a single bone multicellular unit. Journal of Bone and Mineral Research: The Official Journal of the American Society for Bone and Mineral Research, 24(5), 860–870.
Ryser, M. D., Qu, Y., & Komarova, S. V. (2012). Osteoprotegerin in bone metastases: mathematical solution to the puzzle. PLoS Computational Biology, 8(10), e1002703.
Acknowledgments
This work was supported in part by RO1CA143094 (CCL).
Conflict of interest
None
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cook, L.M., Shay, G., Aruajo, A. et al. Integrating new discoveries into the “vicious cycle” paradigm of prostate to bone metastases. Cancer Metastasis Rev 33, 511–525 (2014). https://doi.org/10.1007/s10555-014-9494-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-014-9494-4