
Abstract
Background
Phosphorylated AKT is highly expressed or overexpressed in chemoresistant tumor samples. However, the precise molecular mechanism involved in AKT phosphorylation-related chemoresistance in breast cancer is still elusive. The present research was designed to estimate the effect of AKT phosphorylation on cell viability and chemoresistance in breast cancer.
Methods
We utilized MCF-7 and MDA-MB468 human breast cancer cell lines and developed multidrug-resistant MCF-7/MDR and cisplatin-resistant MDA-MB-468 cells. Immunofluorescence analysis and Western blotting were employed to test the level of glycogen synthase kinase 3 beta (GSK3β), phosphorylated phosphatase and tension homologue (p-PTEN) and phosphorylated AKT (p-AKT) in MCF-7/MDR and MDA-MB468 cells. Xenograft assays in nude mice were performed with MCF-7/MDR cells to verify chemoresistance and the signaling pathway upstream of phosphatidylinositide 3-kinase (PI3K)/AKT.
Results
An increase in GSK3β, p-PTEN and p-AKT expression was strongly induced in MCF-7/MDR and cisplatin-resistant MDA-MB-468 cells, and augmented GSK3β phosphorylation and PTEN inactivation enhanced AKT signaling. The elevation in GSK3β, p-PTEN and p-AKT was associated with cell viability based on a CCK-8 assay. The results of in vivo and in vitro assays indicated that GSK3β knockdown with lentiviral shRNA (shRNA-GSK3β) promoted apoptosis and suppressed the migration of cisplatin-resistant MCF-7/MDR cells, while these effects were reversed by activating p-AKT with the PTEN inhibitor bpV(pic).
Conclusions
AKT phosphorylation mediated by GSK3β and PTEN were correlated with cell viability, migration and apoptosis, which may promote chemoresistance in breast cancer. Furthermore, GSK3β can regulate cell viability through the PTEN/PI3K/AKT signaling pathway and induce chemoresistance, serving as a valuable molecular strategy for breast cancer therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Breast cancer is currently the most prevalent cancer among women in most developed countries [1]. Nearly 22.9% of all female cancers were diagnosed as breast cancer worldwide during the past few years, and the incidence of Chinese cases rose to 12.2% of all recently diagnosed breast cancers in the world [2, 3]. Chemotherapy is the main mode of treatment for breast cancer; however, for nearly half of patients, multidrug resistance (MDR) to chemotherapeutic regimens is a common occurrence during treatment and has become a clinical challenge [4]. Failure to improve treatment success has been attributed to the capacity of breast cancer cells to avoid the effects of conventional chemotherapy. Upon activation, surviving drug-resistant cells can gain the ability to regrow, resulting in tumor relapse and metastasis. Therefore, understanding the potential mechanisms that enable chemotherapeutic resistance of cancer cells, as well as the underlying targets in those pathways, is a key issue on which efforts to improve therapeutic methods should focus. These therapeutic interventions will have a major effect not only on the treatment success rate but also on the tumor recurrence rate [3].
Glycogen synthase kinase 3 beta (GSK3β) is a kinase that regulates apoptosis in response to various stimuli, and it is an important part of the phosphatidylinositol 3-kinase (PI3K)/AKT pathway, the well-studied tumorigenesis mechanism [4]. Studies have demonstrated that the PI3K/AKT pathway plays a key role in the progression and development of most malignancy types. Targeting GSK3β may provide a novel strategy for treating chemoresistant cancers [5]. GSK3β downregulation, for example, resensitized drug-resistant cells to chemotherapy [6, 7]. Additionally, increased GSK3 expression is associated with acquired resistance to paclitaxel in ovarian cancer [5, 7], and inhibition of GSK3β can sensitize human glioblastoma cells to trimetazidine (TMZ) [8]. In our reported research, GSK3β is a negative regulator of the PI3K/AKT signaling pathway, and GSK3β-mediated phosphorylation or deactivated PTEN may bring about deinhibition of PI3K/AKT signaling in macrophage/microglia [9]. These observations and the effects of GSK3β have also been proposed on other multiple cell types [10]. Notably, GSK3β exerts a dual role in chemoresistance, acting as a promoter or a tumor suppressor.
Phosphatase and tensin homolog (PTEN), situated on chromosome 10, is known as one of the most often deleted or mutated genes in various human tumors and functions in transforming phosphatidylinositol-3,4,5-triphosphate (PIP3) into a diphosphate product (PIP2), leading to AKT being negatively regulated [11] and then inhibiting tumor progression by, for example, modulating cell growth, apoptosis, adhesion, migration and invasion. In contrast, when PTEN suppression positively regulates the activity of AKT, many cellular proteins including mTOR, caspase 9, Bad, IKK, and GSK3β [12]. Akt phosphorylates GSK3β on serine 9 and GSK3α on serine 21 to inhibit their kinase activity [12].
It was previously suggested that PTEN mutation in prostate cancer cells induces AKT activation and restrains GSK3β [13]. Here, we explored the role of GSK3β in mediating chemoresistance in MCF-7/MDR breast cancer cells. The results indicated that GSK3β induces PTEN phosphorylation, resulting in activation of AKT in MCF-7/MDR cells. The AKT kinase activates a myriad of downstream targets that advance tumor growth, survival, and chemoresistance.
Methods
Cell culture
The wild-type human breast cancer cell lines MCF-7, MCF-7/MDR and MDA-MB468 were obtained from the Cell Bank of the Institute of Biochemistry and Cell Biology (Shanghai, China) in 2016. The cisplatin-resistant cell line MDA-MB468 was derived from the MDA-MB468 cell line. The short tandem repeat (STR) profiling data were identified to prove the multidrug-resistant MCF-7/MDR and cisplatin-resistant MDA-MB-468 cells derived from the appropriate parent cell line by Shanghai Biowing Applied Biotechnology Co. Ltd. These cells lines were preserved in Dulbecco’s modified Eagle’s medium (Gibco, Waltham, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco, Waltham, USA) at 37 °C in 5% CO2. All cell lines were freshly thawed every 2 months and used within 20 passages. The cell lines were confirmed to be mycoplasma-free through detection of their morphology and maturation profiles. And a periodic mycoplasma detection test must be performed in every cell line manipulated using real time PCR detection as described by Corral-Va´zquez et al. in our laboratory [14].
Real-time quantitative PCR
Trizol reagent (Invitrogen, Carlsbad, USA) was applied to extract total RNA from the cells and tissue specimens. Real-time quantitative PCR (qPCR) was performed in a total volume of 20 μL of SYBR Green PCR Master Mix (Roche, Germany). All reactions were performed in duplicate. The mRNA expression levels of PTEN, AKT and GSK3β were normalized to GAPDH mRNA levels using the 2−ΔΔCT method. The PCR primers were synthesized at Invitrogen, and the sequences for PTEN, AKT, GSK3β and GAPDH were as follows: GAPDH, GCACCGTCAAGGCTGAGAAC and TGGTGAAGACGCCAGTGGA; GSK3β, ACCAATATTTCCTGGGGACA and GTGCCTTGATTTGAGGGAAT; PTEN, TTTGAAGACCATAACCCACCAC and ATTACACCAGTTCGTCCCTTTC; and AKT, GTCATCGAACGCACCTTCCAT and AGCTTCAGGTACTCAAACTCGT.
Western blotting
Cells were washed with ice-cold phosphate-buffered saline, and lysed on ice for 30 min in lysis buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% Triton X-100, 10% glycerol) supplemented with protease and phosphatase inhibitors (2 mM AEBSF, 0.3 μM Aprotinin, 0.13 mM Bestatin, 14 μM E64, 0.01 mM Leupeptin, 5 mM sodium fluoride, 1 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM sodium orthovanadate; P1045, Beyotime Institute of Biotechnology) at 4 °C. The lysis mixture was centrifuged at 13,200 rpm for 10 min at 4 °C, and the supernatant containing cellular proteins was boiled with Laemmli (SDS)-sample buffer. After separation on a 12% SDS-PAGE gel, proteins were transferred onto a nitrocellulose membrane (Millipore, USA). After blocking with 5% nonfat milk, the membrane was incubated with anti-phosphorylated-AKT (P-AKT(T308)) (1:2000, Cell Signaling Technology, Danvers, USA), anti-PTEN (1:1000, Cell Signaling Technology, Danvers, USA), anti-p-PTEN (Thr366) (1:500, Merck Millipore, Billerica, MA, USA), anti-GSK3β (1:1000, Cell Signaling Technology, Danvers, USA) or anti-β-actin (1:8000, Sigma Aldrich) antibodies. The membrane was then washed adequately and incubated with a secondary antibody (Jackson, West Grove, USA). The proteins were observed with ECL detection reagents. Experiments were repeated for three times.
Infection with lentiviral shRNA
Lentiviral shRNA targeting human GSK3β (Santa Cruz Biotechnology, Dallas, USA); mouse bpv(pic) (Santa Cruz Biotechnology, Dallas, USA); and lentivirus containing a control, nontargeting sequence of shRNA (Santa Cruz Biotechnology, Dallas, USA) with titers of ~ 5 × 103 infectious units per μL were purchased from Santa Cruz Biotechnology. Cells were infected with 1.5 μL of virus per well in six-well plates for 48 h, and knockdown of GSK3β was verified by Western blot and qPCR analyses.
Immunofluorescence
Round glass slides were coated with gelatin and laid on the bottom of a 24-well plate wells. Cells were cultivated at a density of 1 × 104 cells per well. Then, the slides were first incubated with primary antibody against mouse GSK3β (1:200, Cell Signaling Technology, Danvers, USA) in a humidified chamber at 4 °C overnight. Goat anti-rabbit secondary antibody (labeled with Alexa488, 1:1000 dilution; Jackson, West Grove, USA) was added and incubated with cells for 1 h at 37 °C. After three washes, nuclei were counterstained with DAPI. Immunostaining was visualized with a confocal laser scanning microscope (SP8, Leica, Germany).
Immunohistochemical staining
Sliced specimens were incubated at 58 °C for ~ 3 h, and subjected to xylene treatment for dewaxing, hydrated through alcohol gradients, and washed with PBS. Antigen was repaired by using microwave repair at 650 W for 20 min and cooled to room temperature. After treatment with 3% H2O2 and normal goat serum to block the activity of endogenous catalase and nonspecific binding for 10 min, the sections were incubated with p-AKT (1:200, Cell Signaling Technology) at 4 °C overnight. Each section was then incubated with biotin labeled goat anti-rabbit immunoglobulin G for 10 min at 37 °C and streptavidin biotin–peroxidase complex methods were employed (Zhongshan Biotechnology Company, Beijing, China). Counterstained with hematoxylin when positive expression was observed in 10 min under microscope. Simultaneously, negative control was performed without primary antibody. Four samples in each group were selected for observing the distribution and change pattern of immunohistochemical staining in the periodontal tissues under a light microscope. Moreover, five visual fields were randomly chosen in the compression side and tension side for analysis by Image-Pro Plus (Media Cybernetics, Silver Spring, USA).
Cell Counting Kit-8 (CCK-8) assay
CCK-8 (Dojindo Molecular Technologies, Kumamoto, Japan) assays were applied to assess cell proliferation following the manufacturer’s instructions. MCF-7/MDR and MCF-7 cells were seeded in 96-well culture plates (3000 cells per well). After treatment for 24 h, 100 µL of 10% CCK-8 reagent diluted in DMEM was supplemented into each well and incubated with the cells for another 1 h. The absorbance was calculated for each well at 450 nm with a Microplate Spectrophotometer (Multiskan™ GO, Thermo Fisher Scientific). The results are displayed as the mean of three more independent experiments.
Wound-healing cell migration assay
MCF-7/MDR and MCF-7 cells were grown to confluence in 6-well plates, and the cell monolayer was then scratched with the narrow end of a sterile 200 μL pipette tip. The medium was promptly replaced to eliminate floating cells and exchanged with DMEM containing 5% FBS. The width of the scratch was measured at two points in each well after initial wounding. The cells were incubated for 24 h at 37 °C in a CO2 incubator, and then, the scratch width was remeasured. The relative motility and migration ability of the cells into the cell-free zone is expressed as the normalized percent change in the scratch width after 24 h.
Nude mice for xenograft assays and evaluation of response to cisplatin
Twenty BALB/c background female nude mice (7–8 weeks old, 18–22 g body weight; Shanghai Experimental Animal Center, Chinese Academy of Sciences, China) were fed and maintained in air conditioned, light-controlled animal facilities. All animal care and experimental protocols were carried out according to the Chinese Animal Management Rules of the Ministry of Health and were authorized by the Animal Ethics Committees of Nantong University. Mice were randomly assigned to five groups (Cisplatin + bpv(pic), Cisplatin + control, Cisplatin + shRNA-GSK3β, Vehicle + bpv(pic), and Vehicle + shRNA-GSK3β groups) and intraperitoneally anesthetized with 10% chloral hydrate (0.2 mL/20 g body weight). MCF-7/MDR (1 × 106) cells per mouse were subcutaneously injected into the left dorsal flank with an abundant blood supply. Each group contained 4 mice. Mice were weighed, and subcutaneous tumor growth was then assessed biweekly by measurement of the tumor diameters with a Vernier caliper. Tumor volume was determined by the ellipsoid volume calculation formula: D × d2/2, where D and d are the longest and shortest diameters, respectively. When tumors were visible (≥ 100 mm3), an i.p. cisplatin injection (Nuo Xin, Jiangsu Hausen Pharmaceutical Co., Ltd. production, Chinese medicine Zhunzi H 20040813) was administered at its optimal dose based on a schedule of three times (5 mg/kg per injection) every seventh day. The vehicle group was injected with equal volumes of saline, using the same injection schedule. The five groups of nude mice were administered negative control shRNA lentivirus (1.5 × 104 infectious units/mL, 0.5 mL), PTEN inhibitor bpV(pic) (1 μM, 0.5 mL) or lentiviral shRNA-GSK3β (1.5 × 104 infectious units/mL, 0.5 mL) with and without drug treatment via intratumoral injection. Mice were killed by anesthesia overdose with subsequent cervical dislocation for tumor removal, and tumors were harvested for immunohistochemical and other analyses.
Statistical analysis
Statistical calculations were performed using the SPSS 17.0 software package (SPSS, Inc., Chicago, IL, USA). Two tailed Student’s t test was used to evaluate differences between two groups. One-way analysis of variance (ANOVA) was carried out to compare the mean results for greater than or equal to three groups. The data are displayed as the mean ± standard deviation (SD), and p < 0.05 was deemed to indicate a statistically significant difference.
Results
Evaluation of chemoresistant MCF-7/MDR cells
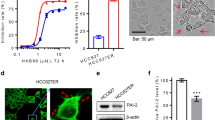
To test the chemoresistance of these cells, we treated them with the indicated concentrations of cisplatin (from 1 to 8.1 nM) and performed CCK-8 assays. As shown in Fig. 1, the 50% inhibitory concentration (IC50) in MCF-7 wild-type cells was 0.7425 ± 0.0183 and in MDA-MB-468 cells was 4.076 ± 0.578; however, the values increased to 1.075 ± 0.031 and 17.28 ± 1.86, respectively, in the corresponding resistant-type cells. The results indicated that MCF-7/MDR and cisplatin-resistant MDA-MB-468 cells were less sensitive to cisplatin chemotherapy.

Effect of cisplatin on the growth of different human breast cancer cell lines. a and c MCF-7/MDR and MCF-7 cells, as well as MDA-MB-468 and cisplatin-resistant MDA-MB-468 cells, were incubated with cisplatin for 48 h. A CCK-8 assay was performed to measure the cell survival rate of both breast cancer cell types exposed to different doses of cisplatin (0, 0.1, 0.3, 0.9, 2.7, 8.1 ng/ml). n = 6; *p < 0.05; **p < 0.01. b and d Colony formation ability and cell growth ability of MCF-7 and MCF-7/MDR cells, as well as MDA-MB-468 and cisplatin-resistant MDA-MB-468 cells, after treatment with cisplatin (0.9 ng/ml). n = 4; *p < 0.05; **p < 0.01
GSK3β, p-PTEN and p-AKT were highly expressed in chemoresistant MCF-7/MDR cells
To investigate whether GSK3β, p-PTEN and p-AKT were highly expressed in chemoresistant-type MCF-7/MDR cells, we performed an immunofluorescence analysis of GSK3β in MCF-7/MDR and MCF-7 cells. The results showed that the expression of GSK3β, p-PTEN and p-AKT was significantly increased in MCF-7/MDR cells in contrast with MCF-7 cells (Fig. 2a, b). Moreover, western blotting was used to analyze the expression levels of GSK3β, p-PTEN and p-AKT (Fig. 2c). We found that the average GSK3β, p-PTEN and p-AKT protein levels in MCF-7/MDR cells were significantly greater than those in MCF-7 cells (Fig. 2d; p < 0.05 or p < 0.01). Similar increases were also found in cisplatin-resistant MDA-MB-468 cells (Supplemental Fig. 1). Together, these results suggested that the expression of p-PTEN and p-AKT was correlated with the increase of GSK3β expression in chemoresistant MCF-7/MDR cells and cisplatin-resistant MDA-MB-468 cells.

Expression level of GSK3β, p-PTEN and p-AKT in chemoresistant MCF-7/MDR cells. a Immunofluorescence analysis of GSK3β in chemoresistant MCF-7/MDR cells. Blue: DAPI; red: GSK3β labeled with Alexa Fluor 594-conjugated secondary antibody. Scale bars, 20 μm. b The fluorescence intensities of GSK3β quantified using ImageJ software are presented. c Western blot image of p-AKT, p-PTEN and GSK3β in wild-type and chemoresistant types of MCF-7/MDR cells. β-Actin was applied as the loading control. d The relative band intensities were quantified using ImageJ software and are presented as the ratio of p-AKT, p-PTEN and GSK3β to β-actin. The values are the mean ± SD (n = 3). Statistical significance was established with a two-tailed t test; *p < 0.05, and **p < 0.01 versus wild-type cells
The increase in GSK3β expression and inactivated PTEN enhanced phosphatidylinositide 3-kinase (PI3K)/AKT signaling
To establish whether the expression level of GSK3β, p-PTEN and p-AKT was correlated with cell viability, we manipulated the GSK3β and p-PTEN level via GSK3β knockdown with lentiviral shRNA (shRNA-GSK3β) to promote apoptosis and suppress the migratory ability of MCF-7/MDR cells, and these effects were reversed by p-AKT activation with the PTEN inhibitor bpV(pic).
First, Western blotting showed that shRNA-GSK3β decreased the level of GSK3β, whereas bpV(pic) decreased the level of PTEN in a dose-dependent manner in MCF-7/MDR cells (Fig. 3a, b). To measure the function of AKT phosphorylation downregulation and upregulation of the viability and proliferation of chemoresistant MCF-7/MDR cells, CCK-8 assays were performed. The CCK-8 assay results showed that GSK3β downregulation significantly suppressed the viability of the chemoresistant MCF-7/MDR cells compared with PTEN downregulation (Fig. 3c, p < 0.05 or p < 0.01). These results indicated that the increase in GSK3β expression and inactivated PTEN-enhanced PI3K/AKT signaling.

Effect of GSK3β downregulation manipulated through PTEN upregulation on cell viability in chemoresistant MCF-7/MDR cells. a Western blot analysis of GSK3β, p-PTEN and p-AKT in chemoresistant MCF-7/MDR cells, which shows the response to lentiviral shRNA-GSK3β and the PTEN inhibitor bpV(pic). β-Actin was used as the loading control. b The relative band intensities were quantified using ImageJ software, and the ratio of GSK3β, p-PTEN and p-AKT to β-actin is presented. The values are the mean ± SD (n = 3; *p < 0.05, and **p < 0.01 versus wild-type cells). c MCF-7/MDR cells were plated in liquid culture and treated with vehicle control, shRNA-GSK3β (1.0 μL/ml of virus), or bpv(pic) (1 μM) for 48 h. CCK-8 assays were then carried out to assess the viability of cells treated with cisplatin (0, 0.1, 0.3, 0.9, 2.7, 8.1 ng/ml) for 48 h to determine the shape of the dose–response curve. d Real-time quantitative PCR analysis of GSK3β, PTEN and AKT mRNA expression levels after each cell treatment. mRNA expression was normalized to that of GAPDH. The means of three independent biological replicates are presented, and error bars represent the SD. All experiments were performed three times. Significant differences are indicated with an asterisk
GSK3β regulated the migration potential of chemoresistant MCF-7/MDR cells
To further investigate the effect of GSK3β on the migration of chemoresistant MCF-7/MDR cells, a wound-healing cell migration assay was performed. The results showed that the number of migratory cells in the GSK3β downregulation group was significantly reduced compared with that in the PTEN inhibitor bpV(pic) group (Fig. 4a, b, p < 0.05). These results revealed that downregulation of AKT phosphorylation correlated with the migratory ability of chemoresistant MCF-7/MDR cells.

Effect of AKT phosphorylation manipulated via lentiviral shRNA-GSK3β and the PTEN inhibitor bpV(pic) on migration in chemoresistant MCF-7/MDR cells. a Representative images of migration assays. Migration assays were performed using a 6-well transwell system. Chemoresistant MCF-7/MDR cells were transfected with GSK-3β shRNA or treated with bpV(pic) for 48 h. b The scratch width was remeasured, and the ability of the cells to migrate into the cell-free zone (relative motility) is presented as the relative fold change in the scratch width after 48 h. n = 4; *p < 0.05; **p < 0.01
GSK3β regulated the chemoresistance potential in nude mice
To observe whether shRNA-GSK3β decreased the tumorigenic properties of breast cancer cell lines, xenograft transplantation into nude mice was performed. Within 10–14 days after subcutaneous injection of MCF-7/MDR cells, palpable subcutaneous tumors developed in all 12 nude mice at the site of inoculation. The dimensions of these tumors ranged from 1.5 to 2.0 cm within 5 weeks postinjection. The growth and size of the tumors were significantly higher in the bpv(pic) group with or without cisplatin treatment than in the control group (Fig. 5a, b, p < 0.01). However, inhibition of GSK3β expression by shRNA-GSK3β decreased the volume of tumors (Fig. 5a, b, p < 0.05 or p < 0.01), with no apparent effects on body weight, suggesting a lack of toxicity (Fig. 5c). In summary, GSK3β augmented the chemoresistance properties of breast cancer cell lines by inducing PTEN phosphorylation.

Effect of AKT phosphorylation manipulated via shRNA-GSK3β and bpv(pic) on the chemoresistant MCF-7/MDR cells transplanted into nude mice with and without cisplatin treatment. a Tumor xenografts in the bpv(pic)-treated group were larger than those in the shRNA-GSK3β- and control-treated groups with and without cisplatin treatment. b Volume of xenograft tumors in the Cisplatin + bpv(pic), Cisplatin + control, Cisplatin + shRNA-GSK3β, Vehicle + bpv(pic), and Vehicle + shRNA-GSK3b groups respectively. c Weight of xenograft tumors in the control, bpv(pic), and shRNA-GSK3β groups. d Representative immunohistochemical images of p-AKT expression in xenograft tumors. The p-Akt protein was expressed in the cytoplasm or nucleus, and the positive expres-sion was visualized as brownish yellow particles. Cells were considered negative when their staining (tan to light brown) was equal to background or not greater than the staining produced by nonspecific DAB adsorption. Cells were rated weakly positive when they appeared from light to medium brown and were darker than nonspecific DAB staining. Strongly positive cells were stained dark brown to black. e Quantitative analysis showing the mean optical density (MOD) of AKT expression in xenograft tumors; the results are presented as the mean ± SD. n = 4; *p < 0.05; **p < 0.01
AKT was overexpressed in MCF-7/MDR bpv(pic) cell lines and tissues
Here, we tested AKT expression in MCF-7 and MCF-7/MDR cells. As shown in Figs. 1 and 2b, based on Western blot analysis, we found that AKT was overexpressed in MCF-7/MDR cell lines compared with normal cells, showing that AKT may play a vital role in the chemoresistance of breast cancer. Thus, we assessed GSK3β and PTEN expression and studied AKT expression in MCF-7/MDR cells transplanted into nude mice (Fig. 5d, e). Immunohistochemical staining analysis was used to detect the difference between these treated groups. The p-Akt protein was expressed in the cytoplasm or nucleus, and the positive expression was visualized as brownish yellow particles by immunohistochemical staining, as shown in Fig. 5d. By data analysis of mean optical density, it was shown that higher volumes of tumors associated with highly positive AKT levels (Fig. 5e, p < 0.05 or p < 0.01). These results demonstrated that AKT is highly expressed in chemoresistant cancer cell lines and that knockdown of GSK3β decreased the expression of AKT in tumor tissues compared with that in the control group.
Discussion
The widespread malignancy of breast cancer deserves deeper investigation. In recent decades, progress in breast cancer therapy has occurred; however, drug resistance is still the primary clinical obstacle. To improve the current chemotherapy program, a greater understanding of potential chemoresistance mechanisms is indispensable. There is increasing evidence showing that GSK3β is aberrantly activated in different types of cancer, including breast cancer, and thus GSK3β has become a new potential treatment target. Aberrant expression of GSK3β has been reported to be relevant to survival, immortalization, proliferation and invasion of tumor cells, and renders them resistant or insensitive to radiation and chemotherapeutic agents [15]. Based on other studies, GSK3β inhibition permits therapy-resistant colon and pancreatic cancer cells to become susceptible to 5-fluorouracil (5-FU) [16] and renal cell cancer cells to become sensitive to a synthetic multikinase inhibitor (sorafenib) that targets growth and angiogenesis signaling [17]. Generally, these studies propose that GSK3β exists in various cancer types and is involved in multiple molecular pathways used to escape chemotherapy, radiotherapy and targeted therapies.
As a serine/threonine kinase, AKT phosphorylates a host of cellular proteins, including GSK3α, GSK3β, FoxO transcription factors, MDM2, BAD and p27KIP1, to promote survival and cell cycle entry [18]. PTEN is a lipid phosphatase that negatively activates the AKT pathway, slowing tumor progression. MAF1-induced PTEN activation was found to depress AKT/mTOR signaling in liver cancer [19]. Additionally, the tumor-suppressor PTEN was disabled, and the subsequent activation of the PI3K/AKT pathway induced cancer aggressiveness in human prostate cancer [19].
Here, we discovered that the activity of GSK3β was significantly upregulated in MCF-7/MDR cells. In addition, a negative correlation between PTEN and GSK3β was found. The increase of GSK3β then phosphorylates PTEN at Thr366, leading to a loss of PTEN activity. This loss of PTEN function results in activation of Akt, because PIP3 is no longer converted to PIP2. Notably, knockdown of GSK3β abolished the increase of phosphorylation of PTEN, and then promoted apoptosis and suppressed the migration of cisplatin-resistant MCF-7/MDR cells, confirming that inactivation of PTEN is dependent on GSK3β. It was also indicated that GSK3β knockdown with lentiviral shRNA (shRNA-GSK3β) promoted apoptosis and suppressed the migration of cisplatin-resistant MCF-7/MDR cells, while these effects were reversed by activating p-AKT with the PTEN inhibitor bpV(pic). Therefore, we proposed that GSK3β induces PTEN phosphorylation, resulting in AKT activation in MCF-7/MDR cells that leads to increased migratory and invasive properties and the development of chemoresistance, probably contributing to cancer progression.
Our next step was to investigate the molecular mechanisms associated with PTEN repression. It has been reported that up-regulating GSK3β inactivated PTEN through phosphorylation, then promoting PI3K/Akt signaling on multiple cell types [9, 10, 20]. The biological activity of PTEN toward anionic lipid substrates is considered to be dependent on its phosphorylation status at various residues between Ser-362 and Ser-385, located in the PTEN C-terminal tail, with phosphorylation maintaining PTEN in an inactive state sites of phosphorylation. Previous studies indicated that the sites of phosphorylation, Ser-385 and Ser-370, may be phosphorylated by protein kinase CK2 (17, 18, 23), and Thr-366 may be phosphorylated by GSK3β on the multiple cell types. We performed Western blotting to analyze alterations in the expression of GSK3β in MCF-7 cells and MCF-7/MDR cells. Aberrant activation of PTEN signaling was found following an increase in GSK3β. Consistent with our findings, previous reports showed that GSK3β may be a positive regulator of cancer cell proliferation and survival [21, 22], further supporting GSK3β as a therapeutic target against cancer. A previous report demonstrated that GSK3β-dependent signal transduction cascades, which underlie the PTEN/PI3K/AKT axis, may modulate the protection of oligodendrocytes via HDAC inhibition [9]. Recent reports suggested that GSK3β could be a novel target for treatment of human leukemia and pancreatic, colon, bladder and renal cancer [23,24,25] and plays an important role in human breast cancer. Excessive GSK3β expression has been shown to be related to several indicators of poor prognosis, and breast cancer patients with GSK3β expression in the highest quartile (246 of 1686 cases) exhibited a 2.7- and 1.7-fold increased risk of distant relapse at 5 and 10 years after tumor resection, respectively [26].
Typically, GSK3β is recognized as a latent downstream gene product of AKT because its mRNA and protein levels were both increased with PTEN. Furthermore, AKT facilitated GSK3β phosphorylation, which is more likely to suppress cytoplasmic PTEN, while PTEN is a negative regulator of the PI3K/AKT signaling pathway [9]. Therefore, GSK3β-mediated phosphorylation/restraint of PTEN may result in derepression of PI3K/AKT signaling, leading to significant attenuation of cell proliferation, invasion and migration.
Interestingly, GSK3β is also regulated by the activity of AKT, and activated GSK3β (the nonphosphorylated state) is reported to modulate cell cycle and apoptosis [27]. AKT can prevent the apoptotic activity of GSK3β through its phosphorylation [28]. It was suggested that AKT can suppress the expression of GSK3β, which further increases the expression of PTEN, and it functions in converting PIP3 into a PIP2, thereby negatively regulating the activity of AKT through a positive feedback loop. Whether this is a general mechanism conserved in multidrug-resistant breast cancer remains to be further established.
In brief, the results of this study reveal the signaling pathway triggered by GSK3β, subsequently altering the activation of PI3K/AKT. GSK3β induces PTEN phosphorylation, resulting in activation of AKT in MCF-7/MDR cells. AKT activates a myriad of downstream products that promote tumor growth, survival, and chemoresistance (Fig. 6). This supports the therapeutic potential of GSK3β against breast cancer and supplies novel potential targets for the control of chemoresistance in breast cancer.

Illustration of the mechanism underlying negative GSK3β regulation of the activity of AKT. PI3K converts phosphatidyl (4,5)-biphosphate (PIP2) to phosphatidylinositol (3,5)-triphosphate (PIP3), thus promoting the activation of the prosurvival kinase AKT and leading to chemotherapeutic resistance. PTEN counterbalances this effect by converting PIP3 back to PIP2. GSK3β activation then phosphorylates PTEN at Thr366, resulting in a loss of PTEN activity. Because PIP3 is no longer being converted to PIP2, this missing of PTEN function leads to increased AKT activation. Activated AKT also phosphorylates GSK3β, thereby depressing its chemoresistance effect. Knockdown of GSK3β prevents the chemoresistance effects of AKT, whereas the PTEN inhibitor bpV(pic) exerted an additional effect over that of AKT
References
Hong W, Dong E (2014) The past, present and future of breast cancer research in China. Cancer Lett 351(1):1–5. https://doi.org/10.1016/j.canlet.2014.04.007
Fan L, Strasser-Weippl K, Li JJ, St Louis J, Finkelstein DM, Yu KD, Chen WQ, Shao ZM, Goss PE (2014) Breast cancer in China. Lancet Oncol 15(7):e279–e289. https://doi.org/10.1016/S1470-2045(13)70567-9
Ghebeh H, Al-Khaldi S, Olabi S, Al-Dhfyan A, Al-Mohanna F, Barnawi R, Tulbah A, Al-Tweigeri T, Ajarim D, Al-Alwan M (2014) Fascin is involved in the chemotherapeutic resistance of breast cancer cells predominantly via the PI3K/Akt pathway. Br J Cancer 111(8):1552–1561. https://doi.org/10.1038/bjc.2014.453
O’Driscoll L, Clynes M (2006) Biomarkers and multiple drug resistance in breast cancer. Curr Cancer Drug Targets 6(5):365–384
Luo J (2009) Glycogen synthase kinase 3beta (GSK3beta) in tumorigenesis and cancer chemotherapy. Cancer Lett 273(2):194–200. https://doi.org/10.1016/j.canlet.2008.05.045
Grassilli E, Ianzano L, Bonomo S, Missaglia C, Cerrito MG, Giovannoni R, Masiero L, Lavitrano M (2014) GSK3A is redundant with GSK3B in modulating drug resistance and chemotherapy-induced necroptosis. PLoS ONE 9(7):e100947. https://doi.org/10.1371/journal.pone.0100947
Hilliard TS, Gaisina IN, Muehlbauer AG, Gaisin AM, Gallier F, Burdette JE (2011) Glycogen synthase kinase 3beta inhibitors induce apoptosis in ovarian cancer cells and inhibit in vivo tumor growth. Anticancer Drugs 22(10):978–985. https://doi.org/10.1097/CAD.0b013e32834ac8fc
Pyko IV, Nakada M, Sabit H, Teng L, Furuyama N, Hayashi Y, Kawakami K, Minamoto T, Fedulau AS, Hamada J (2013) Glycogen synthase kinase 3beta inhibition sensitizes human glioblastoma cells to temozolomide by affecting O6-methylguanine DNA methyltransferase promoter methylation via c-Myc signaling. Carcinogenesis 34(10):2206–2217. https://doi.org/10.1093/carcin/bgt182
Wang G, Shi Y, Jiang X, Leak RK, Hu X, Wu Y, Pu H, Li WW, Tang B, Wang Y, Gao Y, Zheng P, Bennett MV, Chen J (2015) HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3beta/PTEN/Akt axis. Proc Natl Acad Sci USA 112(9):2853–2858. https://doi.org/10.1073/pnas.1501441112
Cohen P, Goedert M (2004) GSK3 inhibitors: development and therapeutic potential. Nat Rev Drug Discov 3(6):479–487. https://doi.org/10.1038/nrd1415
Vazquez F, Matsuoka S, Sellers WR, Yanagida T, Ueda M, Devreotes PN (2006) Tumor suppressor PTEN acts through dynamic interaction with the plasma membrane. Proc Natl Acad Sci USA 103(10):3633–3638. https://doi.org/10.1073/pnas.0510570103
Farina AK, Bong YS, Feltes CM, Byers SW (2009) Post-transcriptional regulation of cadherin-11 expression by GSK-3 and beta-catenin in prostate and breast cancer cells. PLoS ONE 4(3):e4797. https://doi.org/10.1371/journal.pone.0004797
Ohigashi T, Mizuno R, Nakashima J, Marumo K, Murai M (2005) Inhibition of Wnt signaling downregulates Akt activity and induces chemosensitivity in PTEN-mutated prostate cancer cells. Prostate 62(1):61–68. https://doi.org/10.1002/pros.20117
Corral-Vazquez C, Aguilar-Quesada R, Catalina P, Lucena-Aguilar G, Ligero G, Miranda B, Carrillo-Avila JA (2017) Cell lines authentication and mycoplasma detection as minimun quality control of cell lines in biobanking. Cell Tissue Banking 18(2):271–280. https://doi.org/10.1007/s10561-017-9617-6
Domoto T, Pyko IV, Furuta T, Miyashita K, Uehara M, Shimasaki T, Nakada M, Minamoto T (2016) Glycogen synthase kinase-3beta is a pivotal mediator of cancer invasion and resistance to therapy. Cancer Sci 107(10):1363–1372. https://doi.org/10.1111/cas.13028
Grassilli E, Narloch R, Federzoni E, Ianzano L, Pisano F, Giovannoni R, Romano G, Masiero L, Leone BE, Bonin S, Donada M, Stanta G, Helin K, Lavitrano M (2013) Inhibition of GSK3B bypass drug resistance of p53-null colon carcinomas by enabling necroptosis in response to chemotherapy. Clin Cancer Res 19(14):3820–3831. https://doi.org/10.1158/1078-0432.CCR-12-3289
Kawazoe H, Bilim VN, Ugolkov AV, Yuuki K, Naito S, Nagaoka A, Kato T, Tomita Y (2012) GSK-3 inhibition in vitro and in vivo enhances antitumor effect of sorafenib in renal cell carcinoma (RCC). Biochem Biophys Res Commun 423(3):490–495. https://doi.org/10.1016/j.bbrc.2012.05.147
Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129(7):1261–1274. https://doi.org/10.1016/j.cell.2007.06.009
Li Y, Tsang CK, Wang S, Li XX, Yang Y, Fu L, Huang W, Li M, Wang HY, Zheng XF (2016) MAF1 suppresses AKT-mTOR signaling and liver cancer through activation of PTEN transcription. Hepatology 63(6):1928–1942. https://doi.org/10.1002/hep.28507
Ning K, Miller LC, Laidlaw HA, Watterson KR, Gallagher J, Sutherland C, Ashford ML (2009) Leptin-dependent phosphorylation of PTEN mediates actin restructuring and activation of ATP-sensitive K+ channels. J Biol Chem 284(14):9331–9340. https://doi.org/10.1074/jbc.M806774200
Ougolkov AV, Fernandez-Zapico ME, Savoy DN, Urrutia RA, Billadeau DD (2005) Glycogen synthase kinase-3beta participates in nuclear factor kappaB-mediated gene transcription and cell survival in pancreatic cancer cells. Can Res 65(6):2076–2081. https://doi.org/10.1158/0008-5472.CAN-04-3642
Wang Z, Smith KS, Murphy M, Piloto O, Somervaille TC, Cleary ML (2008) Glycogen synthase kinase 3 in MLL leukaemia maintenance and targeted therapy. Nature 455(7217):1205–1209. https://doi.org/10.1038/nature07284
De Toni-Costes F, Despeaux M, Bertrand J, Bourogaa E, Ysebaert L, Payrastre B, Racaud-Sultan C (2010) A new alpha5beta1 integrin-dependent survival pathway through GSK3beta activation in leukemic cells. PLoS ONE 5(3):e9807. https://doi.org/10.1371/journal.pone.0009807
Ougolkov AV, Fernandez-Zapico ME, Bilim VN, Smyrk TC, Chari ST, Billadeau DD (2006) Aberrant nuclear accumulation of glycogen synthase kinase-3beta in human pancreatic cancer: association with kinase activity and tumor dedifferentiation. Clin Cancer Res 12(17):5074–5081. https://doi.org/10.1158/1078-0432.CCR-06-0196
Naito S, Bilim V, Yuuki K, Ugolkov A, Motoyama T, Nagaoka A, Kato T, Tomita Y (2010) Glycogen synthase kinase-3beta: a prognostic marker and a potential therapeutic target in human bladder cancer. Clin Cancer Res 16(21):5124–5132. https://doi.org/10.1158/1078-0432.CCR-10-0275
Quintayo MA, Munro AF, Thomas J, Kunkler IH, Jack W, Kerr GR, Dixon JM, Chetty U, Bartlett JM (2012) GSK3beta and cyclin D1 expression predicts outcome in early breast cancer patients. Breast Cancer Res Treat 136(1):161–168. https://doi.org/10.1007/s10549-012-2229-8
Namba T, Kodama R, Moritomo S, Hoshino T, Mizushima T (2015) Zidovudine, an anti-viral drug, resensitizes gemcitabine-resistant pancreatic cancer cells to gemcitabine by inhibition of the Akt-GSK3beta-Snail pathway. Cell Death Dis 6:e1795. https://doi.org/10.1038/cddis.2015.172
Dey G, Bharti R, Dhanarajan G, Das S, Dey KK, Kumar BN, Sen R, Mandal M (2015) Marine lipopeptide Iturin A inhibits Akt mediated GSK3beta and FoxO3a signaling and triggers apoptosis in breast cancer. Sci Rep 5:10316. https://doi.org/10.1038/srep10316
Funding
This study was supported by the Chinese Natural Science Foundation (Grant 81471257), Natural Science Foundation of Jiangsu Province of China (Grant BK20161283), Jiangsu Province “Six Summit Talent” Foundation (2016-YY-061), and Nantong science and technology project (MS22016066) and sponsored by Qing Lan Project (to G.W.).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors of this manuscript have no conflicts of interest to report.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors. All animal care and experimental protocols were carried out according to the Chinese Animal Management Rules of the Ministry of Health and were authorized by the Animal Ethics Committees of Nantong University research program protocol #NT-16-086. Original data used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10549_2019_5239_MOESM1_ESM.tif
Supplemental Fig. 1 Expression level of p-AKT, p-PTEN and GSK3β in MDA-MB-468 cells and cisplatin-resistant MDA-MB-468 cells. Western blot analysis of p-AKT (A), p-PTEN (B) and GSK3β (C) in wild-type and cisplatin-resistant MDA-MB-468 cells. The relative band intensities were quantified using ImageJ software and are presented as the ratio of p-AKT, p-PTEN and GSK3β to β-actin, and finally normalized to control. The values are the mean ± SD (n = 3). Statistical significance was established with a two-tailed t-test; *p < 0.05, and **p< 0.01 versus wild-type cells. Supplementary material 1 (TIFF 234 kb)
Rights and permissions
About this article

Cite this article
Gao, C., Yuan, X., Jiang, Z. et al. Regulation of AKT phosphorylation by GSK3β and PTEN to control chemoresistance in breast cancer. Breast Cancer Res Treat 176, 291–301 (2019). https://doi.org/10.1007/s10549-019-05239-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-019-05239-3