
Abstract
Purpose
Invasive lobular carcinoma (ILC) is a histological subtype of breast cancer that is predominantly estrogen receptor alpha (ER)-positive (+) and is thus treated with endocrine therapies. Herein, we sought to understand the molecular underpinnings of the 4-hydroxytamoxifen (4OHT) resistance in ILC by assessing the potential role of the epithelial-to-mesenchymal transition transcription factor (EMT-TF) SNAIL (SNAI1).
Methods
Using a series of breast cancer cell lines, we measured the basal, estrogen and 4OHT-induced expression of SNAIL and other EMT-TF family members by quantitative reverse transcription-polymerase chain reaction and immunoblotting. Chromatin immunoprecipitation experiments were performed to assess ER binding to the SNAIL promoter. Cell proliferation, cell cycle and apoptosis were assessed in 2D cultures. 3D growth was assessed in Matrigel and Collagen I cultures.
Results
Estrogen and 4OHT induced SNAIL expression, but not that of the other EMT-TF family members SLUG (SNAI2) and SMUC (SNAI3), with the 4OHT effect being specific to the lobular but not the ductal subtype. We observed estrogen and 4OHT-induced ER recruitment to the SNAI1 promoter and high endogenous basal levels of SNAIL and several EMT-TFs in ILC cell lines. While SNAIL knockdown had a minor impact on the 4OHT partial agonism in estrogen-depleted conditions, it led to a surprising increase in cell proliferation in full serum. In complementary experiments, inducible SNAI1 overexpression caused decreased proliferation, associated with a cell cycle arrest in G0/G1. Additionally, apoptosis was observed in BCK4 cells.
Conclusion
These data suggest a previously unrecognized role for SNAIL in ILC, substantiating a context-dependent behavior for this EMT-TF.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Breast cancer is the most prevalent malignancy for women in the USA, impacting an estimated 252,710 women in 2017 [1, 2]. The two major histological subtypes of breast cancer are Invasive Lobular Carcinoma (ILC) and Invasive Ductal Carcinoma (IDC) [3,4,5,6,7]. ILC is characterized by the loss of the epithelial marker E-cadherin, which leads to the characteristic growth of ILC tumor cells throughout the stroma in discohesive linear cords within dense extracellular matrix (ECM) [8, 9]. Other unique aspects of ILC include its difficulty to be detected by imaging modalities, likely due to its diffuse growth pattern, and increased metastases to the gastrointestinal tract, peritoneum and ovaries [10,11,12,13]. These histological and clinical differences underscore the importance of understanding and treating ILC as a distinct disease and highlight the need to explore the mechanisms of its unique biology.
As ILCs are predominantly estrogen receptor alpha (ER) positive (+), patients are treated with endocrine therapies such as Selective Estrogen Receptor Modulators (SERMs), Aromatase Inhibitors (AIs) and Selective Estrogen Receptor Degraders (SERDs) [14, 15]. Endocrine therapy for breast cancer has remained one of the most successful types of targeted therapy among all cancers [16]. However, recurrence remains a critical issue in breast cancer as tumor cells evade treatment and regrow at primary and metastatic sites [17, 18]. Despite exhibiting favorable prognostic and predictive factors for endocrine therapy such as high ER expression and low proliferation rates, patients with ILC experience a higher frequency of long-term recurrences, often arising many years post-primary lesion diagnosis [15, 19,20,21,22]. A better understanding of the mechanistic responses of ILC tumors to endocrine therapies is critically needed to improve the outcomes of patients with ILC.
The SERM Tamoxifen exhibits mixed agonist/antagonist activities and has been well characterized for its potential to select resistant cell populations with increased tumorigenic phenotypes, often driven by increased PI3K/MAPK signaling [23,24,25]. More specifically in the lobular subtype, our lab recently identified de novo tamoxifen resistance in MDA-MB-134-VI cells [26], showing that these cells recognize SERMs including 4-hydroxytamoxifen (4OHT, active tamoxifen metabolite) as partial agonists that can drive cell proliferation. As part of our efforts to understand the underlying mechanisms of this phenotype, our gene expression analyses identified SNAI1 (encoding the transcription factor SNAIL) as the top 4OHT-induced gene in these cells [26]. SNAIL, and its related family members SLUG and SMUC, are transcription factors (TFs) that regulate epithelial-to-mesenchymal transition (EMT) [18, 27]. In breast cancer, high expression of EMT-TFs is associated with increased tumor grade, progression and metastasis [28,29,30,31,32]. Consistent with this, SNAIL expression has been shown to promote cellular resistance to programmed cell death, and to alter response to genotoxic stress [33,34,35].
The few studies focused on SNAIL in ILC [36, 37] so far have been limited to descriptive expression analyses in clinical samples without any functional or mechanistic interrogation. Based on the known association of SNAI1/SNAIL with aggressive tumor phenotypes, we hypothesized that the induction of SNAI1 (and/or other EMT-TFs) by SERMs could mediate the endocrine resistance programs in ILC cells. Here we show that the levels of SNAIL (but not those of the closely related family members SLUG and SMUC) are regulated by 4OHT, which is associated with 4OHT-mediated recruitment of ER to the SNAI1 promoter. This phenomenon was strongly context-dependent as it was only seen in ILC but not in IDC cell lines. While stable knockdown of SNAIL proved to be challenging, reflecting tight regulation of its expression in ILC cells, transient inhibition led to only a minor impact on 4OHT partial agonism, along with an unexpected increase in cell proliferation in full serum. Conversely, inducible SNAIL overexpression caused a decrease in 2D and 3D growth, suggesting previously unrecognized and hormone-independent roles for SNAIL in tumorigenesis that warrant further investigation in future studies.
Materials and methods
Cell culture and plasmids
MCF-7, T47D, MDA-MB-134-VI and MDA-MB-330 cell lines were purchased from the American Tissue Culture Collection (ATCC), and Sum44PE cells were purchased from Asterand. BCK4 were obtained from the Jacobsen Laboratory [38]. All cell lines were tested to be mycoplasma free every 6 months using MycoAlert™ Mycoplasma Detection Kit (Lonza#LT07-418) and were authenticated by the University of Arizona Genetics Core by Short Tandem Repeat (STR) DNA profiling. Authenticated cells were kept in continuous culture for < 6 months. Cells were cultured as previously described [26, 39], except for the use of 5% Fetal Bovine Serum (FBS) (Gibco#26-140) for MCF-7 and BCK4 cells. Cells were deprived of exogenous steroid hormones as previously described [26]. Estradiol (E2) (Sigma#E2758) and 4-hydroxytamoxifen (4OHT) (Sigma#H6278) were dissolved in Dimethylsulfoxide (DMSO) (ATCC#4-X-5) and used at the indicated conditions. For inducible SNAIL overexpression, an entry clone containing SNAI1 ORF (a kind gift from Dr. Timothy Burns) was cloned into pINDUCER20 (ref [40]; Addgene Plasmid#44-012) using Gateway™ LR Clonase™ II and standard manufacturer’s protocol (Invitrogen#11-791-100). Stable cell lines were generated by lentiviral transduction as previously described [40] and maintained in 1.25 mg/mL Geneticin (Thermo Fisher Scientific#10131-035). To induce overexpression, cells were treated with 0.5 µg/mL Doxycycline (Sigma#D9891-1G) 1 day after plating.
Reverse phase protein array (RPPA), immunoblotting and chromatin immunoprecipitation (ChIP) assays
RPPA assays were performed as previously described [26]. Briefly, MDA-MB-134-VI and Sum44PE cells were seeded at 70–80% confluence in full serum or estrogen-deprived conditions and treated with 1 nM E2 or vehicle (Ethanol) for 24 h. Samples were collected in MD Anderson RPPA lysis buffer and assessed at the Functional Proteomics Core of MD Anderson. For immunoblotting, proteins were extracted using either MD Anderson RPPA or RIPA buffer and assayed using the Odyssey Infrared Imaging system (LiCor). The primary antibodies used are listed in Supplementary Table 1. ChIP assays were performed in two independent experiments as previously described using IgG or ERα (Santa Cruz HC-20#sc-543) antibodies [26, 41]. ChIP Quantitative Reverse Transcription Polymerase Chain Reactions (qRT-PCR) were performed using input samples diluted 1:5 and samples diluted 1:3 with 5 µL/technical replicate well using the primers listed in Supplementary Table 2. Data were analyzed using fold enrichment (FE) over IgG controls. The intensities of the bands in the immunoblots were quantified using ImageJ. Detailed methods are included in Supplementary Text.
qRT-PCR
mRNA was isolated from triplicate samples using either manufacturer’s protocol of the illustra RNAspin Mini Kit (GE Healthcare#25-0500-72) or NucleoSpin RNA Kit (Takara Clontech#740955.250). 250–500 ng of mRNA were converted into cDNA using 1X iScript (BioRad#1708891) manufacturer’s protocol. Samples were assessed by qRT-PCR using SsoAdvanced™ Universal SYBR® Green Supermix (BioRad#1725274) using the primers listed in Supplementary Table 3. All qRT-PCRs were performed in two to three independent experiments with data normalized to the housekeeping gene RPLP0.
Transient knockdown assays
Cells were reverse transfected using Lipofectamine™ RNAiMAX following manufacturer’s protocol (Thermo Fisher Scientific#13778-150) with 10 nM final concentration of ON-TARGETplus Non-Targeting Control Pool (Dharmacon#D-001810-10) or Human SNAI1 Pool (Dharmacon#L-010847-01) siRNA in Opti-MEM® I (Gibco#31985-070) for 24–48 h prior to downstream applications.
2D and 3D growth assays
For 2D assays, cells were plated in technical replicates of 3–6 at 15,000 (ILC; all experiments except Supplementary Fig. 5 at 5000) or 5000 (IDC) cells/well and proliferation was assessed as recently described [26] using FluoReporter™ Blue Fluorometric dsDNA Quantitation Kit (Invitrogen#F2962). Data were captured using VictorX Software on a PerkinElmer plate reader and plotted after correction to background media fluorescence. Nonlinear regressions fitted to each data set were used to test differences in rates of growth. Matrigel and Collagen I embedding assays were done as previously described [39]. Details are included in Supplementary Text.
Mammosphere assay
Mammospheres were generated and processed as previously described [39, 42]. Briefly, cells were cultured in triplicate wells of 6-well Ultra Low Attachment Plates (Corning#3471) in mammosphere media (1:1 DMEM/Ham’s F-12 media, 20 ng/mL bFGF (BD Biosciences#354060), 20 ng/mL EGF (BD Biosciences#354001), B27 (Gibco#17504), 2.5 mL Penicillin/Streptomycin, and 4 µg/mL Heparin (Sigma#H4784)). Detailed methods are included in Supplementary Text.
Cell cycle and apoptosis assays
Cells were seeded at 300,000/well in 6-well plates in triplicates and incubated with Doxycycline for 4 days. Cells were harvested, washed with PBS and stained with Hoechst for cell cycle analysis or with Annexin V/PI using APC-Annexin V (BD Biosciences; #550474) for apoptosis analysis following the manufacturer’s protocol. Samples were acquired on an LSR II Flow cytometer (BD Biosciences) and analyzed using BD FACSDiva software (BD Biosciences).
Results
SNAI1 expression is induced by E2 and 4OHT in ER + ILC cells
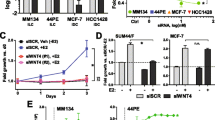
We recently reported that E2 induces the expression of a large number of genes at the RNA level in the ER + ILC cell lines MDA-MB-134-VI and Sum44PE, including the EMT-TF SNAI1 [26]. We first validated this finding in an extended panel of ER + ILC (including BCK4 and MDA-MB-330) and IDC (MCF7 and T47D) cells by qRT-PCR following E2 deprivation and E2 treatment (Fig. 1a). This analysis showed significant E2-mediated induction of SNAI1 in MDA-MB-134-VI and in MCF-7, with a trend for induction in Sum44PE, BCK4 and T47D cells, consistent with a previous report [43]. Importantly, this induction was specific to SNAI1 and not observed for the other EMT-TFs SNAI2 and SNAI3 (Supplementary Fig. 1a).

4OHT Acts as an Agonist in ER + ILC Cells. aSNAI1 qRT-PCR assessment of ILC (red) and IDC (blue) cells was performed at 24 h post-deprivation/treatments (vehicle 0.1% DMSO). Quantifications are a representative single experiment ± STDEV of technical triplicates, with similar results in two additional independently performed experiments. Asterisks depict significance compared to vehicle from One Way ANOVA followed by Tukey’s posttest. b RPPA analysis of MDA-MB-134-VI and Sum44PE cells in Full Serum (FS) or deprived of exogenous estrogens (Cont) followed by treatment with 1 nM E2 for 24 h. Top ten E2-upregulated and downregulated proteins in MDA-MB-134-VI cells are displayed. Log2 fold change (FC) for SNAIL is depicted in the table. c SNAIL immunoblots were performed in MDA-MB-134-VI and Sum44PE cells at 24 h post-deprivation/treatments. Representative images are displayed (top) along with quantification of band intensities (bottom). Graphs represent mean ± SEM relative to vehicle from three independent experiments. Asterisks depict significance compared to vehicle from one-way ANOVA followed by Dunnett’s posttest (*p < 0.05, **p < 0.005, ***p < 0.0005)
To assess whether E2 can induce SNAI1 (and potentially other EMT-related genes) at the protein level, we performed reverse phase protein arrays (RPPA) on MDA-MB-134-VI and Sum44PE cell lines maintained either in full serum (FS), or switched to charcoal-stripped serum (E2-deprived [Cont]) and subsequently treated with 1 nM E2 for 24 h (E2). This analysis revealed that E2 also induces the expression of SNAIL protein in MDA-MB-134-VI cells, with some modest induction also in Sum44PE (Fig. 1b), mirroring the induction of mRNA levels. Of note, other EMT-related proteins on the RPPA such as P-cadherin, Fibronectin and c-kit were not significantly induced by E2 (Supplementary Fig. 1b). Antibodies against the other EMT-TFs SLUG (encoded by SNAI2) and SMUC (encoded by SNAI3) were not included in the RPPA.
Our previous microarray and NanoString analyses had additionally identified SNAI1 induction by the SERM 4OHT, which exhibits partial agonist activity in human ILC cell lines [31]. Assessment of SNAI1 levels by qRT-PCR in our panel of estrogen-deprived ER + ILC and IDC cell lines revealed induction of SNAI1 by 4OHT only in two of the ILC cell lines, MDA-MB-134-VI and Sum44PE, and not in any of the IDC cells (Fig. 1a). Immunoblot analysis in these two cell lines confirmed that E2 and 4OHT lead to induction of SNAIL protein as well (Fig. 1c, Supplementary Fig. 2). Collectively, these data demonstrate that 4OHT induces the expression of the ER target SNAIL, a phenotype unique to a subset of human ILC cell lines.
ER is recruited to the promoter of SNAI1
SNAI1 is an established ER target gene in ER + breast cancer cells [44, 45], but has not previously been reported to be induced by 4OHT. To assess whether the induction of SNAI1 by ER occurs directly, we first assessed ER binding at estrogen response elements (EREs) directly upstream of the SNAI1 promoter. Data mined from the Ross-Innes ER ChIP database [46] revealed ER binding to an ERE, located approximately 1 kbp upstream of the SNAI1 transcriptional start site, in multiple ER + breast cancer cell lines, as well as in primary and metastatic lesions (Fig. 2a). We next performed ER ChIP in E2-deprived MDA-MB-134-VI cells after treatment with E2 or 4OHT for 8 h, followed by qRT-PCR using primers flanking the ERE site. The specificity of the ChIP was confirmed by the observed ER binding to the promoter of its classical target GREB1 but not to a non-functional ERE (NFERE) used as a negative control. Both E2 and 4OHT induced ER recruitment to the ERE upstream of the SNAI1 promoter at this time point (Fig. 2b). We simultaneously performed immunoprecipitations of ER on our ChIP lysates to assess the efficiency of the pulldowns and also confirmed the upregulation of SNAI1 with mRNA analyses run concurrently with our ChIP experiment (Supplementary Fig. 3a, b). This experiment was additionally performed at two earlier time points, 45 min and 3 h, though significant recruitment of ER was only observed at the 8 h time point in response to 4OHT (Supplementary Fig. 3c, d). These data established the recruitment of ER to the SNAI1 promoter in ILC cells, which is enhanced in the presence of E2 and 4OHT.

ER is Recruited to the Promoter of SNAI1. a Conserved EREs upstream of the SNAI1 transcriptional start site (TSS) from in silico analyses of publicly available ER ChIP data were identified. Build hg18 was used. The general ER binding region is underlined. Arrows indicate the position of designed ChIP qRT-PCR primers and the TSS, and an excerpt shows the ERE in the ChIP qRT-PCR primer region with base pairs from the ER motif shown in red font and spacer base pairs in black font. The primer region is 1 kbp upstream of the TSS. b ER ChIP was performed in MDA-MB-134-VI cells after a period of 3 days of estrogen deprivation and treatment with vehicle (0.1% DMSO), E2, or 4OHT for 8 h. ChIP qRT-PCR was performed and displayed as mean ± STDEV of technical triplicates relative to vehicle IgG control for region of interest SNAI1, positive E2 control GREB1, and negative control NFERE. Statistics were performed using one-way ANOVA followed by Tukey’s posttest with comparison relative to vehicle control displayed with asterisks (*p < 0.05, **p < 0.005, ***p < 0.0005). Data are representative of two independently performed experiments
EMT-TF programs are upregulated in some ILC cell lines
Although we did not observe significant E2-regulation of other EMT-related genes, it was possible that the baseline levels of EMT-TFs were higher in ILC vs. IDC cell lines. To directly investigate this, we measured mRNA expression of SNAIL, other EMT-TFs, and their downstream targets in a panel of ER + ILC and IDC cell lines. All ILC cell lines utilized were first confirmed to have expression of ER and the ILC-hallmark loss of E-cadherin protein, with the exception of MDA-MB-330, which does express E-cadherin but has loss of adherens junctions due to a mutation in alpha catenin (Supplementary Fig. 4) [47]. Upregulation of SNAI1, as well as an additional EMT-TF, TWIST1, was consistently seen across several ILC cell lines as opposed to IDC cells (Fig. 3a; top panel). We additionally probed the mRNA expression of the EMT-TF downstream targets CDH2 (N-cadherin), FN1 (Fibronectin) and VIM (Vimentin) and observed similar patterns of upregulation in ILC cell lines relative to IDCs (Fig. 3a; bottom panel). Furthermore, we measured the endogenous protein expression of SNAIL and TWIST1 and observed higher levels in the ILC cell lines MDA-MB-134-VI and Sum44PE compared to other cell lines (Fig. 3b, c). As SNAIL and TWIST1 have previously been shown to have very short half-lives (20–45 min) in cells, we additionally treated cells with the proteasome inhibitor MG132 and observed markedly stabilized levels for these proteins [48]. Taken together, these data implicate the existence of an EMT-like program in the majority of the commonly used ILC cell line models.

EMT-TF-Related Genes are Upregulated in ILC. a mRNA and b protein expression of two EMT-TFs and downstream EMT targets in ILC (red) and IDC (blue) ER + cells. Combined qRT-PCR data from three independent experiments are shown relative to MCF-7 as means ± SEM. One-way ANOVA followed by Tukey’s posttest is displayed with asterisks comparing cells to MCF-7 expression of a gene of interest. Immunoblot is representative of a set of three independently performed experiments ± 5 µM MG132 treatment for 2 h. c Quantification of band intensities from (b). Graphs represent means ± SEM relative to vehicle from three independent experiments. Asterisks depict significance from t-tests followed by correction for multiple comparisons using the Holm–Sidak method (*p < 0.05, **p < 0.005, ***p < 0.0005)
Transient SNAI1 knockdown leads to increased proliferation
We next set out to determine the functional consequences of SNAIL inhibition in ILC cell lines. Our repeated attempts at generating cells with stable SNAI1 knockdown were unsuccessful; however, we were able to decrease SNAIL levels using transient siRNA approaches. We initially knocked down SNAI1 in MDA-MB-134-VI cells using siRNAs following E2-deprivation and assessed cell proliferation over 14 days. This transient SNAI1 inhibition led to a surprising increase in E2-induced growth (Supplementary Fig. 5a) and a trend toward a mild decrease in 4OHT partial agonism (Supplementary Fig. 5b), although no statistically significant differences were observed between the EC50s for either treatment. We next transiently knocked down SNAI1 in full serum in MDA-MB-134-VI cells, which exhibit the highest levels of endogenous SNAIL expression. We confirmed successful SNAI1 knockdown, which was sustained over the 6-day experimental course (Fig. 4a). In agreement with the effect of SNAI1 inhibition in E2-induced growth following E2 deprivation, SNAI1 siRNA also resulted in increased proliferation of cells in full serum (Fig. 4b), a surprising result given the previously described role of SNAIL as a driver of cell proliferation in other contexts [49]. We also checked the effect of transient SNAI1 knockdown on the expression of the downstream EMT-TF target genes and observed that only FN1 was significantly decreased while there was no effect on VIM or CDH2 (Fig. 4c).

Transient SNAI1 Knockdown Leads to Decreases in Few EMT-TF Targets and Increases Proliferation. Transient knockdown of SNAI1 was performed in 2D plates with Scramble or SNAI1 siRNA. a qRT-PCR confirmation of transient knockdown of SNAI1 throughout the experiment was completed at days two, four and six with Student’s t-test evaluations shown. Data represent three independently repeated experiments. b Representative 2D proliferation data corrected to background media fluorescence and shown relative to day one values. Nonlinear regression exponential growth curves were fitted, and a comparison of growth rates was performed; significance between Scramble and SNAI1 siRNA groups is displayed. c Representative qRT-PCR data of probed downstream EMT-TF targets are displayed as means relative to Scramble ± STDEV of technical triplicates with one-way ANOVA followed by Tukey’s posttest displayed by asterisks (*p < 0.05, **p < 0.005, ***p < 0.0005). Data represent two independently repeated experiments. MDA-MB-134-VI: no transfection reagent or siRNA. Nontransfected: transfection reagent only; no siRNA
SNAIL overexpression inhibits 2D and 3D proliferation of ILC cells
To complement the loss-of-function studies described above, we generated stable, Doxycycline (Dox)-inducible SNAIL overexpressing models from ILC cell lines with low endogenous expression, specifically BCK4, MDA-MB-330 and Sum44PE cells. Treatment of these cell lines with Dox induced SNAIL protein expression and suppressed cell growth (Fig. 5a), corroborating the increased cell growth results from the loss-of-function studies in MDA-MB-134-VI cells. Given our observation of 4OHT-induced SNAIL expression, we asked whether ectopic overexpression of SNAIL was sufficient to elicit a partial agonist response to 4OHT treatment in BCK4, MDA-MB-330 and Sum44PE cells and only observed a small but not significant effect in MDA-MB-330 cells (Supplementary Fig. 6). Instead, SNAIL overexpression generally repressed proliferation in the presence of 4OHT, consistent with its effects in full serum. Interestingly, a study by Vega et al. has linked SNAIL overexpression to decreased proliferation and cell cycle arrest [50]. We therefore assessed the cell cycle profiles of our inducible SNAIL overexpressing cells (Supplementary Fig. 7a). While cell cycle distribution of BCK4 control cells was not affected by treatment with Dox (Supplementary Fig. 7b), inducible overexpression of SNAIL led to arrest in the G0/G1 phase of the cell cycle and decreased percentages of BCK4 cells in the S and G2/M phases (Fig. 5b), with similar effects in MDA-MB-330 and Sum44PE cells (Supplementary Fig. 7c, d). In addition, we also assayed apoptosis and observed a marked reduction in the percentage of viable cells upon Dox treatment only in BCK4 cells, concomitant with cleavage of PARP (Supplementary Fig. 7e, f).

SNAIL Overexpression Causes Decreased Proliferative, Invasive and Stem-Like Phenotypes. Inducible SNAIL overexpression ILC BCK4, MDA-MB-330 and Sum44PE cells were treated to ± Dox in a 2D environments followed by b evaluation of cell cycle profiles for BCK4 cells at day 4. Data in b are representative of two independently performed experiments with three technical replicates per experiment ± STDEV, and asterisks depict significance from t-tests followed by correction for multiple comparisons using the Holm–Sidak method (*p < 0.05). In addition, cells were probed for phenotypic changes in c 3D environments, or d in a mammosphere assay. Scale bars represent 50 µm. Proliferation curves and protein were assessed as previously. Data in a are a representative experiment of three independently performed experiments with six technical replicates per time point ± STDEV, displayed relative to day 2. Data in d are three independently performed experiment means ± SEM, each performed with technical triplicates. One-way ANOVA followed by Tukey’s posttest is displayed with asterisks comparing ± Dox treatment (*p < 0.05, ***p < 0.0005, ****p < 0.0001). Images from c and d represent phase images at day 24 or day 17, respectively, 20X magnification
We have recently reported a comprehensive phenotypic characterization of human ILC cell lines in 2D and 3D cultures [39]. To assess the effects of SNAIL in these biological phenotypes, we next assayed the effect of SNAIL overexpression on 3D growth and invasion ability plating the cells in 3D ECM gels: (i) seeding on top of Matrigel, (ii) embedding within Matrigel or (iii) embedding within Collagen I, conditions that mimic the stroma-rich growth of ILC cells in vivo. We then measured the formation of colonies with or without induction of SNAIL over a period of 24 days. While BCK4 and MDA-MB-330 did not form robust colonies in any environment, with or without SNAIL induction, colonies formed by Sum44PE cells were suppressed upon induction of SNAIL in all three environments (Fig. 5c). Finally, we asked whether SNAIL regulated ILC mammosphere formation, an assay that has been used to assess stem cell-like properties of cancer cell lines [51]. As observed in Matrigel and collagen matrices, BCK4 and MDA-MB-330 were unable to form robust mammospheres, but instead formed loosely attached multi-cellular structures (Supplementary Fig. 8). In Sum44PE cells, induction of SNAIL led to significant reduction in mammosphere formation (Fig. 5d). Taken together, overexpression of SNAIL inhibited 2D growth and EMT-like phenotypes in the ILC cell line models, a somewhat surprising result based on the previously described roles of SNAIL in other biological systems [27,28,29,30,31,32, 48]. Nevertheless, the data from both our loss-of-function and overexpression studies complemented each other, suggesting a unique, context-dependent molecular role for SNAIL in ILC.
Discussion
Endocrine therapies have served as an arsenal of targeted treatments against ER + breast cancers, but not without the rise of various resistance mechanisms. While the majority of ILCs respond very well to endocrine therapy, there is increasing evidence that tamoxifen treatment does not yield the desired outcome in a subset of patients [52]. The Cancer Genome Atlas (TCGA) ILC working group was among the first to publish a clear molecular distinction between ILC and IDC, including significant differences in PTEN, PI3K, FOXA1 and GATA3 mutations/activation between ILC and IDC [4]. However, limited studies have addressed potential differences in ER action, including estrogen and antiestrogen response [26, 41, 52,53,54,55].
Here, we identified SNAIL as a potential molecular target of 4OHT in tamoxifen-resistant ILC. Our data indicate that 4OHT can act as a partial agonist with respect to SNAIL induction in ILC cell lines. We found that this agonist behavior was limited to two of our ER + ILC cell lines, MDA-MB-134-VI and Sum44PE. Lack of response in MDA-MB-330 and BCK4 could indicate that these lines represent different histological and/or molecular subtypes within ILC, but further studies will need to delineate this in more detail. Our gene expression data are complemented by results from ChIP assays showing direct binding of ER to the promoter region of SNAI1, thereby providing additional support for the hypothesis that 4OHT can act as an agonist in ILC as opposed to an antagonist.
Our study was hampered by technical limitations in efficiently regulating SNAIL expression, potentially reflecting a very tight regulation of its levels in ILC cells. Nevertheless, results from our transient knockdown and inducible overexpression of SNAIL suggest that there is a negative correlation between SNAIL levels and 2D and 3D growth. These data were surprising, since the majority of studies have described SNAIL as a positive regulator of growth and invasive properties, including in breast cancer [27,28,29,30,31,32, 48]. There is, however, prior precedence for SNAIL overexpression inhibiting cell cycle progression—Vega et al. showed that SNAIL overexpression can cause cell cycle arrest and therefore protection from apoptosis [50]. It is reasonable to postulate that SNAIL overexpression in ILC cells might allow the cells to enter a quiescent state in which energy is conserved, and cells are able to evade therapy for extended periods of time, indicative of tumor dormancy [56]. Future studies will be needed to further test this hypothesis and to determine whether these findings could be related to the observed late recurrences in patients with ER + ILC [8, 15, 19,20,21,22]. Alternatively, SNAIL could be tightly regulated in a temporal context, something not addressed by these studies. Though SNAIL is induced by binding of ER to the SNAI1 promoter circa 8 h post-4OHT treatment and subsequent induction of expression and protein can be observed within a day of this induction, it is possible that this is then repressed at a later time point to allow for the overall 4OHT proliferation phenotype observed [26]. This temporal regulation of SNAIL will be pursued in the future studies. Finally, it will also be critical to study potential contribution of other EMT genes, even if not directly regulated by SERMs, as especially TWIST1 that has been described to be highly expressed in ILC and its non-obligate precursor, Lobular Carcinoma In Situ (LCIS) [37].
In conclusion, we have shown that in some ILC models, 4OHT can act as an ER agonist with respect to regulation of SNAIL, a classical EMT-TF. SNAIL levels are tightly regulated in ILC cell lines, but inducible overexpression causes growth inhibition, associated with cell cycle arrest and apoptosis. These unexpected and hormone-independent quiescent phenotypes could contribute to tumor dormancy and therapeutic resistance, a hypothesis that need to be tested in the future studies. Such studies are required to better understand the etiology of ILC, with the ultimate goal to personalize treatment for this patient population.
References
Howlader N, Krapcho NA, Miller M, Bishop D, Kosary CL et al SEER Cancer Stat Rev. 1975–2014. 2017 April 2017; Based on November 2016 SEER data submission]
Society AC (2017) Cancer Facts and Figures 2017
Barroso-Sousa R, Metzger-Filho O (2016) Differences between invasive lobular and invasive ductal carcinoma of the breast: results and therapeutic implications. Ther Adv Med Oncol 8(4):261–266
Ciriello G et al (2015) Comprehensive molecular portraits of invasive lobular breast cancer. Cell 163(2):506–519
Christgen M et al (2016) Lobular breast cancer: clinical, molecular and morphological characteristics. Pathol Res Pract 212(7):583–597
Desmedt C et al (2017) Transcriptomic and genomic features of invasive lobular breast cancer. Semin Cancer Biol 44:98–105
Malhotra GK et al (2010) Histological, molecular and functional subtypes of breast cancers. Cancer Biol Ther 10(10):955–960
Arpino G et al (2004) Infiltrating lobular carcinoma of the breast: tumor characteristics and clinical outcome. Breast Cancer Res 6(3):R149–R156
McCart Reed AE et al (2015) Invasive lobular carcinoma of the breast: morphology, biomarkers and ‘omics. Breast Cancer Res 17(12):12
Johnson K, Sarma D, Hwang ES (2015) Lobular breast cancer series: imaging. Breast Cancer Res 17(94):94
Sobinsky JD et al. (2013) Unusual metastatic patterns of invasive lobular carcinoma of the breast. Case Rep Oncol Med 2013:986517
Shakoor MT et al (2014) Unique presentations of invasive lobular breast cancer: a case series. Int J Biomed Sci 10(4):287–293
Mathew A et al (2017) Distinct pattern of metastases in patients with invasive lobular carcinoma of the breast. Geburtshilfe Frauenheilkd 77(6):660–666
Lumachi F et al (2011) Endocrine therapy of breast cancer. Curr Med Chem 18(4):513–522
Chen Z et al (2017) Invasive lobular carcinoma of the breast: a special histological type compared with invasive ductal carcinoma. PLoS One 12(9):e0182397
Jordan VC (2014) Tamoxifen as the first targeted long-term adjuvant therapy for breast cancer. Endocr Relat Cancer 21(3):R235–R246
Milani A et al (2014) Overcoming endocrine resistance in metastatic breast cancer: current evidence and future directions. World J Clin Oncol 5(5):990–1001
Smith BN, Bhowmick NA (2016) Role of EMT in metastasis and therapy resistance. J Clin Med. 5(2):17
Pestalozzi BC et al (2008) Distinct clinical and prognostic features of infiltrating lobular carcinoma of the breast: combined results of 15 International Breast Cancer Study Group clinical trials. J Clin Oncol 26(18):3006–3014
Narbe U et al (2014) Invasive lobular carcinoma of the breast: long-term prognostic value of Ki67 and histological grade, alone and in combination with estrogen receptor. Springerplus 3:70
Engstrøm MJ et al (2015) Invasive lobular breast cancer: the prognostic impact of histopathological grade, E-cadherin and molecular subtypes. Histopathology 66:409–419
Adachi Y et al (2016) Comparison of clinical outcomes between luminal invasive ductal carcinoma and luminal invasive lobular carcinoma. BMC Cancer 16:248
Araki K, Miyoshi Y (2017) Mechanism of resistance to endocrine therapy in breast cancer: the important role of PI3K/Akt/mTOR in estrogen receptor-positive, HER2-negative breast cancer. Breast Cancer (Tokyo Japan) 25:392–401
Chang M (2012) Tamoxifen resistance in breast cancer. Biomol Ther (Seoul) 20(3):256–267
Ali S et al (2016) Molecular mechanisms and mode of tamoxifen resistance in breast cancer. Bioinformation 12(3):135–139
Sikora MJ et al (2014) Invasive lobular carcinoma cell lines are characterized by unique estrogen-mediated gene expression patterns and altered tamoxifen response. Cancer Res 74(5):1463–1474
de Herreros AG et al (2010) Snail family regulation and epithelial mesenchymal transitions in breast cancer progression. J Mammary Gland Biol Neoplasia 15(2):135–147
Martin TA et al (2005) Expression of the transcription factors snail, slug, and twist and their clinical significance in human breast cancer. Ann Surg Oncol 12(6):488–496
Peinado H, Olmeda D, Cano A (2007) Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer 7(6):415–428
Moody SE et al (2005) The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell 8(3):197–209
Elloul S et al (2005) Snail, Slug, and Smad-interacting protein 1 as novel parameters of disease aggressiveness in metastatic ovarian and breast carcinoma. Cancer 103(8):1631–1643
Dubois-Marshall S et al (2011) Two possible mechanisms of epithelial to mesenchymal transition in invasive ductal breast cancer. Clin Exp Metastasis 28(8):811–818
Chen WJ et al (2010) Multidrug resistance in breast cancer cells during epithelial-mesenchymal transition is modulated by breast cancer resistant protein. Chin J Cancer 29(2):151–157
Kajita M, McClinic KN, Wade PA (2004) Aberrant expression of the transcription factors snail and slug alters the response to genotoxic stress. Mol Cell Biol 24(17):7559–7566
Proia TA et al (2011) Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell 8(2):149–163
McCart Reed AE et al (2016) An epithelial to mesenchymal transition programme does not usually drive the phenotype of invasive lobular carcinomas. J Pathol 238(4):489–494
Morrogh M et al (2012) Cadherin-catenin complex dissociation in lobular neoplasia of the breast. Breast Cancer Res Treat 132(2):641–652
Jambal P et al (2013) Estrogen switches pure mucinous breast cancer to invasive lobular carcinoma with mucinous features. Breast Cancer Res Treat 137(2):431–448
Tasdemir N et al (2018) Comprehensive phenotypic characterization of human invasive lobular carcinoma cell lines in 2D and 3D cultures. Cancer Res 78(21):6209–6222
Meerbrey KL et al (2011) The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc Natl Acad Sci USA 108(9):3665–3670
Sikora MJ et al (2016) WNT4 mediates estrogen receptor signaling and endocrine resistance in invasive lobular carcinoma cell lines. Breast Cancer Res 18(1):92
Shaw FL et al (2012) A detailed mammosphere assay protocol for the quantification of breast stem cell activity. J Mammary Gland Biol Neoplasia 17(2):111–117
Harrell JC, Shroka TM, Jacobsen BM (2017) Estrogen induces c-Kit and an aggressive phenotype in a model of invasive lobular breast cancer. Oncogenesis 6(11):396
Scherbakov AM et al (2012) The relationships between snail1 and estrogen receptor signaling in breast cancer cells. J Cell Biochem 113(6):2147–2155
Wang M et al (2013) AIB1 cooperates with ERalpha to promote epithelial mesenchymal transition in breast cancer through SNAI1 activation. PLoS ONE 8(6):e65556
Ross-Innes CS et al (2012) Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481(7381):389–393
Christgen M, Derksen P (2015) Lobular breast cancer: molecular basis, mouse and cellular models. Breast Cancer Res 17:16
Zhou BP et al (2004) Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol 6(10):931–940
Lundgren K, Nordenskjold B, Landberg G, Hypoxia (2009) Snail and incomplete epithelial-mesenchymal transition in breast cancer. Br J Cancer 101(10):1769–1781
Vega S et al (2004) Snail blocks the cell cycle and confers resistance to cell death. Genes Dev 18(10):1131–1143
Mani SA et al (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133(4):704–715
Metzger Filho O et al (2015) Relative effectiveness of letrozole compared with tamoxifen for patients with lobular carcinoma in the BIG 1–98 trial. J Clin Oncol 33(25):2772–2779
Riggins RB et al (2008) ERRgamma mediates tamoxifen resistance in novel models of invasive lobular breast cancer. Cancer Res 68(21):8908–8917
Stires H et al (2018) Integrated molecular analysis of Tamoxifen-resistant invasive lobular breast cancer cells identifies MAPK and GRM/mGluR signaling as therapeutic vulnerabilities. Mol Cell Endocrinol 471:105–117
van Agthoven T et al (2015) Breast cancer anti-estrogen resistance 4 (BCAR4) drives proliferation of IPH-926 lobular carcinoma cells. PLoS ONE 10(8):e0136845
Sosa MS, Bragado P, Aguirre-Ghiso JA (2014) Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer 14(9):611–622
Acknowledgements
The authors thank Donald DeFranco, Ph.D., Adrian Lee, Ph.D., and Carola Neumann, M.D., at the University of Pittsburgh, as well as Adam Feinberg, Ph.D. at Carnegie Melon University for intellectual discussion and guidance with this work, and Jennifer Xavier, Ph.D. at the University of Pittsburgh for her critical reading and editing of the manuscript. Additionally, the authors extend gratitude to the laboratories of Adrian V. Lee, Carola Neumann, Mei Zhang, Yi Huang and Steffi Oesterreich in the Women’s Cancer Research Center at the University of Pittsburgh for their support and suggestions.
Funding
Research funding for this project was provided in part by a Susan G. Komen Scholar award (SO; SAC160073) and the Breast Cancer Research Foundation (SO). EAB was supported by a Department of Pharmacology and Chemical Biology John S. Lazo Fellowship, and a National Institutes of Health Ruth L. Kirschstein award [1F31CA203055-01]. NT was supported by a Department of Defense Breakthrough Fellowship Award [BC160764]. MJS was supported by a Department of Defense Postdoctoral Fellowship [BC110619], and a National Institutes of Health Pathway to Independence award [K99 CA193734]. KL was supported by a National Institutes of Health Ruth L. Kirschstein award [1F30CA203154-01]. TFB has received research funding for EMT studies from an American Lung Association Award (LCD 257864), V Foundation Scholar Award, Sidney Kimmel Foundation (SKF-15-099) and a Doris Duke Charitable Foundation Clinical Scientist Award (2015097), a Pittsburgh LUNG SPORE CDA P50CA090440 and American Cancer Society Research Scholar Grant (132939-RSG-18-185-01 TBG). This project used UPMC Hillman Cancer Center Core Facilities, supported by and NCI award [P30CA047904].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr. Timothy F. Burns serves on an Advisory Board for AbbVie, Inc., and Dr. Oesterreich is a member of the External Scientific Advisory Board Panel of NSABP. The authors declare that they have no conflict of interest.
Research involving human and animal rights
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bossart, E.A., Tasdemir, N., Sikora, M.J. et al. SNAIL is induced by tamoxifen and leads to growth inhibition in invasive lobular breast carcinoma. Breast Cancer Res Treat 175, 327–337 (2019). https://doi.org/10.1007/s10549-019-05161-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-019-05161-8