
Abstract
Purpose
To evaluate whether adding humanized monoclonal insulin growth factor-1 receptor (IGF-1R) antibody (dalotuzumab) to mammalian target of rapamycin (mTOR) inhibitor (ridaforolimus) plus aromatase inhibitor (exemestane) improves outcomes in patients with estrogen receptor (ER)-positive advanced/metastatic breast cancer.
Methods
This randomized, open-label, phase II trial enrolled 80 postmenopausal women with high-proliferation (Ki67 index staining ≥15%), ER-positive breast cancer that progressed after a non-steroidal aromatase inhibitor (NCT01605396). Randomly assigned patients were given oral ridaforolimus 10 mg QD 5 ×/week, intravenous dalotuzumab 10 mg/kg/week, and oral exemestane 25 mg/day (R/D/E, n = 40), or ridaforolimus 30 mg QD 5 ×/week and exemestane 25 mg/day (R/E; n = 40). Primary end point was progression-free survival (PFS).
Results
Median PFS was 23.3 weeks for R/D/E versus 31.9 weeks for R/E (hazard ratio 1.18; 80% CI 0.81–1.72; P = 0.565). Grade 3–5 adverse events were reported in 67.5% of patients in the R/E arm and 59.0% in the R/D/E arm. Stomatitis (95.0 vs. 76.9%; P = 0.021) and pneumonitis (22.5 vs. 5.1%; P = 0.027) occurred more frequently in the R/E than the R/D/E arm; hyperglycemia (27.5 vs. 28.2%) occurred at a similar rate.
Conclusions
R/D/E did not improve PFS compared with R/E. Because the PFS reported for R/E was similar to that reported for everolimus plus exemestane in patients with advanced breast cancer, it is possible that lower-dose ridaforolimus in the R/D/E arm (from overlapping toxicities with IGF1R inhibitor) contributed to lack of improved PFS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A key mechanism of endocrine resistance in breast cancer is aberrant signaling through the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) signaling pathway [1, 2]. The kinase mTOR (comprising mTORC1 and mTORC2) is downstream of the PI3K/AKT pathway and can be activated by mitogens, nutrients, and growth factor receptor signals [3]. mTOR plays a central role in normal cell growth and development, including a pivotal role in the development and progression of cancer, making it an attractive target for anticancer therapy [4].
Ridaforolimus (formerly known as deforolimus, AP23573, or MK-8669) is a non-prodrug analog of rapamycin that has been shown to inhibit mTOR with high potency and specificity [5, 6]. However, the efficacy of single-agent mTOR inhibition is limited by feedback upregulation of PI3K/AKT signaling. This effect is mediated by insulin receptor substrate 1 (IRS1), an adaptor protein that, upon ligand binding, propagates insulin growth factor 1 receptor (IGF1R) signaling by facilitating receptor-mediated activation of PI3 K. When mTORC1 is active, p70 S6 kinase phosphorylates the adapter protein of IGF1R, insulin receptor substrate-1 (IRS1), leading to its degradation. A decrease in IRS1 levels reduces IGF1R signaling and blocks activation of the PI3K pathway. Conversely, inhibition of mTORC1 prevents this negative feedback loop, resulting in sustained signaling through IGF1R/IRS1 and activation of the PI3K pathway through AKT [4, 7, 8]. Inhibiting both mTOR and IGF1R with targeted agents has been shown to result in additive or synergistic antitumor activity [4, 9].
Dalotuzumab (MK-0646) is a humanized monoclonal antibody that targets IGF1R with high affinity and inhibits receptor autophosphorylation, downstream AKT phosphorylation, and cell proliferation [10]. By abrogating feedback activation of AKT caused by ridaforolimus, it can potentially lead to more effective antitumor activity [4, 11].
The likelihood of antitumor activity is suggested by the findings of a trial investigating ridaforolimus and dalotuzumab combination therapy in which patients with high-proliferation breast tumors demonstrated prolonged progression-free survival (PFS) and better tolerability with lower doses of ridaforolimus [12]. Ridaforolimus 30 mg (recommended dose) plus dalotuzumab engendered surprisingly high levels of toxicity, specifically grade 3 stomatitis, in a phase II study [12]. In a subsequent analysis, in which patients were sequentially enrolled into single-arm cohorts and treated with ridaforolimus 20 mg then ridaforolimus 10 mg in combination with dalotuzumab 10 mg/kg/week, the toxicity profiles were similar for both doses. However, the incidence of grade 3 stomatitis was greatly reduced for the lower ridaforolimus 10 mg plus dalotuzumab dose, resulting in better tolerability and patients remaining on therapy longer.
The present study was designed to assess whether the addition of an IGF1R inhibitor to the established combination of an mTOR inhibitor plus aromatase inhibitor would result in further gains in efficacy. Our hypothesis was that triplet therapy with ridaforolimus, dalotuzumab, and exemestane (R/D/E) would be more effective than doublet therapy with ridaforolimus and exemestane (R/E).
Patients and methods
Study design and patients
This was a randomized, open-label, multicenter, phase II trial in postmenopausal women with estrogen receptor (ER)-positive breast cancer whose disease had progressed after treatment with a non-steroidal aromatase inhibitor (ClinicalTrials.gov identifier: NCT01605396; protocol number PN064). Written informed consent was obtained from all participants. The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. Independent ethics committees reviewed and approved the protocol and applicable amendments for each institution.
Patients with histologically confirmed metastatic or locally advanced ER-positive/human epidermal growth factor receptor 2 (HER2)-negative, high-proliferation (Ki67 staining index ≥15%) breast cancer were randomly assigned 1:1 to receive either R/D/E or R/E. Other eligibility criteria included adequate organ function, Eastern Cooperative Oncology Group performance status 0 or 1, and disease previously refractory to letrozole or anastrozole, with ≥1 confirmed measurable metastatic lesion on computed tomography (CT) or magnetic resonance imaging (MRI) as per Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST v1.1) [13]. Patients were excluded if they were receiving any other systemic treatment for cancer; had participated in a study of an investigational agent within 30 days; had previously received rapamycin or rapamycin analogs or treatment with IGF1R inhibitors, PI3K inhibitors, or other experimental agents that target PI3K, AKT, or mTOR; were receiving long-term high-dose corticosteroids; had symptomatic or progressing brain metastases, significant or uncontrolled cardiovascular disease, or poorly controlled diabetes; were positive for human immunodeficiency virus or had active hepatitis B or C; or had known psychiatric or substance abuse disorders that would interfere with study compliance.
Treatments
In the R/D/E arm, patients received oral ridaforolimus 10 mg for five consecutive days followed by two consecutive days off each week (qd 5 ×/week) repeated weekly, intravenous dalotuzumab 10 mg/kg/week, and oral exemestane 25 mg/day. Patients in the R/E arm received ridaforolimus 30 mg qd 5 ×/week plus exemestane 25 mg/day. After the first cycle (with each cycle lasting 28 days) and in the absence of grade 2 or higher stomatitis, the ridaforolimus dose could be increased to 20 or 40 mg qd 5 ×/week in the R/D/E or the R/E arm, respectively. For both arms, temporary and permanent dose reductions were permitted to manage adverse events (AEs) according to specified dose-modification guidelines.
End points and assessments
The primary efficacy end point was PFS (time from randomization to progressive disease or death, whichever occurred earlier) in the intention-to-treat population (ITT) by central review. An additional supportive analysis of PFS was performed according to investigator review.
Secondary efficacy end points included the percentage reduction from baseline to week 16 in the sum of target lesion sizes (the difference between the sum, at baseline and at week 16, of the greatest diameter or volume of each target lesion), objective response rate (ORR; the proportion of patients whose best response was partial response or complete response according to enhanced RECIST v1.1 criteria [13]), and OS (time from randomization to death from any cause).
Bidimensional diagnostic anatomic imaging (using CT or MRI) was used for the assessment of disease status at baseline and every 8 weeks during treatment and was analyzed by independent central review.
Adverse events were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0 (https://evs.nci.nih.gov/ftp1/CTCAE/About.html). Serious AEs (SAEs) were defined as any AEs that would be life threatening or that would result in death, persistent or significant disability, hospitalization, congenital anomaly/birth defects, new cancer, overdose, or other events according to medical judgment. Stomatitis, pneumonitis, hyperglycemia, and hearing loss were prespecified as events of clinical interest. Stomatitis and pneumonitis are known side effects of rapamycin and its analogs. New onset or the worsening of preexisting hyperglycemia has been reported in clinical trials of dalotuzumab and ridaforolimus [14, 15], while hearing loss (though rare) has been reported in clinical trials of monoclonal antibodies targeting IGF1R [16,17,18]. For stomatitis, pneumonitis, and hyperglycemia, the incidence was calculated by combining related terms, as follows: stomatitis—stomatitis, aphthous stomatitis, mouth ulceration, mucosal inflammation, and tongue ulceration; pneumonitis—pneumonitis, allergic alveolitis, pneumonia, and interstitial lung disease; hyperglycemia—hyperglycemia, blood glucose increased, diabetes mellitus, diabetes mellitus with inadequate control, diabetic ketoacidosis, glucose tolerance impaired, type 1 diabetes mellitus, and type 2 diabetes mellitus.
Statistical analysis
This study was designed to randomly assign approximately 42 patients into each treatment group (R/D/E and R/E) at a 1:1 ratio, which would give 80% power to demonstrate that R/D/E was superior to R/E at an overall one-sided 10% α-level if the underlying hazard ratio (HR) was 0.5 (corresponding to an approximate 10.6 months’ improvement in median PFS, from 10.6 to 21.2 months). The sample size was event driven with a target of 38 PFS events, and the calculation was carried out based on the following assumptions: (1) PFS followed an exponential distribution; (2) median PFS in the R/E arm was ~10.6 months; (3) total study enrollment was ~9.4 months; and (4) cumulative dropout rate was ~10% at 1 year. Given the assumed median PFS in the R/E group, a statistically significant result could occur when the observed HR was ~0.66 or less.
Efficacy analyses were performed on the ITT population (all randomly assigned patients). The PFS curve and the median PFS for each treatment group was estimated using a non-parametric Kaplan–Meier method. For the primary hypothesis comparing PFS in the R/D/E and the R/E arms, R/D/E would be considered superior to R/E if the upper limit of the two-sided 85.6% confidence interval (CI) for the HR was <1. The ORR was compared using the Miettinen and Nurminen method [19], and OS was estimated using a Kaplan–Meier method with significance determined by a Cox proportional hazards model. No multiplicity adjustment was made for secondary end points because there was a single primary end point and other efficacy analyses were considered supportive, explanatory, or both.
The safety population consisted of all randomly assigned patients who received ≥1 dose of study treatment. P values and 95% CIs for between-treatment differences in the percentage of patients with events were calculated using the Miettinen and Nurminen method [19].
Results
Demographics and baseline characteristics
A total of 80 postmenopausal women from 31 sites in 15 countries were randomly assigned to either R/D/E (n = 40) or R/D (n = 40). Seventy-two trial centers participated in this study (four in Belgium, four in Columbia, three in the Czech Republic, two in Denmark, three in France, two in Germany, one in Israel, two in Italy, nine in South Korea, four in Peru, four in Portugal, four in Spain, three in Sweden, four in Taiwan, and 23 in the United States). Baseline characteristics were generally balanced between treatment groups (Table 1).
Disposition
Database lock was triggered when 38 PFS events had occurred (April 29, 2014), at which time 71 of the 80 enrolled patients (88.8%) had discontinued from the study and nine remained on treatment (Fig. 1). The median duration on ridaforolimus was 56 days (range 6–304; mean 81) for patients in the R/D/E arm and 121.5 days (range 13–292) for patients in the R/E arm. Four patients in the R/E arm increased the ridaforolimus dose per protocol to 40 mg/kg after completing the first cycle of treatment; no patients in the R/D/E arm increased the ridaforolimus dose. The most common reason for discontinuing the study was, according to investigator review, progressive disease (57.5% in the R/D/E arm and 67.5% in the R/E arm), whereas relatively few patients discontinued because of an AE (five patients [12.5%] in the R/D/E arm and three patients [7.5%] in the R/E arm).

CONSORT diagram of patient disposition through the trial. Each patient was counted once based on the latest corresponding disposition record. Patients for whom a disposition record did not exist at the time of reporting were recorded as unknown. R/D/E, ridaforolimus 10 mg qd 5 ×/week, dalotuzumab 10 mg/kg/week, and exemestane 25 mg/day; R/E, ridaforolimus 30 mg qd 5 ×/week and exemestane 25 mg/day
Safety
The safety population comprised 39 patients in the R/D/E arm (1 allocated patient did not receive study medication) and 40 patients in the R/E arm. A higher percentage of patients discontinued treatment in the R/D/E arm than the R/E arm because of AEs [12.5% (n = 5) vs. 7.5% (n = 3)] and withdrawal of consent [10% (n = 4) vs. 5% (n = 2)] (Fig. 1).
All patients experienced ≥1 AE, with more patients in the R/E arm than the R/D/E arm experiencing grade 3–5 AEs (67.5 vs. 59%; Table 2). Dose modifications due to AEs were required for fewer patients in the R/D/E arm than in the R/E arm (10.3 vs. 50%; difference −39.7%; 95% CI −56.7 to −20.4) (Table 2). Drug-related AEs were experienced by 92.3 and 100% (−7.7% difference; 95% CI −20.4 to 1.5) of patients in the R/D/E arm and the R/E arm, respectively (Table 2). The most commonly reported drug-related AEs for patients treated with R/D/E versus R/E were stomatitis (74.4 vs. 90.0%; grade ≥ 3, 23.1 vs. 25.0%), decreased appetite (33.3 vs. 22.5%; grade ≥ 3, 5.1 vs. 2.5%), dysgeusia (35.9 vs. 12.5%; no grade ≥ 3), asthenia (23.1 vs. 25.0%; grade ≥ 3, 2.6 vs. 2.5%), diarrhea (15.4 vs. 25.0%; grade ≥ 3, 5.1 vs. 0%), hyperglycemia (20.5 vs. 17.5%; grade ≥ 3, 5.1 vs. 5.0%), fatigue (20.5 vs. 10.0%; grade ≥ 3, 0 vs. 2.5%), and rash (15.4 vs. 15.0%; no grade ≥ 3) (Table 3).
More SAEs occurred in the R/E arm than the R/D/E arm [15% (n = 6) vs. 2.6% (n = 1); −12.4% difference, 95% CI −27.1 to 0.2] (Table 2). In the R/E arm, the SAEs with at least a possible association with drug (as assessed by the treating physician) were grade 5 myocardial infarction (during the safety follow-up period), grade 3 hematochezia, grade 3 decreased appetite, grade 3 asthenia, grade 2 sinus tachycardia, and grade 2 ophthalmic herpes zoster (one patient each). One patient experienced a drug-related SAE of grade 3 esophagitis in the R/D/E arm. Of the 13 deaths in the study, three were attributed to fatal AEs: one death unrelated to treatment in each of the R/D/E and the R/E arms from disease progression and 1 exemestane treatment-related myocardial infarction in the R/E arm (Table 3).
AEs of clinical interest occurred less frequently in the R/D/E arm than the R/E arm and were primarily graded 1/2 in severity (Table 3).
Efficacy
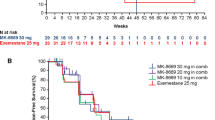
The median PFS based on independent central radiology review was 23.3 weeks (95% CI 8.71–38.43) for the R/D/E arm versus 31.9 weeks (95% CI 16.00–39.29) for the R/E arm (HR 1.18; 80% CI 0.81–1.72; P = 0.565) (Fig. 2). A secondary analysis based on local investigator evaluation yielded a median PFS of 15.3 weeks (95% CI 9.29–31.29) for the R/D/E arm versus 32.0 weeks (95% CI 23.57–39.29) for the R/E arm (HR 1.58; 80% CI 1.13–2.19; P = 0.075). Sensitivity analyses including discontinuation as a PFS event yielded a median PFS (based on independent radiology review) of 11.43 weeks (95% CI 7.86–22.57) for the R/D/E arm, and 23.57 weeks (95% CI 15.86–32.00) for the R/E arm.

Kaplan–Meier plot of progression-free survival (PFS) by independent radiology review in the intent-to-treat primary analysis population for patients receiving either ridaforolimus/dalotuzumab/exemestane (R/D/E) or ridaforolimus/exemestane (R/E)
The mean (± standard deviation [SD]) percentage change from baseline in sum of target lesion diameters at week 16 (by independent radiology review) was −19.3 ± 20.4% in the R/D/E arm (n = 15) and −10.7 ± 28.5% in the R/E arm (n = 32). By local investigator assessment, the 2 arms were very similar [−16.3 ± 29.1% (n = 20) vs. −17.7 ± 31.5% (n = 33) in the R/D/E and R/E arms, respectively]. Imaging was not available for central review for 5 patients in the R/D/E arm and one patient in the R/E arm.
More patients in the R/E arm than the R/D/E arm, based on independent radiology review, had an ORR [25% (n = 10) vs. 15% (n = 6), difference −10.0%; 95% CI 27.8–8.0; P = 0.267]. Based on local investigator assessment, the ORR was 22.5% versus 12.5% in the R/E and R/D/E arms, respectively (difference −10.0%; 95% CI 27.2–7.1; P = 0.242).
Median OS could not be calculated for either of the treatment arms. Seven patients (17.5%) in the R/D/E arm and 6 patients (15%) in the R/E arm had died by the time of this report.
Discussion
This study tested the hypothesis that triple therapy with ridaforolimus, dalotuzumab, and exemestane (R/D/E) would be more beneficial than double therapy with ridaforolimus and exemestane (R/E); however, no improvement in PFS was found in postmenopausal women with ER-positive/HER2-negative high-proliferation breast cancer. Furthermore, the PFS hazard ratio for the R/D/E arm over the R/E arm suggested a trend favoring the latter—patients treated with R/D/E were more likely to experience disease progression. Although the median PFS in the R/D/E arm was 23 weeks based on independent radiology review, and the median duration on ridaforolimus was substantially shorter at 56 days (8 weeks), the secondary PFS analysis based on local investigator evaluation yielded a median PFS of 15.3 weeks, which is closer to the mean treatment duration of 81 days (11.6 weeks) for ridaforolimus in this treatment arm. Furthermore, inclusion of discontinuation as a PFS event in sensitivity analyses also yielded a PFS (based on independent radiology review) closer to the mean duration of treatment for ridaforolimus. Discontinuation due to AEs was reported in 12.5% of patients. Another 15% of patients discontinued because of physician decision or withdrawal by patient; it is possible that tolerability concerns might also have contributed to the decision to discontinue treatment in at least some of these patients.
Patients with high-proliferation breast cancer were included in the study because of in vitro data supporting the clinical evaluation of combined IGF1R/mTOR inhibition in this population. In preclinical studies, the combination acted synergistically to significantly inhibit the growth of tumor cell lines that expressed IGF1R, and it was more effective than the single agents at suppressing tumor growth in a human xenograft model [4]. In a phase I trial, the ridaforolimus/dalotuzumab combination resulted in 3 partial responses in 11 patients (27%) who had ER-positive, high-proliferation (Ki67 ≥ 15%) breast cancer [4]. Thus it was reasonable to assume that the addition of dalotuzumab to ridaforolimus plus exemestane would increase efficacy. However, the responses observed in the phase I trial occurred with ridaforolimus doses of 20 mg qd 5 ×/week and 40 mg qd 5 ×/week, which were higher than the 10 mg qd 5 ×/week ridaforolimus dose used in the R/D/E arm in our study [4].
IGF1R inhibitors disrupt growth hormone feedback and may cause insulin resistance and subsequent hyperinsulinemia and hyperglycemia [20]. Thus, IGF1R inhibitors are often administered in conjunction with metformin to counter insulin resistance. One hypothesis for the outcomes in the current study could be hyperinsulinemia induced by dalotuzumab in the absence of metformin. The similar proportion of hypoglycemia AEs reported in the R/D/E and R/E arms suggest that this was not the case. Recently, the phase II Investigation of Serial Studies to Predict Your Therapeutic Response With Imaging And moLecular Analysis 2 (I-SPY-2) trial explored the IGF1R inhibitor ganitumab in combination with metformin in patients with stage 2/3 breast cancer and one of three tumor profiles: HER2-negative, hormone receptor-positive/HER2-negative, and hormone receptor-negative/HER2-negative [21]. Ganitumab/metformin did not meet the predefined criteria for graduation to a larger trial (≥85% bayesian predictive probability of success), nor did the addition of metformin stabilize HbA1c levels [21].
In addition, a phase II study of the anti-IGF1R antibody cetuximab in patients with ER-positive breast cancer whose cancer progressed on endocrine therapy has shown that the copies of IGF1R mRNA are low in endocrine-resistant patients, and IGF1R expression was not associated with clinical outcomes [22]. Although IGF1R expression was not evaluated in our study, all patients had cancer that progressed on at least one line of prior therapy and could be considered endocrine resistant. Therefore, endocrine-resistant tumors may have higher expression of the insulin receptor than the IGFR1, which could explain the relative lack of efficacy of IGFR1 inhibitors. However, experimental models have suggested that mTORC1 inhibition can overcome insulin receptor activation [15]; this effect may be limited by off-target toxicity which then limits the achievable dose of the mTOR inhibitor given in combination. We hypothesize that the lack of additional efficacy benefit in the triple therapy R/D/E arm in our study over the R/E arm was possibly attributed to the reduced dose of ridaforolimus required to ameliorate the effect of overlapping toxicities between dalotuzumab and ridaforolimus. These results, combined with those presented in the current study, suggest that more specific biomarker studies need to be conducted to identify potential tumor subgroups that benefit from IGF1R inhibition.
The PFS reported for the R/E arm in this study (8.0 months) is similar to that reported in the BOLERO 2 study that evaluated the mTOR inhibitor everolimus in combination with exemestane in patients with advanced breast cancer (7.8 months) [23]. Everolimus plus exemestane is an approved systemic treatment for patients whose disease has progressed after therapy with a non-steroidal aromatase inhibitor [24]. Our results indicate that ridaforolimus plus exemestane achieved a level of efficacy similar to that reported for the everolimus/exemestane combination, further supporting the concept of combined mTOR and antiestrogen therapy for patients with ER-positive breast cancer.
Clearly, dose and schedule matter; these factors should be considered when designing clinical trials with mTOR inhibitors. A higher number of drug-related toxicities was reported in the R/E arm than in the R/D/E arm, possibly reflecting the suboptimal dose of ridaforolimus in the triple combination. In particular, the incidence rates of stomatitis and pneumonitis were significantly lower in the R/D/E arm than in the R/E arm, and, because these AEs are recognized class effects of mTOR inhibitors [25, 26], the difference is likely to reflect the lower dose of ridaforolimus used in the R/D/E combination. Hence, we cannot rule out that the lack of improved benefit with the triple combination is related to relative underdosing of the mTOR inhibitor.
Careful management of common toxicities, in particular stomatitis (the most frequent toxicity with this combination), may allow higher doses of ridaforolimus to be used in combinations with IGF1R inhibitors in the future [27]. In this regard, and in retrospect, conducting this trial in multiple sites simultaneously rather than in the experienced clinical trial sites that participated in the phase I trial [4] might have resulted in a lack of experience in identifying and managing AEs, resulting in dose interruptions and ultimately in lower than optimal dose exposure. This likely contributed to the lack of difference observed between the treatment arms despite the encouraging proof-of-concept results from preclinical and phase I studies [4].
In conclusion, the combination of ridaforolimus (10 mg qd 5 ×/week), dalotuzumab, and exemestane did not improve PFS compared with ridaforolimus (30 mg qd 5 × /week) plus exemestane in postmenopausal women with ER-positive breast cancer. The incidence rates of AEs were lower in the R/D/E arm than in the R/E arm for most categories of AEs, likely because of the higher dose of ridaforolimus administered in the R/E arm. The choice of dose of ridaforolimus in the R/D/E triple combination to minimize the risk for overlapping toxicities with dalotuzumab possibly contributed to the lack of improved PFS when the IGF1R inhibitor was added to the mTOR/aromatase inhibitor combination, highlighting the need for proper management of toxicologic profiles of mTOR inhibitors in the design of future clinical trials. Our study further supports the activity of an mTOR inhibitor in combination with an aromatase inhibitor as shown in the BOLERO study [28], and it supports that the addition of an IGF1R inhibitor—while showing a better toxicologic profile—ultimately demonstrated inferior efficacy.
References
Johnston SR (2006) Clinical efforts to combine endocrine agents with targeted therapies against epidermal growth factor receptor/human epidermal growth factor receptor 2 and mammalian target of rapamycin in breast cancer. Clin Cancer Res 12:1061s–1068s
Burstein HJ (2011) Novel agents and future directions for refractory breast cancer. Semin Oncol 38(suppl 2):S17–S24
Quek R, Wang Q, Morgan JA et al (2011) Combination mTOR and IGF-1R inhibition: phase I trial of everolimus and figitumumab in patients with advanced sarcomas and other solid tumors. Clin Cancer Res 17:871–879
Di Cosimo S, Sathyanarayanan S, Bendell JC et al (2015) Combination of the mTOR inhibitor ridaforolimus and the anti-IGF1R monoclonal antibody dalotuzumab: preclinical characterization and phase I clinical trial. Clin Cancer Res 21:49–59
Elit L (2006) Drug evaluation: aP-23573—an mTOR inhibitor for the treatment of cancer. IDrugs 9:636–644
Rivera VM, Squillace RM, Miller D et al (2011) Ridaforolimus (AP23573; MK-8669), a potent mTOR inhibitor, has broad antitumor activity and can be optimally administered using intermittent dosing regimens. Mol Cancer Ther 10:1059–1071
Sun SY, Rosenberg LM, Wang X et al (2005) Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res 65:7052–7058
Baselga J, Semiglazov V, van Dam P et al (2009) Phase II randomized study of neoadjuvant everolimus plus letrozole compared with placebo plus letrozole in patients with estrogen receptor-positive breast cancer. J Clin Oncol 27:2630–2637
Kurmasheva RT, Dudkin L, Billups C, Debelenko LV, Morton CL, Houghton PJ (2009) The insulin-like growth factor-1 receptor-targeting antibody, CP-751,871, suppresses tumor-derived VEGF and synergizes with rapamycin in models of childhood sarcoma. Cancer Res 69:7662–7671
Broussas M, Dupont J, Gonzalez A et al (2009) Molecular mechanisms involved in activity of h7C10, a humanized monoclonal antibody, to IGF-1 receptor. Int J Cancer 124:2281–2293
O’Reilly KE, Rojo F, She QB et al (2006) mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 66:1500–1508
Baselga J, Morales SM, Awada A et al (2013) A phase 2 study of ridaforolimus (RIDA) and dalotuzumab (DALO) in estrogen receptor positive (ER+) breast cancer [Abstract]. Cancer Res 73(24):TPS110
Eisenhauer EA, Therasse P, Bogaerts J et al (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45:228–247
Ellis PM, Shepherd FA, Laurie SA et al (2014) NCIC CTG IND.190 phase I trial of dalotuzumab (MK-0646) in combination with cisplatin and etoposide in extensive-stage small-cell lung cancer. J Thorac Oncol 9:410–413
Fierz Y, Novosyadlyy R, Vijayakumar A, Yakar S, LeRoith D (2010) Mammalian target of rapamycin inhibition abrogates insulin-mediated mammary tumor progression in type 2 diabetes. Endocr Rel Cancer 17:941–951
Ray-Coquard I, Haluska P, O’Reilly S et al (2013) A multicenter open-label phase II study of the efficacy and safety of ganitumab (AMG 479), a fully human monoclonal antibody against insulin-like growth factor type 1 receptor (IGF-1R) as second-line therapy in patients with recurrent platinum-sensitive ovarian cancer. J Clin Oncol 31:5515
Ryan PD, Neven P, Dirix LY et al (2016) Safety of the anti-IGF-1R antibody CP-751,871 in combination with exemestane in patients with advanced breast cancer. Cancer Res 69(2):2136
Okusaka T, Ikeda M, Fukutomi A et al (2014) Safety, tolerability, pharmacokinetics and antitumor activity of ganitumab, an investigational fully human monoclonal antibody to insulin-like growth factor type 1 receptor, combined with gemcitabine as first-line therapy in patients with metastatic pancreatic cancer: a phase 1b study. Jpn J Clin Oncol 44:442–447
Miettinen O, Nurminen M (1985) Comparative analysis of two rates. Stat Med 4:213–226
Haluska P, Shaw HM, Batzel GN et al (2007) Phase I dose escalation study of the anti insulin-like growth factor-I receptor monoclonal antibody CP-751,871 in patients with refractory solid tumors. Clin Cancer Res 13:5834–5840
Yee D, Paoloni M, Van’t Veer L et al (2016) The evaluation of ganitumab/metformin plus standard neoadjuvant therapy in high-risk breast cancer: results from the I-SPY 2 trial. Presented at: San Antonio Breast Cancer Symposium; December 6–10, 2016; San Antonio, TX. Abstract P6-11-04
Gradishar WJ, Yardley DA, Layman R et al (2016) Clinical and translational results of a phase II, randomized trial of an anti-IGF-1R (cixutumumab) in women with breast cancer that progressed on endocrine therapy. Clin Cancer Res 22:301–309
Yardley DA, Noguchi S, Pritchard KI et al (2013) Everolimus plus exemestane in postmenopausal patients with HR breast cancer: BOLERO-2 final progression-free survival analysis. Adv Ther 30:870–884
National Comprehensive Cancer Network, Inc (2014) NCCN Clinical Practice Guidelines in Oncology. Breast Cancer. version 1. 2014. https://www.nccn.org/professionals/physician_gls/f_guidelines.asp. Accessed 22 Nov 2016
Boers-Doets CB, Raber-Durlacher JE, Treister NS et al (2013) Mammalian target of rapamycin inhibitor-associated stomatitis. Future Oncol 9:1883–1892
Martins F, de Oliveira MA, Wang Q et al (2013) A review of oral toxicity associated with mTOR inhibitor therapy in cancer patients. Oral Oncol 49:293–298
Pilotte AP, Hohos MB, Polson KM, Huftale TMn, Treister N (2011) Managing stomatitis in patients treated with Mammalian target of rapamycin inhibitors. Clin J Oncol Nurs 15:E83–E89
Baselga J, Campone M, Piccart M et al (2012) Everolimus in postmenopausal hormone receptor-positive advanced breast cancer. N Engl J Med 366:520–529
Acknowledgements
We thank the patients and their families for their participation and support during the study as well as the study center staff and principal investigators. Editorial support was provided by Tim Ibbotson, PhD, of ApotheCom and was funded by Merck & Co., Inc., Kenilworth, NJ.
Funding
This work was supported by a grant from Merck & Co., Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
JC has received fees for lectures and consulting from Roche, Celgene, Novartis, and Eisai. HSR has received research support from Merck & Co., Inc. and Novartis. ART received grants from Merck & Co., Inc. for conducting this study. AD, CKG, EI, MBJ, DJM, and ZW are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ and may hold stock or stock options in the company. MF has received grants from Merck Sharp & Dohme Corp. and AstraZeneca, grants and non-financial support from Novartis, and F. Hoffmann-La Roche, and non-financial support from Merck Sharp & Dohme Corp. NA, JB, JLB, MC, LE, AM, JR, SMM, OT, and KHP have nothing to disclose.
Ethical approval
The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. Independent ethics committees reviewed and approved the protocol and applicable amendments for each institution.
Informed consent
Written informed consent was obtained from all participants.
Rights and permissions
About this article

Cite this article
Rugo, H.S., Trédan, O., Ro, J. et al. A randomized phase II trial of ridaforolimus, dalotuzumab, and exemestane compared with ridaforolimus and exemestane in patients with advanced breast cancer. Breast Cancer Res Treat 165, 601–609 (2017). https://doi.org/10.1007/s10549-017-4375-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-017-4375-5