
Abstract
Detection of circulating tumor cells (CTCs) in whole blood from metastatic cancer patients by the CellSearch™ CTC Test (Veridex LLC, Warren, NJ, USA) has been shown to have clinical relevance. In addition to enumeration, there is great interest in molecular characterization of these CTCs. We aimed to establish a robust method to perform mRNA expression analysis of multiple genes by a real-time reverse transcriptase (RT)-PCR on small numbers of CTCs enriched from whole blood by the CellSearch™ system. Despite the 4 log depletion of leukocytes after CellSearch enrichment, the CTC-enriched fractions still contained leukocytes, in particular B-lymphocytes, which severely interfered with our CTC-specific gene expression profiling. After extensive washing and leukocyte-specific depletion by anti-CD45 coated magnetic beads prior to CellSearch™ enrichment, the number of leukocytes present in the enriched fraction was still high (range 60–929). However, by using a set of genes with no or minor expression by leukocytes, we succeeded to perform quantitative gene expression profiling specific for as little as one breast cancer CTC present in a CTC-enriched environment typically containing over 800 contaminating leukocytes. Our method allows molecular characterization specific for as little as one CTC, and can be used to expand the understanding of the biology of metastasis and, potentially, to improve patient management.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Circulating cells with the characteristics of tumor cells of epithelial origin have been demonstrated in blood and bone marrow of prostate, melanoma, colon, esophageal, head and neck, lung and breast cancer patients (reviewed in [1–11]). These cells have not only been shown in patients with metastatic disease, but also in those whose tumors are apparently localized [12, 13]. The development and optimization of new technologies to identify and characterize such cells, and the establishment of the association of their presence with potentially clinical significance are highly relevant.
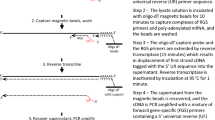
A recently developed technology to quantify the number of CTCs in whole blood of cancer patients is the use of the CellSearch™ CTC Test (Veridex LLC, Warren, NJ), so far the only US Food and Drug Administration (FDA) approved diagnostic test [14] to automate the detection and enumeration of CTCs for monitoring disease progression and therapy efficacy in metastatic prostate [15], colorectal [16] and breast [17] cancer. After enrichment using magnetic beads coated with EpCAM-specific antibodies, isolated cells are stained with fluorescent monoclonal antibodies specific for epithelial cells (CK-8/18/19), for leukocytes (CD45) and for their nuclei with a nuclear staining dye (DAPI), and subsequently enumerated by a semi-automated fluorescence microscope.
In addition to enumeration, there is great interest in molecular characterization of CTCs to understand fundamental issues such as their genetic composition, and to establish association of these genetic profiles with patient treatment outcomes. However, although this system allows capture of CTCs in blood of cancer patients by selectively isolating EpCAM-positive cells followed by quantification of DAPI- and CK-8/18/19-positive cells, there are still considerable quantities of contaminating leukocytes (DAPI+/CD45+) present after enrichment. This contamination, together with the fact that CTCs detected by CellSearch™ CTC Test appear to occur in small numbers in humans (frequently fewer than 5 CTCs per 7.5 ml blood in metastatic breast cancer patients [17]), forms a pitfall that can not be ignored when one is interested in a molecular characterization method specific for CTCs.
The purpose of this study was to optimize a robust method to perform mRNA expression analysis of up to 96 genes by real-time RT-PCR, thus conveniently fitting a 96-well plate, on as little as one CTC in an environment containing large quantities of contaminating leukocytes.
Materials and methods
Breast cancer cell lines and healthy blood donors
The MDA-MB-231, CAMA-1 and SK-BR-3 human breast cancer cell lines used in this study were all obtained from American Type Culture Collection (Manassas, VA). Extensive analysis of nearly 150 polymorphic microsatellite markers had shown that all cell lines are unique and monoclonal [18]. Clinical blood samples were obtained from breast cancer patients with metastatic disease and blood samples from healthy controls from laboratory volunteers and the Sanquin Blood Bank South-west Region (Rotterdam, The Netherlands). This study was approved by the Erasmus MC Institutional Review Board (METC 2006-248), and all donors gave their written informed consent.
Flow cytometry: absolute number of leukocytes and lymphocyte subsets in whole blood
An aliquot of 100 μl whole blood was incubated with 10 μl of the following fluorochrome conjugated antibodies: CD56 PE (DAKO, Glostrup, Denmark), CD45 FITC, CD19 PE-Cy7, CD14 APC, CD3 APC-Cy7 (all BD Biosciences) and 1 μg/ml 7-amino-actinomycin D (7AAD, Sigma–Aldrich, St. Louis, MO). After a 15 min incubation at room temperature erythrocytes were lysed with 2 ml NH4CL containing 100 μl flow count fluorospheres of known concentration (Beckman Coulter Inc, Miami, FL), which were added to obtain absolute counts. Samples were run on a FACSCanto flow cytometer (BD Biosciences) within 1 h and dead cells (7AAD positive) were excluded from analysis. Cell types could be identified with the following definitions: leukocytes (CD45+), mature myeloid (CD45+, SSChigh, CD14−), monocytes (CD45+, CD14+), basophils (CD45dim, SSClow), lymphocytes (CD45+, CD14−, SSClow), B lymphocytes (CD19+), T lymphocyte (CD3+) and NK lymphocytes (CD56+, CD3−).
Flow cytometry: absolute number of leukocytes and lymphocyte subsets in CellSearch-enriched samples
After the CellSearch procedure enriched samples were placed in a magnet (MagCellect, R&D systems, Abingdon, UK) for 10 min and supernatant was discarded. After removing the tube from the magnet the cell pellet was re-suspended in 100 μl PBS and incubated for 15 min with the same monoclonal antibody cocktail as described for whole blood samples. After washing with 2 ml PBS the tube was placed in a magnet and supernatant discarded. Cells were re-suspended in 500 μl PBS and 100 μl Flow-Count fluorospheres (Beckman–Coulter Inc, Miami, FL).
Leukocyte depletion of whole blood with anti-CD45 coated magnetic beads
EDTA blood (5.25 ml) was diluted with CellSearch buffer (3 ml) and mixed with 1 ml anti-CD45 coated magnetic beads (Dynabeads, Invitrogen, Breda, The Netherlands) and after a 30 min incubation with gently tilting and rotation placed in a magnet for 10 min. A volume of 7.5 ml blood containing unbound cells was transferred into a CellSearch tube and run on the CellTrack™ AutoPrep Analyzer (Veridex LLC, San Diego, USA) according to the manufacturer’s instructions.
Enumeration of epithelial cells spiked in whole blood
CellSave™ (Veridex LLC, San Diego, USA) EDTA blood samples (7.5 ml) of various healthy blood donors were quantitatively spiked with 20 μl cell suspension containing 1–150 cultured human breast cancer cells. To determine the actual viable cell number of epithelial cells to be spiked, a 100 μl aliquot of the cultured cells was incubated with 10 μl of 1 μg/ml 7AAD and 100 μl of FlowCount fluorospheres. After a 15 min incubation at room temperature, 2 ml PBS was added and samples were run on a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA). At least 10,000 beads were acquired to estimate the number of 7AAD negative (viable) cells. After serial dilution, spiking efficiency was controlled by counting the exact number of cells in 20 μl drops in triplicate under a phase contrast light microscope. To establish CTC recovery, samples were processed on the CellTrack™ AutoPrep Analyzer (Veridex LLC) with the CellSearch™ CTC profile Kit (Veridex LLC) and CTC counts were determined on the CellSpotter™ Analyzer (Veridex LLC) according to the manufacturer’s instructions.
RNA isolation
For gene expression studies 7.5 ml EDTA blood samples of the same healthy donors were in parallel with the enumeration studies spiked identically as described above and enriched on the CellTrack™ AutoPrep Analyzer (CellSearch CTC profile kit). After removal of the supernatant using a MagCellect Magnet (R&D Systems, Minneapolis, USA), the CellSearch-enriched cells were lysed by adding 250 μl of Qiagen RNeasy RLT Lysis buffer (Qiagen BV, Venlo, The Netherlands). The RNA lysate was immediately stored at −80°C. Cultured cells were also directly spiked in Qiagen RNeasy RLT Lysis buffer to serve as positive control. Subsequent RNA isolation was performed with the RNeasy (Micro) Kit (Qiagen) according to the manufacturer’s instructions.
cDNA synthesis, pre-amplification and real-time PCR (qRT-PCR)
Two different cDNA synthesis methods (High Capacity cDNA Archive kit from Applied Biosystems (ABI), Nieuwerkerk a/d IJssel, The Netherlands; RevertAid™ H Minus First Strand cDNA Synthesis Kit from Fermentas, St. Leon-Rot, Germany) and three different linear pre-amplification methods (TaqMan™ PreAmp from ABI suitable for multiplexing up to 100 gene expression targets; Whole Transcriptome Ovation RNA Amplification from NuGEN, Bemmel, The Netherlands; Full spectrum RNA Amplification from System Bioscience, Uden, The Netherlands) were utilized according to the manufacturer’s instructions. The resulting pre-amplified cDNA preparations were analyzed by real-time PCR in a 20 μl reaction volume in a Mx3000P™ Real-Time PCR System (Stratagene, Amsterdam, The Netherlands), using TaqMan® Gene Expression Assays in combination with TaqMan Universal PCR Master Mix No AmpErase UNG (ABI) according to the manufacturer’s instructions. PCR efficiency of each gene-specific real-time PCR session was validated with a standard curve constructed from a simultaneously run serially diluted cDNA pool of human breast fibroblasts and cell-lines. Negative controls included samples without reverse transcriptase and samples where total RNA and cDNA was replaced with genomic DNA. Quantitative values were obtained from the threshold cycle (Ct) at which the increase in TaqMan probe fluorescent signal associated with an exponential increase of PCR products reached the fixed threshold value of 0.02, which was in all cases, at least tenfold the standard deviation of the background signal.
Reference genes and data normalization
Unless stated otherwise, levels of HMBS, HPRT1 and GUSB were used to control sample loading and RNA quality, as described previously [19]. Bone marrow stromal cell antigen 1 (BST1) and protein tyrosine phosphatase receptor type C (PTPRC coding for CD45) were the control genes for leukocyte background and keratin 19 (KRT19) and tumor-associated calcium signal transducer 1 (TACSTD1 coding for EpCAM) for CTC quantification. After verification of equal PCR efficiency, the relative expression levels were quantified by using the delta threshold cycle (Ct) method, which is the difference in the target gene Ct minus the average Ct of the appropriate control genes.
Statistical analysis
Statistical analyses were performed with SPSS 15.0, Analyse-it 2.11 and GenEx Pro version 4.3.6 software for real-time PCR expression profiling. Pearson’s correlation was used to determine the degree of linear relationship between two variables. The strengths of the associations between continuous variables was tested with the non-parametric Spearman rank correlation test (r s). Gene expression levels in the various fractions were compared using the non-parametric one-tailed Wilcoxon’s test to test the null hypothesis and the Mann–Whitney U test and two-tailed paired t-test to identify genes with significantly different expression levels in between groups. A false discovery rate (FDR) control of 10% was applied to correct for multiple testing [20] and principal component analysis (PCA) to reduce multidimensional data sets to lower dimensions. All statistical tests were two-sided with P < 0.05 considered as statistically significant.
Results and discussion
Homogeneous and linear pre-amplification
The purpose of this study was to optimize a sensitive method to perform mRNA expression analysis of up to 96 genes including controls (thus conveniently fitting a 96-well plate) specific for CTCs present in an environment containing large quantities of contaminating leukocytes. Our first challenge was to find a method that would enable us to measure gene transcripts by real-time RT-PCR in total RNA isolated from as little as one cell (~10 pg RNA) in a linear and homogeneous (unbiased) manner. In view of our ultimate goal to molecularly characterize CTCs, any gene expression assay showing as a non-homogeneously amplified outlier in our tests could not be used for further gene expression analysis because the data would not be truly representative for the original sample. Therefore, our criteria to meet were high sensitivity combined with a minimum number of non-homogeneously amplified gene assays. For that, two different cDNA synthesis methods (a: high Capacity cDNA archive and b: revertaid H minus first strand cDNA synthesis) and three different linear pre-amplification methods (1: TaqMan PreAmp, 2: whole transcriptome (WT) Ovation RNA amplification and 3: full spectrum RNA amplification) were compared. To validate the linear and homogeneous nature of the various pre-amplification methods, we performed comparative tests between amplified and non-amplified cDNA with the use of total RNA from various sources. Next, the sensitivity (call rate) was calculated from the percentage of real-time PCR assays that required less than 35 cycles (considered as cycle detection threshold) for one or two cells. Thus, a call rate of for example 70% implicates that 70% of the genes can be detected reliably within 35 cycles while 30% of the genes are not detectable within 35 cycles. The homogeneity of amplification was set at a cut-off of 1 Ct, i.e., for an assay to be considered homogeneously amplified, the number of cycles that were required after pre-amplification should be within one Ct range of the number of cycles that were required for the non-amplified material.
The results of analyzing our data with the as such defined call rate, using 22 TaqMan Gene Expression Assays in combination with RNA isolated from two different cell lines and 32 assays in combination with RNA isolated from two different breast tumors, are shown in Table 1. Of the three linear pre-amplification methods (PreAmp, WT Ovation, Full Spectrum), the call rate obtained with total RNA isolated from one or two cells (i.e.,~10–20 pg total RNA) in combination with the gene specific PreAmp method from ABI with cDNA synthesized with the RevertAid H Minus First Strand cDNA Synthesis Kit from Fermentas was the highest and was significantly higher than the call rate obtained with cDNA generated with the High Archive cDNA synthesis kit (one-tailed Wilcoxon P = 0.028, n = 108). In addition, the homogeneity of amplification was superior for the PreAmp method from ABI (delta Ct amplified versus non-amplified within one Ct range for over 70% of 59 assays analyzed in two different breast tumors, while this was only 9% for the 59 assays analyzed with the WT ovation method and 31% for the 59 assays analyzed with the Full Spectrum method).
Based on these results, we continued with the RevertAid H Minus First Strand cDNA Synthesis Kit to synthesize cDNA and the TaqMan PreAmp method to amplify this cDNA. To further validate the sensitivity plus the linear and homogeneous nature of this cDNA, we performed similar comparative tests between amplified and non-amplified cDNA from various sources (breast tumor specimens, cell lines, healthy blood donors). Representative results are shown in Fig. 1a and b. These experiments showed that, following 14 pre-amplification cycles, all genes were linearly amplified in up to 30 real-time PCR cycles and that most genes could reliably be quantified at the level equivalent to one cell (Fig. 1a, red arrow in left panel). Only quantification of the less abundant transcripts, typically requiring over 30 PCR cycles, such as MAGEA3, TERT, KRT20 in this example (Fig. 1a, genes in the top of the right panel) appeared less reliable for assessment of gene transcript levels in low cell numbers such as expected in minimal residual disease. Secondly, the data implied that for 9 out of 144 different genes (6%) quantification might not be representative of the original sample (Fig. 1b; 9 assays outside the ±1 Ct range depicted by the red and blue dotted lines). Because the latter was not merely associated with transcript abundance (all outliers were found midrange from 23 to 32 Ct and not at the extreme Ct-values that are associated with high and low transcript abundance), these data demonstrated that reliability and reproducibility must be checked beforehand for each individual real-time PCR assay.


a Linearity and sensitivity. Expression levels of 32 genes were analyzed in cDNA generated from total RNA isolated from a primary human breast tumor. Prior to real-time PCR cDNA was pre-amplified in 14 cycles with the PreAmp method from ABI as described in the “Materials and methods” section, using the same TaqMan Gene Expression Assays that were used for the real-time PCR. Linearity and sensitivity of the PreAmp method was assessed by measuring transcript levels in cDNA that was serially diluted down to the equivalent of less than one cell. In the left panel Ct-values obtained after pre-amplification are shown, the right panel shows the Ct-values obtained with RNA equivalent to 1,000 cells without pre-amplification. b Homogeneous amplification. Expression levels of 144 genes were analyzed in cDNA generated from total RNA from cultured MDA-MB-231 cells. Prior to real-time PCR cDNA was pre-amplified in 14 cycles with the PreAmp method from ABI as described in the “Materials and methods” section, using the same TaqMan Gene Expression Assays that were used for the real-time PCR. Comparative tests between pre-amplified and non pre-amplified cDNA from RNA equivalent to ~2,000 cells were performed to establish the degree of homogeneous amplification of the pre-amplified material. Black dotted assays within the boundary set with red and blue dotted lines (delta Ct amplified versus non-amplified within one Ct range) are considered to be homogeneously amplified. Red dotted assays indicate an underestimation of gene expression levels after pre-amplification, blue dotted assays an overestimation. c Overall reproducibility of our method. Reproducibility of the whole procedure was assessed by performing all steps that were needed to establish gene expression levels of 75 genes in two independently performed CellSearch experiments. Each dot represents the Ct levels of a different TaqMan Gene Expression Assay as assessed in two independently performed experiments that involved 7.5 ml whole blood of the same healthy donor (HBD-4). After CellSearch enrichment, but prior to qRT-PCR, the total RNA was diluted to the equivalent of 20 blood cells (~200 pg). d Comparative gene expression analysis with 91 genes measured in one, two and ten breast cancer tumor cells. Expression levels were analyzed with real-time PCR with 91 TaqMan Gene Expression Assays in cDNA generated from total RNA isolated from 1, 2 and 10 cultured CAMA-1 to MDA-MB-231 (MM231) cells. Sample loading and RNA integrity was controlled with three additional universal reference genes (GUSB, HPRT1 and HMBS). Prior to real-time PCR cDNA was pre-amplified in 14 cycles with the PreAmp method from ABI as described in the “Materials and methods” section, using the same TaqMan Gene Expression Assays that were used for the real-time PCR. Unsupervised two-dimensional hierarchical cluster analysis was performed for comparing mRNA profiles of 1, 2 and 10 human breast cancer cells. Each horizontal row represents a gene, and each vertical column corresponds to a sample. Red color indicates a transcription level above the detection level of the particular assay, black color indicates a transcription level below the detection level of the particular assay. Data shown are the averages of two experiments after median normalization of each individual sample across all genes
Sources of variation
Next, we investigated which procedure in our protocol (CellSearch enrichment, RNA isolation, cDNA synthesis, PreAmp, qPCR), was the most susceptible to variation (see Table 2 for an overview). For this, we randomly selected a panel of 75 genes. Although all steps in the procedure added to the within- and between-run variation (Table 2), the resultant Pearson correlation coefficient between the expression levels of the 75 tested genes in independently performed experiments was never below 0.93 (see Fig. 1c for a graphical presentation of the final resultant with respect to Ct values obtained for two independently performed CellSearch experiments where 75 different TaqMan Gene Expression Assays were measured in duplicate). Thus, our method to assess transcript levels showed to be reproducible.
qRT-PCR analysis of 94 genes in one breast cancer tumor cell
Having established the feasibility of our approach, i.e., analyzing a large number of genes by pre-amplified RT-PCR in as few as a single cell, we selected 91 TaqMan Gene Expression Assays that were available in our lab at the time of performing the experiment. This set was supplemented with three universal reference genes (GUSB, HPRT1 and HMBS) to control sample loading and RNA integrity, one no-assay negative control and one no-template (H2O only) negative control. Subsequently, we assessed whether or not our method was robust enough to measure these genes in one cell with similar sensitivity and specificity compared with 10 cells. Figure 1d shows that this was the case: expression profiling obtained with 10 CAMA-1 or 10 MDA-MB-231 cells showed a gene expression profile very similar to the profile obtained with one or two cells (Spearman r s for one cell vs. 10 cells = 0.94 and 0.92, respectively, P < 0.0001). In addition, CAMA-1 and MDA-MB-231 cells clustered together for the 1, 2 and 10 cell samples. Thus, although not all genes were expressed by these two cell types or required for cell type specific clustering, our approach proved to be robust enough to characterize gene expression levels of up to 91 genes and three reference genes in RNA isolated from as little as one cultured human breast tumor cell very comparable to what was achieved with RNA obtained from 10 cultured human breast tumor cells.
Selection of pilot genes to study circulating tumor cells
Having established a method that enabled us to measure up to 94 transcripts present in as little as one cell, our next step was to select a pilot panel of genes to be measured in the CTCs after CellSearch enrichment. However, the main challenge in characterizing CTCs is to deal with the contamination with residual white blood cells that is still present after CTC enrichment. That a portion of the white blood cell population is still present in the CellSearch-enriched fraction became evident when we subjected whole blood of eight healthy individuals to CellSearch enrichment. Not only were we able to measure considerable amounts of gene transcripts in all these EpCAM-based CellSearch-enriched fractions (see for example Fig. 1c), also the variation in the amount of transcripts in-between donor derived white blood cells was enormous, ranging from 13.08 Ct to 19.98 Ct (120-fold difference) for the CD45 marker PTPRC and from 18.71 Ct to 28.34 Ct (800-fold difference) for the proliferation marker MKI67.
To investigate how these contaminating leukocytes might affect our CTC characterization, gene expression levels measured in CellSearch-enriched HBDs were compared with gene expression levels measured in three different human breast cancer reference cell lines. Results shown in Fig. 2 show that, depending on the genes included in the panel, material derived from HBD will contribute to the gene expression profile (in this example for the genes at the left side of the graph) thereby potentially interfering with the assessment of genes expressed by CTC.

Gene expression in a panel of three human breast cancer cell lines and eight HBDs (after EpCAM-based CellSearch enrichment). To evaluate the potential contribution of gene transcript levels derived from eight different HBDs after CellSearch enrichment, transcript levels of a pilot set of genes were compared with levels measured in three human breast cancer reference cell lines [MDA-MB-M231 (MM-231), CAMA-1 (CAMA) and SKBR-3 (SKBR3)]. For this, all cDNA preparations were subjected to pre-amplification and qPCR using a pilot set of 26 TaqMan Gene Expression Assays. The average transcript levels ± SEM for the unspiked HBD (n = 8, RNA from 7.5 ml whole blood after CellSearch enrichment) and the reference cell lines (n = 2 each, RNA from 10 to 100 cells) are plotted as auto scaled ratios of the universal reference gene GUSB to correct for differences in RNA input and quality
Reduction of leukocyte contribution
Our first attempt to reduce the confounding contribution of these white blood cells involved extensive washing and resuspension after CellSearch enrichment of the epithelial cells. If the remaining white blood cells were only trapped in the mass of ferrobeads after CellSearch, this procedure should reduce the amount of non-specifically trapped cells. To address this hypothesis of leukocytes a-specifically trapped in the ferrobead mass, 20 CAMA-1 cells were spiked in duplicate in 7.5 ml whole blood from two different healthy donors and subjected to CellSearch enrichment. This resulted in both cases in a 75% recovery (15 CAMA-1 cells were counted in the cartridge in both cases). In parallel, enriched fractions were analyzed after 5 cycles of extensive washing with PBS and resuspension in new PBS prior to RNA isolation. Although this rigorous washing did result in an up to 20-fold reduction in contaminating leukocytes as assessed by quantitative RT-PCR, a similar and undesirable reduction was seen in the amount of recovered CAMA-1 tumor cells (only 1 out of the 20 spiked CAMA-1 cells was recovered).
We therefore took another approach and attempted to deplete our samples of leukocytes prior to CellSearch enrichment. For this,~80 epithelial tumor cells were spiked in 7.5 ml whole blood of three donors. The preparations were then subjected to leukocyte-specific depletion with anti-CD45 coated magnetic beads, followed by a standard CellSearch enrichment procedure. Although leukocyte-specific depletion prior to CellSearch did reduce the contribution of leukocytes by up to 92% (n = 3, range 63–92%), there was still a considerable number of leukocytes (n = 3, range 60–929) present in the CellSearch-enriched fraction. Thus, it appeared that at least some of the white blood cells were captured by the anti-EpCAM monoclonal antibody and not merely non-specifically trapped in the ferro bead mass.
Origin of concomitantly isolated leukocytes
Because non-specific trapping did not appear the (only) reason for the presence of the leukocytes after CellSearch, we were curious if there was a specific white blood cell population more prone to EpCAM-based CellSearch enrichment and thus the major culprit responsible for the contribution of leukocyte-derived RNA in our CTC-enriched preparations. To address this in more detail, and to get an estimate of the actual number of contaminating leukocytes still present after CellSearch enrichment, we assessed absolute numbers (and percentages) of white blood cell subsets present in 7.5 ml whole blood from two HBDs before and after CellSearch enrichment. The resulting distributions of specific leukocyte blood cell subsets before and after this enrichment are summarized in Table 3. Although the CellSearch procedure resulted in over 99.9% (4 log) depletion of leukocytes (1,381 out of 580 × 105 and 810 out of 600 × 105 leukocytes remained after CellSearch enrichment), there was still a high number of over 800 leukocytes present after EpCam-based enrichment. Apparently, the enrichment particularly favored B lymphocytes (and possibly monocytes) to the detriment of T lymphocytes (Table 3). This specific enrichment of B lymphocytes and depletion of T lymphocytes was confirmed in additional experiments with other HBDs (data not shown).
Profiling whole blood to correct for leukocyte contamination in CellSearch-enriched fractions
Another way that has been used to circumvent this problem of co-assessment of genes derived from contaminating leukocytes is to assess gene expression levels in a fraction of whole blood of the patient that was recovered after the CTCs had been captured. Next, only candidate genes are selected by comparing the corresponding depleted and enriched fractions for minimal expression in the CTC-depleted fraction and significant expression in the CTC-enriched fraction [21]. Also less sophisticated methods such as only selecting markers with high expression in tissue samples from primary tumors and a median 1,000-fold lower expression in normal blood [7] have been used. However, our data in Table 3 show that such approaches may not be accurate for EpCAM-enriched samples because the whole blood cell profile prior to CellSearch is not representative for the blood cell profile after CellSearch. This was confirmed when we analyzed expression of 144 genes by qRT-PCR in six different HBDs before and after CellSearch enrichment. Of these 144 genes, most of them selected in silico, based on their reported low expression in white blood cells (http://cgap.nci.nih.gov/SAGE/AnatomicViewer), 119 genes were still expressed to some extent in these HBDs. After correcting for multiple comparison using a false discovery rate of 10%, expression levels of TACSTD1 (EpCAM, although higher in the HBDs after CellSearch, not statistically significant with P FDR adjusted = 0.018), KRT19, PTPRC (CD45) and BST1 showed no difference between the two groups. But 14 genes (10.9%) were consistently differentially expressed in all six HBDs after CellSearch enrichment (all lower) (P FDR adjusted < 0.012).
Selection of pilot genes not expressed by leukocytes to study circulating tumor cells
Obviously, inclusion of genes more dominantly expressed by the surplus of leukocytes will favor characterization of the leukocytes instead of the few, frequently only one, CTC present in the pool of leukocytes that remain after CellSearch enrichment of the patient’s blood samples. Having established that profiling whole blood to correct for leukocyte contribution would be inappropriate, especially for such sensitive methods like real-time RT-PCR with inclusion of a limited number of up to 96 genes, we aimed for another way to circumvent this problem of co-assessment of genes derived from contaminating leukocytes. This was achieved by focusing on genes that are higher expressed in breast cancer cells compared with levels measured in white blood cells after CellSearch enrichment. For this, three different human breast cancer reference cell lines were spiked in six different HBDs in a final quantity equivalent to approximately one CTC (~10 pg) per ml whole blood. Gene expression levels using a pilot set of 25 TaqMan Gene Expression Assays were normalized on KRT19 and compared with PCA with and without inclusion of the 9 genes shown to be predominantly expressed by the leukocytes (genes on the left side of the panel in Fig. 2). Even with this limited and still explorative set of 25 genes, improvement was clearly noticed in our PCA analysis after exclusion of 9 genes that were predominantly expressed by leukocytes (Fig. 3, compare the left panel were all 25 genes were included with the right panel were 9 of the more leukocyte-specific genes were excluded). Although certainly still not optimal, only by excluding the genes more dominantly expressed by the leukocytes did the HBDs spiked with MDA-MB-231 cells cluster separately from the same HBDs spiked with either SKBR-3 cells (Fig. 3, top panel) or CAMA-1 cells (Fig. 3, bottom panel) and together with the respective unspiked cell line preparations (as demonstrated by the closer location of the closed and open symbols in the right panel).

PCA before (left) and after (right) exclusion of genes expressed by leukocytes. To investigate how contaminating leukocytes might affect CTC characterization, gene expression profiles of cells from three different human breast cancer reference cell lines [MDA-MB-231 (▼), CAMA-1 (■), and SKBR-3 (●)], were compared before (closed symbols) and after (open symbols) spiking these cells in CellSearch-enriched HBDs. For this, cDNA’s of six different HBDs were spiked -after RNA isolation and cDNA synthesis, but prior to pre-amplification and real-time PCR- with cDNA from the three different human breast cancer reference cell lines in a final quantity equivalent to approximately one CTC (~10 pg) per ml whole blood. Gene expression levels using a pilot set of 25 TaqMan Gene Expression Assays were normalized on KRT19 and compared by principal component analysis (PCA) with (left panel) and without (right panel) inclusion of the 9 genes shown to be predominantly expressed by the leukocytes (genes on the left side of the panel in Fig. 2). Reference cells in the absence of HBD: MDA-MB-231, closed triangles; SKBR-3, closed circles; CAMA-1, closed squares. Approximately one reference cell spiked in cDNA obtained from 1 ml CellSearch-enriched blood from a HBD: MDA-MB-231, open triangles; SKBR-3, open circles; CAMA-1, open squares
Finally, as proof of principle, we analyzed CellSearch-enriched blood of ten breast cancer patients with metastatic disease with our multiplex real-time PCR method to molecularly characterize CTCs in large quantities of contaminating leukocytes. For this experiment, 7.5 ml EDTA blood samples were processed in parallel for RNA and CTC count on the CellTrack™ AutoPrep Analyzer with the CellSearch™ CTC profile Kit and CTC counts were determined on the CellSpotter™ Analyzer. From one of the patients we were unable to extract sufficient good quality RNA for a reliable analysis. The results of the remaining nine patients are described in Fig. 4. And indeed, even with this limited and still explorative set of only 16 genes, distinct gene expression patterns were seen in the blood of the five patients with CTCs when compared to the four patients that were devoid of CTCs. In addition, we noticed a discrepancy between the CTC count in the blood of patient-3, i.e., the third green bar in Fig. 4 (no CTCs were detected by the CellSpotter™ Analyzer), and the gene expression pattern associated with this patient (indicative for the presence of CTC), suggesting that our sensitive PreAmp-RT-PCR method may have detected the presence of a CTC missed by the CellSearch counting method. Alternatively, no CTCs were present in the 7.5 ml of blood used for enumeration while the blood sample used for molecular characterization contained at least one CTC.

CTC-specific gene expression levels in clinical samples. To evaluate the usefulness of our method for clinical samples, gene expression levels were measured in CellSearch-enriched preparations from 7.5 ml EDTA blood samples of breast cancer patients with metastatic disease. CTC counts were assessed by use of the CellTrack™ AutoPrep Analyzer with the CellSearch™ CTC profile Kit as described in the “Materials and methods” section. For the gene expression study 7.5 ml EDTA blood samples of the same breast cancer patients were enriched in parallel. After CellSearch enrichment, RNA was isolated and cDNA was prepared as described in the “Materials and methods” section. All cDNA preparations were subjected to pre-amplification and qPCR using a pilot set of 16 TaqMan Gene Expression Assays that had shown not to be expressed in CellSearch enriched preparations of healthy blood donors (see Fig. 2), and three universal reference genes. To evaluate the CTC-specific contribution, the transcript levels were first normalized on the average of the universal reference genes GUSB, HMBS, and HPRT1 (house keeper set) to correct for differences in RNA input and quality, and next on KRT19. Patients are ranked according the number of CTCs counted in the 7.5 ml blood samples, with no detectable CTCs present in the peripheral blood of four patients (closed green bars patients-1 to -4) and 2–342 CTCs in the peripheral blood of the remaining five patients (hatched grey bars patient-5; 2 CTCs, patient-6; 2 CTCs, patient-7; 6 CTCs, patient-8; 12 CTCs, patient-9; 342 CTCs). Note the discrepancy between the CTC count of patient-3 and the gene expression pattern associated with this patient
In summary, our study shows that it is possible to perform mRNA expression analysis of up to 96 genes (thus conveniently fitting a 96-well plate) specific for as little as one cell. If this one cell is a CTC present in an environment containing large quantities of contaminating leukocytes, the only restriction is that the selected genes are not, or below a threshold detection value, i.e., much lower, expressed by these leukocytes. With the restriction of not using genes more dominantly expressed by CellSearch-enriched leukocytes, any gene set specific for any cancer type can be implemented in the method we described. These genes may represent markers to identify the tissue origin of the CTCs, for example by implementing the markers described by Xi et al. [7], as well as more cancer-specific markers such as those useful for drug targeting. The resulting data can be used to further characterize cancer type specific CTCs, thereby potentially improving our insight into biological processes and ultimately patient management.
References
Jiang WG, Martin TA, Mansel RE (2002) Molecular detection of micro-metastasis in breast cancer. Crit Rev Oncol Hematol 43:13–31. doi:10.1016/S1040-8428(01)00181-0
Gilbey AM, Burnett D, Coleman RE, Holen I (2004) The detection of circulating breast cancer cells in blood. J Clin Pathol 57:903–911. doi:10.1136/jcp.2003.013755
Ring A, Smith IE, Dowsett M (2004) Circulating tumour cells in breast cancer. Lancet Oncol 5:79–88. doi:10.1016/S1470-2045(04)01381-6
Zieglschmid V, Hollmann C, Bocher O (2005) Detection of disseminated tumor cells in peripheral blood. Crit Rev Clin Lab Sci 42:155–196. doi:10.1080/10408360590913696
Budd GT, Cristofanilli M, Ellis MJ, Stopeck A, Borden E, Miller MC, Matera J, Repollet M, Doyle GV, Terstappen LW et al (2006) Circulating tumor cells versus imaging—predicting overall survival in metastatic breast cancer. Clin Cancer Res 12:6403–6640. doi:10.1158/1078-0432.CCR-05-1769
Morgan TM, Lange PH, Vessella RL (2007) Detection and characterization of circulating and disseminated prostate cancer cells. Front Biosci 12:3000–3009. doi:10.2741/2290
Xi L, Nicastri DG, El-Hefnawy T, Hughes SJ, Luketich JD, Godfrey TE (2007) Optimal markers for real-time quantitative reverse transcription PCR detection of circulating tumor cells from melanoma, breast, colon, esophageal, head and neck, and lung cancers. Clin Chem 53:1206–1215. doi:10.1373/clinchem.2006.081828
Sleijfer S, Gratama JW, Sieuwerts AM, Kraan J, Martens JW, Foekens JA (2007) Circulating tumour cell detection on its way to routine diagnostic implementation? Eur J Cancer 43:2645–2650
Bertazza L, Mocellin S, Nitti D (2008) Circulating tumor cells in solid cancer: tumor marker of clinical relevance? Curr Oncol Rep 10:137–146. doi:10.1007/s11912-008-0022-y
Pantel K, Brakenhoff RH, Brandt B (2008) Detection, clinical relevance and specific biological properties of disseminating tumour cells. Nat Rev Cancer 8:329–340. doi:10.1038/nrc2375
Steen S, Nemunaitis J, Fisher T, Kuhn J (2008) Circulating tumor cells in melanoma: a review of the literature and description of a novel technique. Proceedings (Bayl Univ Med Cent) 21:127–132
Lang JE, Hall CS, Singh B, Lucci A (2007) Significance of micrometastasis in bone marrow and blood of operable breast cancer patients: research tool or clinical application? Expert Rev Anticancer Ther 7:1463–1472. doi:10.1586/14737140.7.10.1463
Davis JW, Nakanishi H, Kumar VS, Bhadkamkar VA, McCormack R, Fritsche HA, Handy B, Gornet T, Babaian RJ (2008) Circulating tumor cells in peripheral blood samples from patients with increased serum prostate specific antigen: initial results in early prostate cancer. J Urol 179:2187–2191. doi:10.1016/j.juro.2008.01.102
Kahan LS (2004) Medical devices; immunology and microbiology devices; classification of the immunomagnetic circulating cancer cell selection and enumeration system. Final rule. Fed Regist 69:26036–26038
Danila DC, Heller G, Gignac GA, Gonzalez-Espinoza R, Anand A, Tanaka E, Lilja H, Schwartz L, Larson S, Fleisher M et al (2007) Circulating tumor cell number and prognosis in progressive castration-resistant prostate cancer. Clin Cancer Res 13:7053–7058. doi:10.1158/1078-0432.CCR-07-1506
Cohen SJ, Alpaugh RK, Gross S, O’Hara SM, Smirnov DA, Terstappen LW, Allard WJ, Bilbee M, Cheng JD, Hoffman JP et al (2006) Isolation and characterization of circulating tumor cells in patients with metastatic colorectal cancer. Clin Colorectal Cancer 6:125–132. doi:10.3816/CCC.2006.n.029
Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LW et al (2004) Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med 351:781–791. doi:10.1056/NEJMoa040766
Harkes IC, Elstrodt F, Dinjens WN, Molier M, Klijn JG, Berns EM, Schutte M (2003) Allelotype of 28 human breast cancer cell lines and xenografts. Br J Cancer 89:2289–2292. doi:10.1038/sj.bjc.6601448
Sieuwerts AM, Meijer-van Gelder ME, Timmermans M, Trapman AM, Garcia RR, Arnold M, Goedheer AJ, Portengen H, Klijn JG, Foekens JA (2005) How ADAM-9 and ADAM-11 differentially from estrogen receptor predict response to tamoxifen treatment in patients with recurrent breast cancer: a retrospective study. Clin Cancer Res 11:7311–7321. doi:10.1158/1078-0432.CCR-05-0560
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc Ser B Methodol 57:289–300
Smirnov DA, Zweitzig DR, Foulk BW, Miller MC, Doyle GV, Pienta KJ, Meropol NJ, Weiner LM, Cohen SJ, Moreno JG et al (2005) Global gene expression profiling of circulating tumor cells. Cancer Res 65:4993–4997. doi:10.1158/0008-5472.CAN-04-4330
Acknowledgments
This work was supported by the Netherlands Genomic Initiative (NGI)/Netherlands Organisation for Scientific Research (NWO).
Author information
Authors and Affiliations
Corresponding author
Additional information
Anieta M. Sieuwerts and Jaco Kraan contributed equally to this study.
Rights and permissions
About this article
Cite this article
Sieuwerts, A.M., Kraan, J., Bolt-de Vries, J. et al. Molecular characterization of circulating tumor cells in large quantities of contaminating leukocytes by a multiplex real-time PCR. Breast Cancer Res Treat 118, 455–468 (2009). https://doi.org/10.1007/s10549-008-0290-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-008-0290-0