
Abstract
Hyperinsulinemic hypoglycemia (HI) is the most common cause of hypoglycemia in children. Impairment of cellular pathways involved in insulin secretion from pancreatic β-cells, broadly classified as channelopathies and metabolopathies, have been discovered in the past two decades. The increasing use of NGS target panels, combined with clinical, biochemical and imaging findings allows differentiating the diagnostic management of children with focal forms, surgically curable, from those with diffuse forms, more conservatively treated with pharmacological and nutritional interventions. Specific approaches according to the subtype of HI have been established and novel therapies are currently under investigation. Despite diagnostic and therapeutic advances, HI remains an important cause of morbidity in children, still accounting for 26–44% of permanent intellectual disabilities, especially in neonatal-onset patients. Initial insult from recurrent hypoglycemia in early life greatly contributes to the poor outcomes. Therefore, patients need to be rapidly identified and treated aggressively, and require at follow-up a complex and regular monitoring, managed by a multidisciplinary HI team. This review gives an overview on the more recent diagnostic and therapeutic tools, on the novel drug and nutritional therapies, and on the long-term neurological outcomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hyperinsulinemic hypoglycemia (HI) is the most frequent cause of severe and persistent hypoglycemia in the neonatal period and early infancy (1:20,000–1:50,000 live births, and 1:2500 in areas with high rates of consanguinity, such as Saudi Arabia) (Bruining 1990). HI is caused by uncontrolled or excessive insulin secretion for the prevailing glucose levels. Patients present with recurrent episodes of profound hypoglycemia requiring rapid and intensive treatment with high dose glucose infusions and i.v. glucagon to prevent irreversible neurological sequelae (De Leon and Stanley 2007; Kapoor et al 2009a; Arnoux et al 2011; Stanley and Matschinsky 2012; Stanley 2016). Diagnosis is defined by the finding of inappropriate unsuppressed insulin during hypoglycemia and/or by indirect signs of inappropriate insulin excess, such as suppressed circulating non-esterified fatty acids (NEFA), hypoketonemia, hyperglycemic response to glucagon and high glucose demand (De Leon and Stanley 2013; Ferrara et al 2016).
Pathophysiology
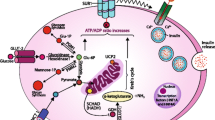
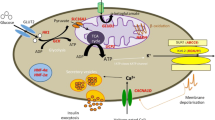
The insulin secretion from pancreatic β-cells is dependent from intracellular glucose metabolism, as summarized in Fig. 1. As soon as glucose enters the β-cells through GLUT1 and GLUT2, it is phosphorylated by glucokinase (GCK) to glucose-6-phosphate (glucose-6-P), then converted to pyruvate through the glycolysis. Pyruvate can enter the mitochondrion and, through its oxidation in the TCA cycle, increases the ATP/ADP ratio, which causes the inactivation of the pancreatic ATP-sensitive potassium (KATP) channels. Closure of KATP channels leads to depolarization of the plasma membrane, activation of voltage-gated calcium channels, elevation of cytosolic Ca2+, and release of insulin into the circulation (Stanley 2016).

Cellular pathways of insulin secretion. Blood glucose enters the β-cell through GLUT1 and GLUT2. It is phosphorylated by GCK to glucose-6-P, then fuels the glycolysis and is converted to pyruvate, which enters into the mitochondrion. Its oxidation in TCA cycle raises the ATP/ADP ratio, which is followed by the inactivation of the KATP channels. Closure of KATP channels leads to depolarization of the plasma membrane, activation of voltage-gated calcium channels and elevation of cytosolic Ca2+ . The rise of the intracellular Ca2+ concentration stimulates insulin secretion from granules and insulin transcription. Gray boxes indicate known genes causing HI
Genetics and histology
In the past, diffuse HI was erroneously labeled as “nesidioblastosis,” presumed to be an embryological anomaly of β-cells proliferating from ductal epithelium (Laidlaw 1938; Yakovac et al 1971). Subsequently, it became clear that nesidioblastosis is a common feature of the pancreas in normoglycemic neonates and infants, therefore the term was abandoned (Rahier et al 1981; Palladino and Stanley 2011). Currently, mutations in 11 genes have been associated with HI (Stanley 2016). Furthermore, INSR mutations have also been reported causing HI (Hojlund et al 2004). However, the causative role of INSR is still debated. They can be broadly categorized into two groups, channelopathies and metabolopathies (Dunne et al 2004), as listed in Table 1. Approximately 300 different loss-of-function mutations in ABCC8 and 30 in KCNJ11, which respectively encode for the SUR1 and Kir6.2 subunits of the KATP channel (channelopathies), account for more than 60% of HI cases (De Leon and Stanley 2017; Kapoor et al 2013; Faletra et al 2013; Flanagan et al 2009; Snider et al 2013). Mutations on these two genes have been associated with two histological aspects of the endocrine pancreas. A diffuse form, affecting all β-cells, inherited as either autosomal recessive or dominant traits, and a focal form, which results from the combination of a paternally inherited germinal mutation and a somatic loss of heterozygosity of the maternal allele in a restricted group of β-cells (De Lonlay et al 1997). Histologically the two forms are different and can be easily recognized (Rahier et al 2011). A third form, defined as atypical HI, has also been described and accounts for approximately 10–15% of patients undergoing pancreatectomy. Patients with atypical form normally seek treatment later in childhood, have unknown genetic cause of disease, and do not exhibit the characteristic histopathological findings of diffuse and focal HI. These forms can present as segmental mosaic forms or extensive
focal forms (Sempoux et al 2011; Capito et al 2011). More recently, analysis of post-operative pancreatic samples strongly reaffirmed the role of islet cell nucleomegaly as the hallmark of diffuse HI, when enlarged nuclei are detected in more than one-third of islets. Conversely, nucleomegaly was eightfold lower both in focal and atypical HI, along with a higher proliferation rate (Han et al 2016). Atypical forms can be associated with somatic mutations of HK1 and GCK (Henquin et al 2013). Alterations outside the β-cells lineage imply that HI may directly affect other pancreatic lineages. Abnormal somatostatin-stained δ-cells have been reported in diffuse HI (Salisbury et al 2015).
Metabolopathies refer to genetic defects in cellular metabolic pathways that, with different mechanisms and inheritance, lead to more rare forms of diffuse HI (Table 1). Metabolopathies include genes involved in fatty acid oxidation (Molven et al 2004), in energy and aminoacid metabolism (Glaser et al 1998; Otonkoski et al 2003; Gonzalez-Barroso et al 2008; Henquin et al 2013; Pinney et al 2013; Stanley et al 1998; Staufner et al 2016), in protein glycosylation (Jaeken et al 1998; Sun et al 2005; Shanti et al 2009; Miller et al 2011; Tegtmeyer et al 2014) as well as in genes encoding for transcription factors (Pearson et al 2007; Flanagan et al 2010) and for insulin receptor (Hojlund et al 2004). More detailed descriptions of the metabolopathies are discussed in the Supplementary material. Moreover, HI has also been associated with several syndromes (Table 2). Details on novel syndromic disorders presenting HI and on molecular mechanisms of HI in patients with BWS are reported as Supplementary material.
Diagnosis
The diagnosis of HI is defined by an unsuppressed detectable plasma insulin level (>2–3 μU/ml) in a critical sample collected at the time of spontaneous hypoglycemia or when plasma glucose is lower than 50 mg/dl in a diagnostic fasting test (Thornton et al 2015). Since plasma insulin concentrations are frequently not elevated, the diagnosis also relies on signs of inappropriate insulin excess, which include suppressed NEFA (<1.7 mM), hypoketonemia (<1.8 mM), a hyperglycemic response to i.m. administration of glucagon (delta glucose >30 mg/dl in 30 min) and on a high glucose demand (>10 mg/kg/min in neonates) (Arnoux et al 2011; De Leon and Stanley 2013; Ferrara et al 2016). Once HI is defined as the cause of hypoglycemia, in parallel with treatment start, the next step is the differentiation between the diffuse and focal forms which have different management and outcome, and hence differential diagnosis becomes a crucial point. In the late 1990s, pancreatic venous sampling (PVS), with measurement of insulin concentration in the pancreatic drainage veins, and selective pancreatic arterial calcium stimulation with hepatic venous sampling (ASVS) were the elective procedures to differentiate between diffuse and focal HI (De Lonlay et al 1999; Stanley et al 2004). In 2005, 18F–DOPA PET/TC imaging overcame the PVS and ASVS, due to higher sensitivity, specificity, and accuracy in localizing focal lesions in the pancreas (Ribeiro et al 2005; Otonkoski et al 2006; Treglia et al 2012; Blomberg et al 2013; Laje et al 2013) or in ectopic sites (Hussain et al 2006). Focal HI is an elective indication to partial pancreatectomy, which allows the complete cure of disease. On the other hand, diffuse HI is first approached with conservative pharmacological treatment, and only when medical measures are ineffective, a near-total pancreatectomy is required. However, this procedure is often associated with later appearance of diabetes and an increased risk of exocrine pancreatic failure (De Lonlay et al 1999; Beltrand et al 2012; Arya et al 2014b; Lord et al 2015), and does not guarantee the remission of hypoglycemia (Arya et al 2014b).
Most of current diagnostic algorithms recommend 18F–DOPA PET/TC only after testing the response to medical therapy, based on the assumption that focal HI is always unresponsive, therefore avoiding the procedure in patients showing a response to first line drugs (Kapoor et al 2009a, b; Arnoux et al 2011; De Leon and Stanley 2017). However, although rarely, focal HI could also be drug-responsive (Touati et al 1998; Loechner et al 2011; Ismail et al 2012; Kapoor et al 2013; Maiorana et al 2014). Since neurological sequelae have been reported in HI patients despite long-term diazoxide therapy (Meissner et al 2003; Avatapalle et al 2013), the indication to perform 18F–DOPA PET/TC is mainly driven by genetic analysis, also in diazoxide-responsive patients (Maiorana et al 2014). Indeed, the recent wide use of next generation sequencing (NGS) targeted panels, which represents a high-throughput technology for rapid genetic screening diagnosis (Ponzi et al 2016), further strengthens this diagnostic approach (Fig. 2). Preoperative diagnosis of focal HI is of great importance for clinical decision-making. Predicting the clinical phenotype of novel variants might be feasible for null mutations, such as nonsense defects, because these could only act in a recessive manner. However, the large number of novel missense mutations may lead to difficult diagnostic interpretation. They could potentially be either recessively or dominantly inherited or might also represent variants of unknown significance. Sensitivity of mutation analysis for predicting focal HI based on findings of a monoallelic recessive KATP mutation was found to be 97%, with a specificity of 90%, that slightly increased in the case of paternal inheritance (Snider et al 2013). In addition, some cases lacking mutations might have a postzygotic mutation of dominant HI genes as GCK (Henquin et al 2013). Finally, the possibility of novel genetic causes of HI is likely. Therefore, accurate and timely prediction of phenotype based on genotype through expression study and parental history is crucial to limit exposure to persistent hypoglycemia in infants and children with HI (Snider et al 2013).

Suggested diagnostic and management algorithm for HI. Once the diagnosis of HI and medical therapy have been established, a metabolic diagnostic work-up is recommended to identify specific inherited disorders which include hyperinsulinism-hyperammonemia (HI-HA) syndrome and SCHAD. As highlighted by the asterisk, the clinical phenotype may be of help in further metabolic testing to diagnose CDGs and ADK deficiency. In patients positive for metabolic biomarkers, further genetic analysis is needed to confirm diagnosis and for genetic counseling. Patients negative for metabolic biomarkers should undergo a NGS screening for known genes causing HI. When genetic analysis is consistent with the suspicion of focal HI, patients should undergo 18F DOPA PET/CT to look for a focal form, which is an elective indication for partial pancreatectomy. Conversely, diffuse forms should be conservatively treated with drug and nutritional therapy. Near-total pancreatectomy should be considered only in the case of unresponsive diffuse HI. Patients with no detectable mutations (wt/wt) should also undergo 18F DOPA PET/CT looking for some atypical forms, potentially treatable by limited pancreatectomy, i.e., extensive focal forms. Wt, wild type; mt, mutant; wt/mt dominant§, already reported in the literature or family tree strongly suggestive of dominant negative mutation; transferrin IEF* and aminoacids for methionine (met)*, to be performed in selected cases, according to the clinical phenotype; U, urinary; DZX, diazoxide
Current treatment
Treatment of HI includes the emergency management of hypoglycemia and the long-term therapy.
At diagnosis, the goal of the emergency therapy is to promptly restore normoglycemia using concentrated glucose infusion. Given the high glucose demand, continuous i.v. glucagon infusion (1–2 mg/day) is helpful in maintaining normoglycemia, reducing fluids overload especially in neonates. The main goal of the long-term treatment is the prevention of neurological damage. This is obtained by maintaining normoglycemia, tailoring the optimal treatment regimen according to the patient characteristics and type of hyperinsulinism. Diazoxide is the first line drug to be attempted and is considered the mainstay long-term therapy (Aynsley-Green et al 2000). Diazoxide opens the KATP channel via SUR1 binding, thus reducing insulin secretion. It is administered perorally with a dose range of 5–15 mg/kg/day. Common side effects include hypertrichosis and fluid retention, that might cause an increased risk of heart failure, especially in pre-term newborns (Welters et al 2015). Infants on intravenous fluids are especially at risk and are likely to require intensive diuretic therapy (hydrochlorothiazide and furosemide) to control diazoxide-induced fluid retention. Diazoxide-responsiveness is defined as the ability to wean i.v. glucose infusion, while maintaining normoglycemia on a normal feeding schedule and an age-based fasting after at least 5 days from therapy initiation at a maximal dose (Arnoux et al 2011; De Leon and Stanley 2017).
In diazoxide-unresponsive patients, the second line drug is octreotide, a somatostatin analogue (Glaser et al 1989). Although not officially approved for HI (off-label use), octreotide is administered subcutaneously 3–4 times/day at the dosage of 5–20 μg/kg/day. Octreotide reduces insulin secretion with multiple mechanisms, by activating KATP channels, affecting intracellular translocation of Ca2+ and through a direct inhibition of insulin transcription via activation of protein kinase A (Welters et al 2015). Side effects include tachyphylaxis, site injection nodules, diarrhea and gallstones. Tachyphylaxis is probably due to down-regulation of the β-cell somatostatin receptors and often limits the efficacy of the drug. Necrotizing enterocolitis has been reported in neonates (Laje et al 2010; Reck-Burneo et al 2008; Hawkes et al 2016). Octreotide has also been associated with elevated liver enzymes, both severe and transient (Avatapalle et al 2012; Demirbilek et al 2014).
Calcium channel antagonists, such as nifedipine or amlodipine, have been anecdotally attempted in a few HI patients (Muller et al 2004). There is general consensus that these drugs are not considered as indicated because of the lack of effectiveness (Guemes et al 2017).
Unresponsive or partially responsive HI may require frequent glucose enriched oral feedings, with frequent or continuous enteral feedings (Arnoux et al 2010). Low protein diet with a reduced leucine intake, is recommended in the leucine-sensitive hyperinsulinism-hyperammonemia syndrome (Zammarchi et al 1996; De Lonlay et al 2001; Hsu et al 2001; Kelly et al 2001).
Novel drug therapies
Long-acting somatostatin-analogues
In the last few years, long-acting somatostatin-analogues (LAR-octreotide and lanreotide) have been successfully used in children over 1 year of age responsive to octreotide (Modan-Moses et al 2011; Le Quan Sang et al 2012; Kuhnen et al 2012). These analogues act through specific receptors (SST2) with KATP-dependent and independent mechanisms. The use of standard octreotide, which requires multiple subcutaneous administrations, is burdensome for long-term treatment, whereas long-acting somatostatin-analogues can be administered every 28 days, with striking advantages for patients and families. LAR-octreotide is administered i.m., its concentration raises in 7 days and remains stable for 4 weeks; lanreotide is administered deeply subcutaneously, its concentration raises quickly in a few hours and then progressively declines within the following 4 weeks. The dosage of long-acting somatostatin-analogues is equal to the cumulative dose of 30 days of octreotide (Astruc et al 2005; Modan-Moses et al 2011; Le Quan Sang et al 2012). Compared to octreotide, long-acting somatostatin-analogues allow a similar or even better glycemic control with the advantage of a single administration every 4 weeks (Kuhnen et al 2012), meaning a shift from 90 to 120 injections per month to 1 injection per month, with major improvement in quality of life. These drugs are considered for use in older infants responsive to short-acting octreotide aiming to reduce the clinical burden (less injections, easier for kindergarten, school, vacations, daily life).
Sirolimus
To avoid an extensive surgical approach in drug-unresponsive patients, sirolimus (rapamycin) has been recently attempted (Senniappan et al 2014). Sirolimus, a mTOR pathway inhibitor, is an immunosuppressive drug with current indications for post-kidney transplant immunosuppression and with an expanding use in pancreatic neuroendocrine tumors, insulinoma, leukemia, lymphangioleiomyomatosis, and tuberous sclerosis (Boulay et al 2004; Boucier et al 2009; Kulke et al 2009; Yao et al 2010; Krueger et al 2013). The rationale for its use in HI was based on the observation that patients treated with sirolimus often presented hyperglycemia. This side effect is due to the inhibitory effect on the PI3K/AKT/mTor pathway, a nutrient sensor involved in numerous cellular processes, including cell metabolism, growth, proliferation, apoptosis, response to oxidative stress, and insulin secretion (Leibowitz et al 2008). So far, some case studies reported on the successful use of sirolimus, sometimes in combination with octreotide. However, efficacy was limited in the face of important side effects, which include immunosuppression, cytopenia, stomatitis, increased infections, transaminases elevation, and pancreatic insufficiency (Szymanowski et al 2016). Furthermore, a recent report showed very poor results in patients with severe HI, highlighting the risk of long-term severe side effects and pointing out the need to only use sirolimus in the context of a controlled clinical trial (Banerjee et al 2017).
Future promising therapies
Exendin-(9–39)
Exendin-(9–39) is a reverse agonist of glucagon-like peptide−1 (GLP-1) receptor. GLP-1 is an incretin hormone which stimulates insulin secretion in response to ingested nutrients. Chronic subcutaneous infusion of exendin-(9–39) prevented fasting hypoglycemia in SUR1−/− mice (De Leon et al 2008). Moreover, in vivo and in vitro studies demonstrated that exendin-(9–39) inhibited aminoacid-induced insulin secretion, in SUR1−/− mice and in pancreatic islet isolated from neonates with KATP-HI, respectively (De Leon et al 2008). Glutamine may mediate protein-sensitivity of KATP-HI via the “amplification” pathway of the GLP-1 receptor (Li et al 2004; De Leon et al 2008). A short-term trial conducted on 9 adolescents-adults with KATP-HI allowed a significant improvement of blood glucose levels along with a reduction of the insulin/glucose ratio (Calabria et al 2012). Similar effects have also been observed in pediatric patients, where exendin-(9–39) prevented aminoacid-induced hypoglycemia (NCT00897676, http://www.clinicaltrials.gov). A trial on the effect on glucose requirements is ongoing in infants (NCT00835328).
Subcutaneous glucagon
The use of glucagon for long-term treatment is hampered by its short half-life and because of the need of parenteral administration. Subcutaneous glucagon (via portable subcutaneous pump at a dosage of 0.026–0.8 mg/kg/day) was successfully used in a few diazoxide-unresponsive children, allowing reduction or discontinuation of central glucose infusion (Mohnike et al 2008). In patients treated with octreotide, its dosage was considerably reduced, avoiding pancreatectomy or subsequent resurgery. To prevent crystallization, an experimental glucagon technospheres suspension in aqueous solution was successfully used in 3 of these children (Mohnike et al 2008). More recently, another study reported that continuous subcutaneous glucagon infusion allowed restoration of normoglycaemia, attenuating weight gain and improving developmental skills in a patient with atypical diffuse HI with mosaic ABCC8 mutations, still unresponsive after two subtotal pancreatectomies (Neylon et al 2013). A proof-of-concept clinical trial is ongoing in infants with diazoxide-unresponsive HI (NCT02937558).
Anti insulin receptor antibodies
Antibodies for allosteric inhibition of insulin receptor corrected fasting hypoglycemia in SUR1−/− mice by reducing hepatic insulin sensitivity and insulin signaling in muscle, liver, and fat tissue (Patel et al 2013) and prevented insulin-induced hypoglycemia in normal volunteers (Nath et al ENDO 2015 ). A trial to evaluate the effect of a single dose in HI adult patients is currently ongoing (NCT02604485).
Small molecules
Rescue of KATP channel activity could be beneficial for the treatment of HI, especially if these channels retained regulatory properties or could be activated by KATP channel agonists. Obtaining a successful enhancement of KATP channel activity could eventually have an impact on clinical treatment of HI (Powell et al 2011). In vitro studies with small molecules acting as chaperones to correct KATP trafficking defects (carbamazepine and sulfonylureas) showed rescue of SUR1 protein only when mutations affected the TMD0 domain, with normal co-expression of Kir6.2 (Martin et al 2016).
Novel nutritional interventions
Ketogenic diet
Nutritional interventions could provide an important contribution in maintaining normoglycemia in HI, particularly in patients with poor drug responsiveness. Ketogenic diet (KD) is a nutritional regimen with over 60% of the total caloric intake provided by lipids. The main indication of KD in children is the treatment of refractory epilepsy (Bough and Rho 2007; Kossoff et al 2009). However, KD is also the elective treatment of GLUT1 deficiency, a disorder in which the impaired glucose transport across the blood-brain barrier causes neuroglycopenia, with consequent epilepsy, developmental delay and movement disorders (De Vivo et al 1991; Pearson et al 2013). In GLUT1 deficiency, neuroglycopenia and CNS energy failure are bypassed by KD, which provides ketone bodies as an alternative energy source for the brain (De Vivo et al 1991). In HI, insulin excess induces severe hypoglycemia with consequent neuroglycopenia. Moreover, the availability of alternative energetic substrates for the CNS is further reduced, because of the concomitant inhibition of gluconeogenesis, glycogenolysis, and lipolysis. Based on the similarities of brain metabolism perturbation shared by GLUT1 deficiency and HI, we successfully utilized KD in a patient with severe drug-resistant GCK-HI, presenting recurrent hypoglycemia, refractory epilepsy, and mild intellectual disability (Maiorana et al 2015). While maintaining blood ketones between 2 and 5 mmol/L, KD fully resolved neuroglycopenic signs, with disappearance of epilepsy, improvement of EEG and cognitive abilities. Definitely, the availability of an alternative cerebral energy source improved neurodevelopment, avoiding the need of near-total pancreatectomy (Maiorana et al 2015). Although based on a single experience, KD could represent an effective treatment to support brain function in selected cases of unresponsive HI.
Galactose therapy in PGM1-CDG
Phosphoglucomutase 1 (PGM1)-CDG has been recently reported as a novel disease, bridging CDGs and glycogen storage diseases (Morava 2014; Tegtmeyer et al 2014). PGM1 catalyzes the transfer of phosphate between glucose-1-P and glucose-6-P. Patients with recessive mutations in PGM1 manifest a complex phenotype characterized by hepatopathy, bifid uvula, growth retardation, myopathy, cardiomyopathy, coagulation and endocrine alterations. Patients show a combination of fasting ketotic hypoglycemia, with post-prandial hyperinsulinemic hypoglycemia. Although not fully supported by strong evidence, the proposed mechanisms were impaired carbohydrate metabolism for the first condition, and a lower glucose threshold for insulin secretion caused by the increased glucose-6-P for the latter. Therapy with oral galactose at the dosage of 1 g/kg/day improved hypoglycemia, coagulation, and endocrine abnormalities as well as transferrin glycosylation pattern (Morava 2014; Tegtmeyer et al 2014).
Long-term neurological outcome
Hyperinsulinemic hypoglycemia is an important cause of brain injury in children, leading to long-term neurological impairment with intellectual disability, epilepsy, blindness and cerebral palsy. In HI, the major vulnerability of brain in early life is related to the lack of energy substrate for cerebral activity, which is mainly dependent on glucose, accounting for 40–60% of basal metabolic rate (Holliday 1971; Goyal et al 2014). Moreover, insulin excess further impairs CNS energy failure by suppressing the availability of alternative substrates. The topographic distribution of brain lesions is determined by patients age, reflecting the physiological timing of brain maturation: posterior white matter in neonates, basal ganglia in infants, and parieto-temporal cortex in children (Gataullina et al 2013). As shown in Table 3, most reports on the neurological outcome in large HI series, which often include non-homogenous cohorts (genotypic and phenotypic heterogeneity, combination of medical and surgical treatments, patients treated before the systematic use of octreotide, ages of subjects, and source of data, i.e., parental interview or psychometric tests), showed that developmental delay ranges from 26% to 44%, and epilepsy accounts for 18–25% (Menni et al 2001; Meissner et al 2003; Steinkrauss et al 2005; Avatapalle et al 2013). In a more recent study conducted on KATP-HI patients treated with both diazoxide or somatostatin-analogues, mild developmental delay was observed in 38% of children (Salomon-Estebanez et al 2016). Remarkably, the proportion of children with neurodevelopmental outcomes was similar in subjects with spontaneous disease remission when compared to those with persistent HI and, surprisingly, 75% of the most severe KATP-patients had normal neurodevelopment (Salomon-Estebanez et al 2016). Another recent study, focused on the neurocognitive outcome in subjects treated with near-total pancreatectomy, 48% of patients showed neurobehavioral signs (i.e., speech delay, learning disabilities, seizures, physical disabilities, ADHD, autism), combined with a high risk of developing diabetes (36%) and no outcome differences between patients diagnosed before and after 2004, when 18F DOPA PET imaging was undertaken (Lord et al 2015). We retrospectively evaluated a cohort of 50 patients followed at the Bambino Gesù Children’s Hospital in Rome and found a rate of developmental delay similar to the report of Menni (2001), with a predominance of mild delay (28%, of whom 12% were borderline and 16% mild). Epilepsy was less frequently recorded (8%) and prevailed in patients with hyperinsulinism-hyperammonemia syndrome, a condition with a higher incidence of epilepsy and neurological disabilities (Bahi-Buisson et al 2008). The incidence of drug-unresponsiveness was lower in our series, but the strict monitoring at follow-up based on serial use of the continuous glucose monitoring system (Maiorana et al 2014) combined with individualized pharmacological and nutritional interventions, might have contributed to a more favorable neurological outcome (Table 3 and Supplementary material). Definitely, despite diagnostic and therapeutic advances, HI still represents an important cause of brain injury. Therefore, based on the principle that hypoglycemia in the first days of life greatly contribute to the poor outcomes (Lord et al 2015), we recommend to increase the awareness of neonatologists and pediatricians to improve early diseases recognition, allowing a rapid and aggressive treatment, before the appearance of irreversible neurological damage (Thornton et al 2015). At follow-up, patients require regular and strict monitoring, with serial use of continuous glucose monitoring (Maiorana et al 2014). Longitudinal evaluations should also include neurodevelopmental testing to identify children who need intervention and therapy (De Leon and Stanley 2017).
Conclusions
In conclusion, HI is the most frequent form of hypoglycemia in infancy, still causing long-term neurological sequelae. The increasing use of NGS will be of great help for the diagnosis, and the use of whole exome or genome sequencing should be encouraged for patients negative at the NGS, with the aim to discover new genes causing HI. New disease pathways have recently been discovered and novel drug and nutritional therapies are now available. Maintenance of normoglycemia is the mainstay of therapy to protect brain development, especially in early life. A multidisciplinary team is essential to diagnose, treat, and follow children with HI.
References
Alexander S, Ramadan D, Alkhayyat H et al (2005) Costello syndrome and hyperinsulinemic hypoglycemia. Am J Med Genet A 139:227–230
Alkhayyat H, Christesen HB, Steer J, Stewart H, Brusgaard K, Hussain K (2006) Mosaic turner syndrome and hyperinsulinaemic hypoglycaemia. J Pediatr Endocrinol Metab 19:1451–1457
Arnoux JB, De Lonlay P, Ribeiro MJ et al (2010) Congenital hyperinsulinism. Early Hum Dev 86:287–294
Arnoux JB, Verkarre V, Saint-Martin C et al (2011) Congenital hyperinsulinism: current trends in diagnosis and therapy. Orphanet J Rare Dis 6:63
Arya VB, Flanagan SE, Schober E, Rami-Merhar B, Ellard S, Hussain K (2014a) Activating AKT2 mutation: hypoinsulinemic hypoketotic hypoglycemia. J Clin Endocrinol Metab 99:391–394
Arya VB, Senniappan S, Demirbilek H et al (2014b) Pancreatic endocrine and exocrine function in children following near-total pancreatectomy for diffuse congenital hyperinsulinism. PLoS One 9:e98054
Astruc B, Marbach P, Bouterfa H et al (2005) Long-acting octreotide and prolonged-release lanreotide formulations have different pharmacokinetic profiles. J Clin Pharmacol 45:836–844
Avatapalle B, Padidela R, Randell T, Banerjee I (2012) Drug-induced hepatitis following use of octreotide for long-term treatment of congenital hyperinsulinism. BMJ Case Rep. 2012
Avatapalle HB, Banerjee I, Shah S et al (2013) Abnormal neurodevelopmental outcomes are common in children with transient congenital Hyperinsulinism. Front Endocrinol (Lausanne) 4:60
Aynsley-Green A, Hussain K, Hall J et al (2000) Practical management of hyperinsulinism in infancy. Arch Dis Child Fetal Neonatal Ed 82:F98–F107
Bahi-Buisson N, Roze E, Dionisi Vici C et al (2008) Neurological aspects of hyperinsulinism-hyperammonaemia syndrome. Dev Med Child Neurol 50:945–949
Banerjee I, De Leon D, Dunne MJ (2017) Extreme caution on the use of sirolimus for the congenital hyperinsulinism in infancy patient. Orphanet J Rare Dis 12:70
Baujat G, Rio M, Rossignol S, Sanlaville D et al (2004) Paradoxical NSD1 mutations in Beckwith-Wiedemann syndrome and 11p15 anomalies in Sotos syndrome. Am J Hum Genet 74:715–720
Beltrand J, Caquard M, Arnoux JB et al (2012) Glucose metabolism in 105 children and adolescents after pancreatectomy for congenital hyperinsulinism. Diabetes Care 35:198–203
Bereket A, Turan S, Alper G, Comu S, Alpay H, Akalin F (2001) Two patients with kabuki syndrome presenting with endocrine problems. J Pediatr Endocrinol Metab 14:215–220
Bitner-Glindzicz M, Lindley KJ, Rutland P et al (2000) A recessive contiguous gene deletion causing infantile hyperinsulinism, enteropathy and deafness identifies the usher type 1C gene. Nat Genet 26:56–60
Blomberg BA, Moghbel MC, Saboury B, Stanley CA, Alavi A (2013) The value of radiologic interventions and (18)F-DOPA PET in diagnosing and localizing focal congenital hyperinsulinism: systematic review and meta-analysis. Mol Imaging Biol 15:97–105
Bough KJ, Rho JM (2007) Anticonvulsant mechanisms of the ketogenic diet. Epilepsia 48:43–58
Boulay A, Zumstein-Mecker S, Stephan C et al (2004) Antitumor efficacy of intermittent treatment schedules with the rapamycin derivative RAD001 correlates with prolonged inactivation of ribosomal protein S6 kinase 1 in peripheral blood mononuclear cells. Cancer Res 64:252–261
Bourcier ME, Sherrod A, DiGuardo M, Vinik AI (2009) Successful control of intractable hypoglycemia using rapamycin in an 86-year-old man with a pancreatic insulin-secreting islet cell tumor and metastases. J Clin Endocrinol Metab 94:3157–3162
Bruining GJ (1990) Recent advances in hyperinsulinism and the pathogenesis of diabetes mellitus. Curr Opin Pediatr 2:758–765
Calabria AC, Li C, Gallagher PR, Stanley CA, De Leon DD (2012) GLP-1 receptor antagonist exendin-(9-39) elevates fasting blood glucose levels in congenital hyperinsulinism owing to inactivating mutations in the ATP-sensitive K+ channel. Diabetes 61:2585–2591
Capito C, de Lonlay P, Verkarre V et al (2011) The surgical management of atypical forms of congenital hyperinsulinism. Semin Pediatr Surg 20:54–55
De Leon DD, Li C, Delson MI, Matschinsky FM, Stanley CA, Stoffers DA (2008) Exendin-(9-39) corrects fasting hypoglycemia in SUR-1−/− mice by lowering cAMP in pancreatic beta-cells and inhibiting insulin secretion. J Biol Chem 283:25786–25793
De Leon DD, Stanley CA (2007) Mechanisms of disease: advances in diagnosis and treatment of hyperinsulinism in neonates. Nat Nat Clin Pract Endocrinol Metab 3:57–68
De Leon DD, Stanley CA (2013) Determination of insulin for the diagnosis of hyperinsulinemic hypoglycemia. Best Pract Res Clin Endocrinol Metab 27:763–769
De Leon DD, Stanley CA (2017) Congenital hypoglycemia disorders: new aspects of etiology, diagnosis, treatment and outcomes: highlights of the proceedings of the congenital hypoglycemia disorders symposium, Philadelphia April 2016. Pediatr Diabetes 18:3–9
De Lonlay P, Benelli C, Fouque F et al (2001) Hyperinsulinism and hyperammonemia syndrome: report of twelve unrelated patients. Pediatr Res 50:353–357
De Lonlay P, Fournet JC, Rahier J et al (1997) Somatic deletion of the imprinted 11p15 region in sporadic persistent hyperinsulinemic hypoglycemia of infancy is specific of focal adenomatous hyperplasia and endorses partial pancreatectomy. J Clin Invest 100:802–807
de Lonlay-Debeney P, Poggi-Travert F, Fournet JC et al (1999) Clinical features of 52 neonates with hyperinsulinism. N Engl J Med 340:1169–1175
De Vivo DC, Trifiletti RR, Jacobson RI, Ronen GM, Behmand RA, Harik SI (1991) Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N Engl J Med 325:703–709
DeBaun MR, King AA, White N (2000) Hypoglycemia in Beckwith-Wiedemann syndrome. Semin Perinatol 24:164–171
Demirbilek H, Shah P, Arya VB et al (2014) Long-term follow-up of children with congenital hyperinsulinism on octreotide therapy. J Clin Endocrinol Metab 99:3660–3667
Dunne MJ, Cosgrove KE, Shepherd RM, Aynsley-Green A, Lindley KJ (2004) Hyperinsulinism in infancy: from basic science to clinical disease. Physiol Rev 84:239–275
Faletra F, Athanasakis E, Morgan A et al (2013) Congenital hyperinsulinism: clinical and molecular analysis of a large Italian cohort. Gene 521:160–165
Ferrara C, Patel P, Becker S, Stanley CA, Kelly A (2016) Biomarkers of insulin for the diagnosis of hyperinsulinemic hypoglycemia in infants and children. J Pediatr 168:212–219
Flanagan SE, Clauin S, Bellanné-Chantelot C et al (2009) Update of mutations in the genes encoding the pancreatic beta-cell K(ATP) channel subunits Kir6.2 (KCNJ11) and sulfonylurea receptor 1 (ABCC8) in diabetes mellitus and hyperinsulinism. Hum Mutat 30:170–180
Flanagan SE, Kapoor RR, Mali G et al (2010) Diazoxide-responsive hyperinsulinemic hypoglycemia caused by HNF4A gene mutations. Eur J Endocrinol 62:987–992
Flanagan SE, Vairo F, Johnson MB et al (2017) A CACNA1D mutation in a patient with persistent hyperinsulinaemic hypoglycaemia, heart defects, and severe hypotonia. Pediatr Diabetes 18:320–323
Garg N, Bademci G, Foster J 2nd, Sıklar Z, Berberoglu M, Tekin M (2015) MORFAN syndrome: an infantile hypoinsulinemic hypoketotic hypoglycemia due to an AKT2 mutation. J Pediatr 167:489–491
Gataullina S, De Lonlay P, Dellatolas G et al (2013) Topography of brain damage in metabolic hypoglycaemia is determined by age at which hypoglycaemia occurred. Dev Med Child Neurol 55:162–166
Geneviève D, Amiel J, Viot G et al (2004) Atypical findings in kabuki syndrome: report of 8 patients in a series of 20 and review of the literature. Am J Med Genet A 129A:64–68
Gillis D, Krishnamohan A, Yaacov B, Shaag A, Jackman JE, Elpeleg O (2014) TRMT10A dysfunction is associated with abnormalities in glucose homeostasis, short stature and microcephaly. J Med Genet 51:581–586
Glaser B, Kesavan P, Heyman M et al (1998) Familial hyperinsulinism caused by an activating glucokinase mutation. N Engl J Med 338:226–230
Glaser B, Landau H, Smilovici A, Nesher R (1989) Persistent hyperinsulinaemic hypoglycaemia of infancy: long-term treatment with the somatostatin analogue Sandostatin. Clin Endocrinol 31:71–80
Gonzalez-Barroso MM, Giurgea I, Bouillaud F et al (2008) Mutations in UCP2 in congenital hyperinsulinism reveal a role for regulation of insulin secretion. PLoS One 3:e3850
Goyal MS, Hawrylycz M, Miller JA, Snyder AZ, Raichle ME (2014) Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metab 19:49–57
Guemes M, Shah P, Silvera S (2017) Assessment of nifedipine therapy in hyperinsulinemic hypoglycemia due to mutations in the ABCC8 gene. J Clin Endocrinol Metab 102:822–830
Han B, Newbould M, Batra G (2016) Enhanced islet cell nucleomegaly defines diffuse congenital hyperinsulinism in infancy but not other forms of the disease. Am J Clin Pathol 145:757–768
Hawkes CP, Adzick NS, Palladino AA, De Leon DD (2016) Late presentation of fulminant necrotizing enterocolitis in a child with hyperinsulinism on octreotide therapy. Horm Res Paediatr 86:131–136
Henneveld HT, van Lingen RA, Hamel BC, Stolte-Dijkstra I, van Essen AJ (1999) Perlman syndrome: four additional cases and review. Am J Med Genet 86:439–446
Hennewig U, Hadzik B, Vogel M et al (2008) Congenital central hypoventilation syndrome with hyperinsulinism in a preterm infant. J Hum Genet 53:573–577
Henquin JC, Sempoux C, Marchandise J et al (2013) Congenital hyperinsulinism caused by hexokinase I expression or glucokinase-activating mutation in a subset of β-cells. Diabetes 62:1689–1696
Hojlund K, Hansen T, Lajer M et al (2004) A novel syndrome of autosomal-dominant hyperinsulinemic hypoglycemia linked to a mutation in the human insulin receptor gene. Diabetes 53:1592–1598
Holliday MA (1971) Metabolic rate and organ size during growth from infancy to maturity and during late gastation and early infancy. Pediatrics 47(Suppl 2):169+
Hsu BY, Kelly A, Thornton PS, Greenberg CR, Dilling LA, Stanley CA (2001) Protein-sensitive and fasting hypoglycemia in children with the hyperinsulinism/hyperammonemia syndrome. J Pediatr 138:383–389
Hussain K, Challis B, Rocha N et al (2011) An activating mutation of AKT2 and human hypoglycemia. Science 334:474
Hussain K, Cosgrove KE, Shepherd RM et al (2005) Hyperinsulinemic hypoglycemia in Beckwith-Wiedemann syndrome due to defects in the function of pancreatic beta-cell adenosine triphosphate-sensitive potassium channels. J Clin Endocrinol Metab 90:4376–4382
Hussain K, Seppanen M, Nanto-Salonen K et al (2006) The diagnosis of ectopic focal hyperinsulinism of infancy with [18F]- dopa emission tomography. J Clin Endocrinol Metab 91:2839–2842
Ismail D, Kapoor RR, Smith VV, Ashworth M, Blankenstein O, Pierro A, Flanagan SE, Ellard S, Hussain K (2012) The heterogeneity of focal forms of congenital hyperinsulinism. J Clin Endocrinol Metab 97(1):E94–E99
Jaeken J, Matthijs G, Saudubray JM et al (1998) Phosphomannose isomerase deficiency: a carbohydrate-deficient glycoprotein syndrome with hepatic-intestinal presentation. Am J Hum Genet 62:1535–1539
Kalish JM, Boodhansingh KE, Bhatti TR et al (2016) Congenital hyperinsulinism in children with paternal 11p uniparental isodisomy and Beckwith-Wiedemann syndrome. J Med Genet 53:53–61
Kapoor RR, Flanagan SE, Arya VB, Shield JP, Ellard S, Hussain K (2013) Clinical and molecular characterisation of 300 patients with congenital hyperinsulinism. Eur J Endocrinol 168:557–564
Kapoor RR, Flanagan SE, James C, Shield J, Ellard S, Hussain K (2009a) Hyperinsulinemic hypoglycemia. Arch Dis Child 94:450–457
Kapoor RR, James C, Hussain K (2009b) Advances in the diagnosis and management of hyperinsulinemic hypoglycaemia. Nat Clin Pract Endocrinol Metab 5:101–112
Kelly A, Ng D, Ferry RJ Jr et al (2001) Acute insulin responses to leucine in children with the hyperinsulinism/hyperammonemia syndrome. J Clin Endocrinol Metab 86:3724–3728
Kossoff EH, Zupec-Kania BA, Amark PE et al (2009) Optimal clinical management of children receiving the ketogenic diet: recommendations of the international ketogenic diet study group. Epilepsia 50:304–317
Kuhnen P, Marquard J, Ernert A et al (2012) Long-term lanreotide treatment in six patients with congenital hyperinsulinism. Horm Res Paediatr 78:106–112
Kulke MH, Bergsland EK, Yao JC (2009) Glycemic control in patients with insulinoma treated with everolimus. N Engl J Med 360:195–197
Krueger DA, Wilfong AA, Holland-Bouley K et al (2013) Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann Neurol 74:679–687
Laje P, Halaby L, Adzick NS, Stanley CA (2010) Necrotizing enterocolitis in neonates receiving octreotide for the management of congenital hyperinsulinism. Pediatr Diabetes 11:142–147
Laje P, States LJ, Zhuang H et al (2013) Accuracy of PET/CT scan in the diagnosis of the focal form of congenital hyperinsulinism. J Pediatr Surg 48:388–393
Laidlaw GF (1938) Nesidioblastoma, the islet tumor of the pancreas. Am J Pathol 14:125–134.5
Leibowitz G, Cerasi E, Ketzinel-Gilad M (2008) The role of mTOR in the adaptation and failure of beta-cells in type 2 diabetes. Diabetes Obes Metab 10(Suppl 4):157–169
Le Quan Sang KH, Arnoux JB, Mamoune A et al (2012) Successful treatment of congenital hyperinsulinism with long-acting release octreotide. Eur J Endocrinol 166:333–339
Li C, Buettger C, Kwagh J et al (2004) A signaling role of glutamine in insulin secretion. J Biol Chem 279:13393–13401
Loechner KJ, Akrouh A, Kurata HT et al (2011) Congenital hyperinsulinism and glucose hypersensitivity in homozygous and heterozygous carriers of Kir6.2 (KCNJ11) mutation V290M mutation: K(ATP) channel inactivation mechanism and clinical management. Diabetes 60:209–217
Lord K, Radcliffe J, Gallagher PR, Adzick NS, Stanley CA, De Leon DD (2015) High risk of diabetes and neurobehavioral deficits in individuals with surgically treated hyperinsulinism. J Clin Endocrinol Metab 100:4133–4139
Maiorana A, Barbetti F, Boiani A et al (2014) Focal congenital hyperinsulinism managed by medical treatment: a diagnostic algorithm based on molecular genetic screening. Clin Endocrinol 81:679–688
Maiorana A, Manganozzi L, Barbetti F et al (2015) Ketogenic diet in a patient with congenital hyperinsulinism: a novel approach to prevent brain damage. Orphanet J Rare Dis 10:120
Martin GM, Rex EA, Devaraneni P et al (2016) Pharmacological correction of trafficking defects in ATP-sensitive potassium channels caused by sulfonylurea receptor 1 mutations. J Biol Chem 291:21971–21983
Meissner T, Rabl W, Mohnike K, Scholl S, Santer R, Mayatepek E (2001) Hyperinsulinism in syndromal disorders. Acta Paediatr 90:856–859
Meissner T, Wendel U, Burgard P, Schaetzle S, Mayatepek E (2003) Long-term follow-up of 114 patients with congenital hyperinsulinism. Eur J Endocrinol 149:43–51
Menni F, de Lonlay P, Sevin C et al (2001) Neurologic outcomes of 90 neonates and infants with persistent hyperinsulinemic hypoglycemia. Pediatrics 107:476–479
Miller BS, Freeze HI, Hoffmann GF, Sarafoglou K (2011) Pubertal development in ALG6 deficiency (congenital disorder of glycosylation type Ic). Mol Genet Metab 103:101–103
Modan-Moses D, Koren I, Mazor-Aronovitch K, Pinhas-Hamiel O, Landau H (2011) Treatment of congenital hyperinsulinism with lanreotide acetate (Somatuline Autogel). J Clin Endocrinol Metab 96:2312–2317
Mohnike K, Blankenstein O, Pfuetzner A et al (2008) Long-term non-surgical therapy of severe persistent congenital hyperinsulinism with glucagon. Horm Res 70:59–64
Molven A, Matre GE, Duran M et al (2004) Familial hyperinsulinemic hypoglycemia caused by a defect in the SCHAD enzyme of mitochondrial fatty acid oxidation. Diabetes 53:221–227
Morava E (2014) Galactose supplementation in phosphoglucomutase-1 deficiency; review and outlook for a novel treatable CDG. Mol Genet Metab 112:275–279
Muller D, Zimmering M, Roehr CC (2004) Should nifedipine be used to counter low blood sugar levels in children with persistent hyperinsulinaemic hypoglycaemia? Arch Dis Child 89:83–85
Munns CF, Batch JA (2001) Hyperinsulinism and Beckwith-Wiedemann syndrome. Arch Dis Child Fetal Neonatal Ed 84:F67–F69
Nath R, Johnson KW, Roessig JM (2015) XOMA 358, a novel treatment for hyperinsulinemic hypoglycemia: safety and clinical pharmacology from the first in human trial. ENDO March 2015
Nellist M, Schot R, Hoogeveen-Westerveld M et al (2015) Germline activating AKT3 mutation associated with megalencephaly, polymicrogyria, epilepsy and hypoglycemia. Mol Genet Metab 114:467–473
Neylon OM, Moran MM, Pellicano A, Nightingale M, O’Connell MA (2013) Successful subcutaneous glucagon use for persistent hypoglycaemia in congenital hyperinsulinism. J Pediatr Endocrinol Metab 26:1157–1161
Otonkoski T, Kaminen N, Ustinov J et al (2003) Physical exercise-induced hyperinsulinemic hypoglycemia is an autosomal-dominant trait characterized by abnormal pyruvate-induced insulin release. Diabetes 52:199–204
Otonkoski T, Näntö-Salonen K, Seppänen M et al (2006) Noninvasive diagnosis of focal hyperinsulinism of infancy with [18F]-DOPA positron emission tomography. Diabetes 55:13–18
Palladino AA, Stanley CA (2011) Nesidioblastosis no longer! It’s all about genetics. J Clin Endocrinol Metab 96:617–619
Patel P, Corbin JA, Goldfine ID, Rubin P, De Leon DD (2013) A unique allosteric insulin receptor monoclonal antibody that prevents hypoglycemia in the SUR-1−/− mouse model of KATP hyperinsulinism. Diabetes 62(suppl1):A–93 abstract
Pearson ER, Boj SF, Steele AM et al (2007) Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Med 4:e118
Pearson TS, Akman C, Hinton VJ, Engelstad K, De Vivo DC (2013) Phenotypic spectrum of glucose transporter type 1 deficiency syndrome (Glut1 DS). Curr Neurol Neurosci Rep 13:342
Pinney SE, Ganapathy K, Bradfield J et al (2013) Dominant form of congenital hyperinsulinism maps to HK1 region on 10q. Horm Res Paediatr 80:18–27
Ponzi E, Maiorana A, Lepri FR, Novelli A, Dionisi-Vici C (2016) Hypoglycemia in children: uncovering the genetic basis of related metabolic disorders by custom gene panel in an Italian cohort. J Inherit Metabol Dis 39(Suppl 1):S146–S147
Powell PD, Bellanné-Chantelot C, Flanagan SE et al (2011) In vitro recovery of ATP-sensitive potassium channels in β-cells from patients with congenital hyperinsulinism of infancy. Diabetes 60:1223–1228
Rahier J, Guiot Y, Sempoux C (2011) Morphologic analysis of focal and diffuse forms of congenital hyperinsulinism. Semin Pediatr Surg 20:3–12
Rahier J, Wallon J, Henquin JC (1981) Cell populations in the endocrine pancreas of human neonates and infants. Diabetologia 20:540–546
Reck-Burneo CA, Parekh A, Velcek FT (2008) Is octreotide a risk factor in necrotizing enterocolitis? J Pediatr Surg 43:1209–1210
Ribeiro MJ, De Lonlay P, Delzescaux TJ et al (2005) Characterization of hyperinsulinism in infancy assessed with PET and 18F-fluoro-L-DOPA. J Nucl Med 46:560–566
Salisbury RJ, Han B, Jennings RE et al (2015) Altered phenotype of β-cells and other pancreatic cell lineages in patients with diffuse congenital hyperinsulinism in infancy caused by mutations in the ATP-sensitive K-channel. Diabetes 64:3182–3188
Salomon-Estebanez M, Flanagan SE, Ellard S et al (2016) Conservatively treated congenital Hyperinsulinism (CHI) due to K-ATP channel gene mutations: reducing severity over time. Orphanet J Rare Dis 11:163
Sempoux C, Capito C, Bellanné-Chantelot C et al (2011) Morphological mosaicism of the pancreatic islets: a novel anatomopathological form of persistent hyperinsulinemic hypoglycemia of infancy. J Clin Endocrinol Metab 96:3785–3793
Senniappan S, Alexandrescu S, Tatevian N et al (2014) Sirolimus therapy in infants with severe hyperinsulinemic hypoglycemia. N Engl J Med 370:1131–1137
Shanti B, Silink M, Bhattacharya K et al (2009) Congenital disorder of glycosylation type Ia: heterogeneity in the clinical presentation from multivisceral failure to hyperinsulinaemic hypoglycaemia as leading symptoms in three infants with phosphomannomutase deficiency. J Inherit Metab Dis 32(Suppl 1):S241–S251
Sheffield BS, Yip S, Ruchelli ED et al (2015) Fatal congenital hypertrophic cardiomyopathy and a pancreatic nodule morphologically identical to focal lesion of congenital hyperinsulinism in an infant with costello syndrome: case report and review of the literature. Pediatr Dev Pathol 18:237–244
Smith VS, Giacoia GP (1985) Hyperinsulinaemic hypoglycaemia in an infant with mosaic trisomy 13. J Med Genet 22:228–230
Snider KE, Becker S, Boyajian L et al (2013) Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab 98:E355–E363
Splawski I, Timothy KW, Sharpe LM et al (2004) Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119:19–31
Stanley CA (2016) Perspective on the genetics and diagnosis of congenital hyperinsulinism disorders. J Clin Endocrinol Metab 101:815–826
Stanley CA, Lieu YK, Hsu BY et al (1998) Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. N Engl J Med 338:1352–1357
Stanley CA, Matschinsky FM (2012) Frontiers in diabetes. In Stanley CA and De Leon DD (eds.) Monogenic hyperinsulinemic hypoglycemia disorders, vol. 21. Karger, Basel, p 1–6
Stanley CA, Thornton PS, Ganguly A et al (2004) Preoperative evaluation of infants with focal or diffuse congenital hyperinsulinism by intravenous acute insulin response tests and selective pancreatic arterial calcium stimulation. J Clin Endocrinol Metab 89:288–296
Staufner C, Lindner M, Dionisi-Vici C et al (2016) Adenosine kinase deficiency: expanding the clinical spectrum and evaluating therapeutic options. J Inherit Metab Dis 39:273–283
Steinkrauss L, Lipman TH, Hendell CD, Gerdes M, Thornton PS, Stanley CA (2005) Effects of hypoglycemia on developmental outcome in children with congenital hyperinsulinism. J Pediatr Nurs 20:109–118
Sun L, Eklund EA, Chung WK, Wang C, Cohen J, Freeze HI (2005) Congenital disorder of glycosylation id presenting with hyperinsulinemic hypoglycemia and islet cell hyperplasia. J Clin Endocrinol Metab 907:4371–4375
Szymanowski M, Estebanez MS, Padidela R et al (2016) mTOR inhibitors for the treatment of severe congenital hyperinsulinism: perspectives on limited therapeutic success. J Clin Endocrinol Metab 101:4719–4729
Tegtmeyer LC, Rust S, van Scherpenzeel M et al (2014) Multiple phenotypes in phosphoglucomutase 1 deficiency. N Engl J Med 370:533–542
Terespolsky D, Farrell SA, Siegel-Bartelt J, Weksberg R (1995) Infantile lethal variant of Simpson-Golabi-Behmel syndrome associated with hydrops fetalis. Am J Med Genet 59:329–333
Thornton PS, Stanley CA, De Leon DD et al (2015) Recommendations from the pediatric Endocrine Society for evaluation and management of persistent hypoglycemia in neonates, infants, and children. J Pediatr 167:238–245
Touati G, Poggi-Travert F, Ogier de Baulny H, Rahier J, Brunelle F, Nihoul-Fekete C, Czernichow P, Saudubray JM (1998) Long-term treatment of persistent hyperinsulinaemic hypoglycaemia of infancy with diazoxide: a retrospective review of 77 cases and analysis of efficacy-predicting criteria. Eur J Pediatr 157(8):628–633
Treglia G, Mirk P, Giordano A, Rufini V (2012) Diagnostic performance of fluorine-18-dihydroxyphenylalanine positron emission tomography in diagnosing and localizing the focal form of congenital hyperinsulinism: a meta-analysis. Pediatr Radiol 42:1372–1379
Welters A, Lerch C, Kummer S et al (2015) Long-term medical treatment in congenital hyperinsulinism: a descriptive analysis in a large cohort of patients from different clinical centers. Orphanet J Rare Dis 10:150
Yao JC, Lombard-Bohas C, Baudin E et al (2010) Daily oral everolimus activity in patients with metastatic pancreatic neuroendocrine tumors after failure of cytotoxic chemotherapy: a phase II trial. J Clin Oncol 28:69–76
Yakovac WC, Baker L, Hummeler K (1971) Beta cell nesidioblastosis in idiopathic hypoglycemia of infancy. J Pediatr 79:226–231
Zammarchi E, Filippi L, Novembre E, Donati MA (1996) Biochemical evaluation of a patient with a familial form of leucine-sensitive hypoglycemia and concomitant hyperammonemia. Metabolism 45:957–960
Acknowledgements
We are grateful to the multidisciplinary HI team at the Bambino Gesù Children’s Hospital: Dr. Fabrizio Barbetti, Dr. Silvia M. Bernabei, Dr. Stefania Caviglia, Dr. Paola Francalanci, Dr. Chiara Grimaldi, Dr. Francesca R. Lepri, Dr. Antonio Novelli, Dr. Milena Pizzoferro, Dr. Emanuela Ponzi, Dr. Marco Spada. We also thank Dr. Vittoria Rufini, Dr. Jaques Rahier (recently deceased), and Dr. Jean de Ville de Goyet. This work has been partly supported by the Ricerca Corrente from the Italian Ministry of Health and by the “Associazione la Vita è un Dono”.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
A. Maiorana and C. Dionisi-Vici declare that they have no conflict of interest.
Additional information
Communicated by: Ertan Mayatepek
Electronic supplementary material
ESM 1
(DOCX 39.4 kb)
Rights and permissions
About this article

Cite this article
Maiorana, A., Dionisi-Vici, C. Hyperinsulinemic hypoglycemia: clinical, molecular and therapeutical novelties. J Inherit Metab Dis 40, 531–542 (2017). https://doi.org/10.1007/s10545-017-0059-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-017-0059-x